

Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Glucose Uptake and Cell Viability
2.2. Cas9 CRISPR Gene Deletion
2.3. Quantitative PCR Assays
2.4. Western Blot
2.5. Amino Acid Supplementation Assay
2.6. Extracellular Acidification Assays
2.7. Fatty Acid Synthesis Assay
3. Results
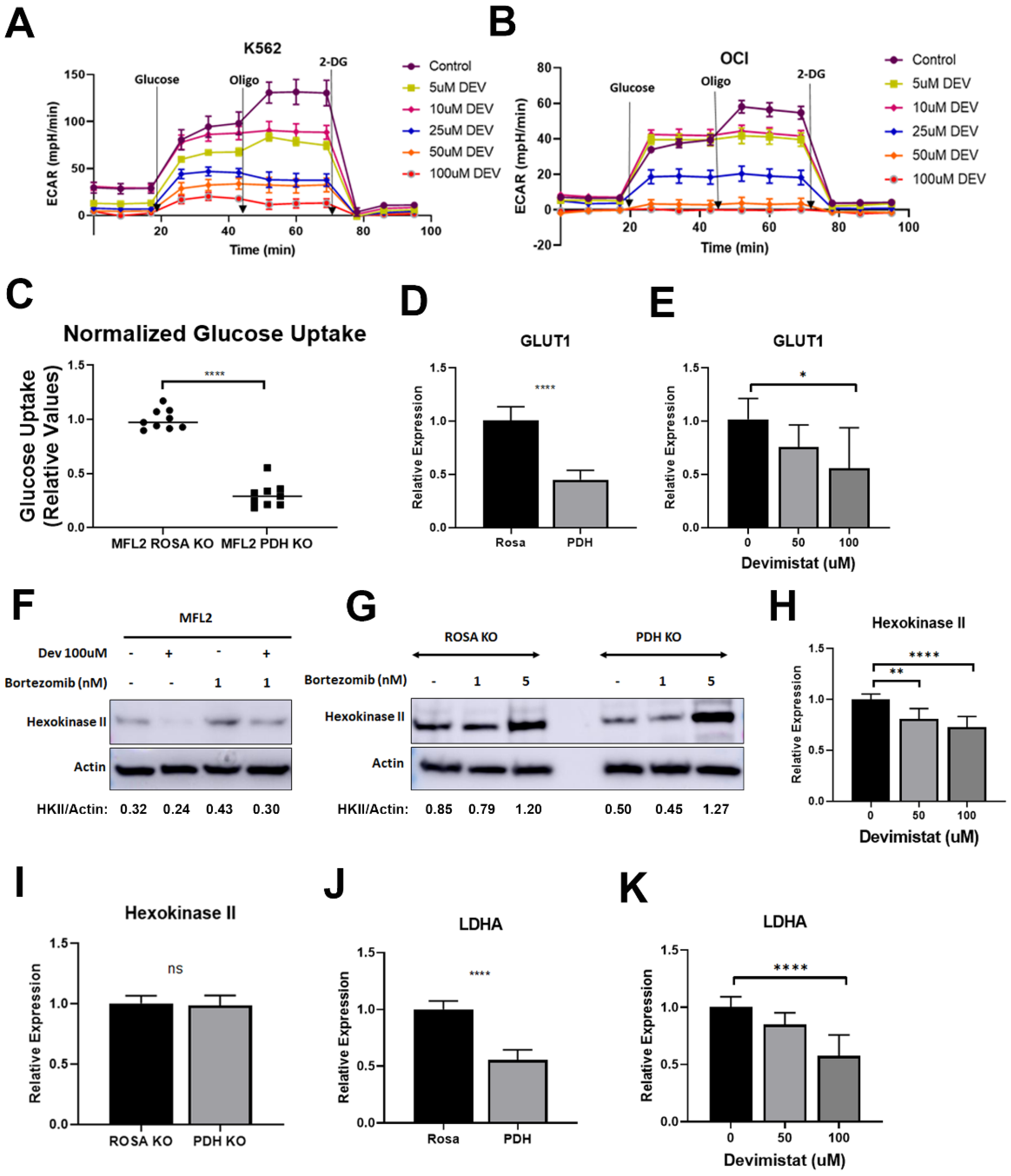
3.1. PDH Inhibition Leads to Decreased Glucose Uptake and Retention but Reliance on Residual Glycolysis
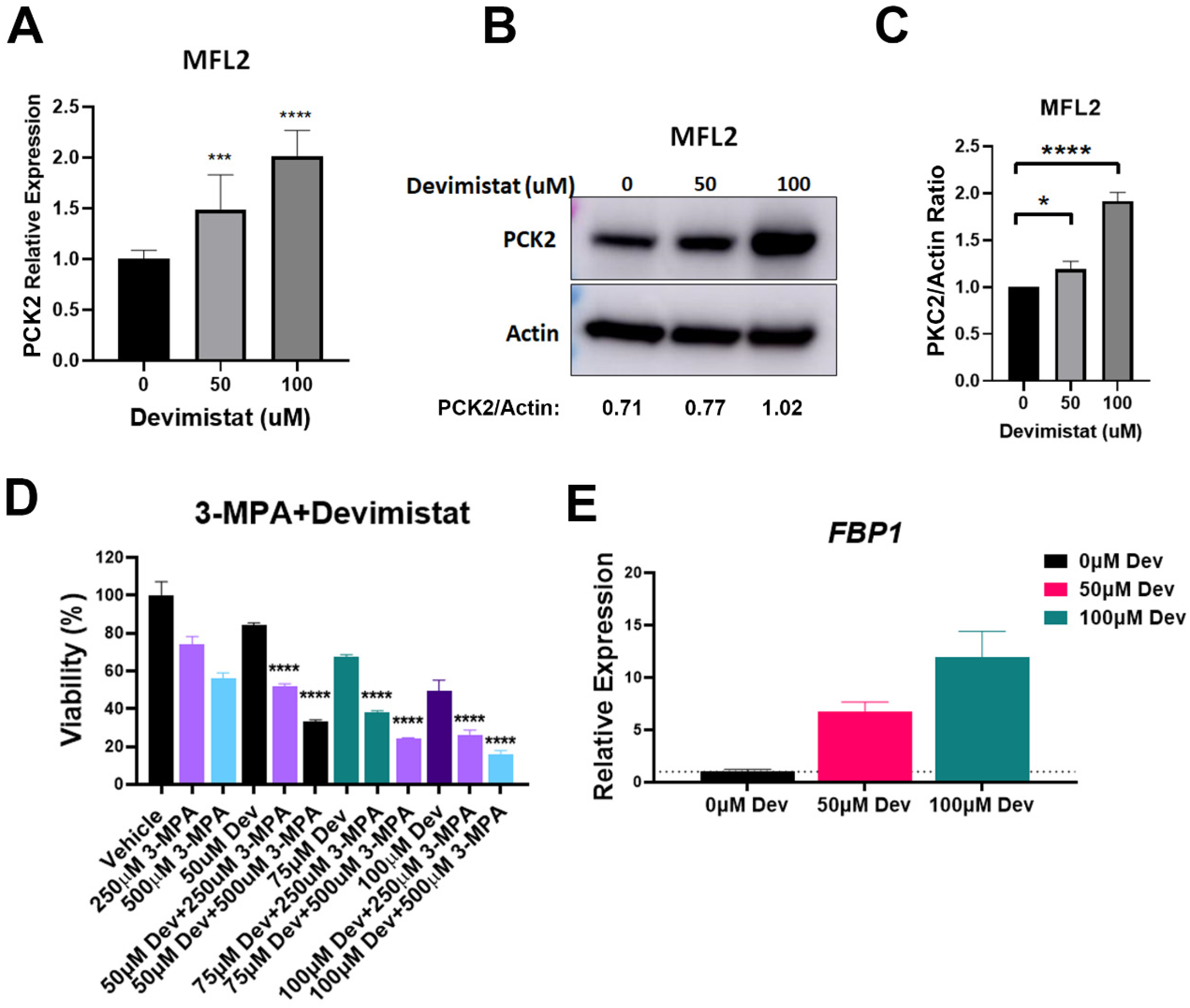
3.2. Gluconeogenesis Is a Source of Resistance to Devimistat
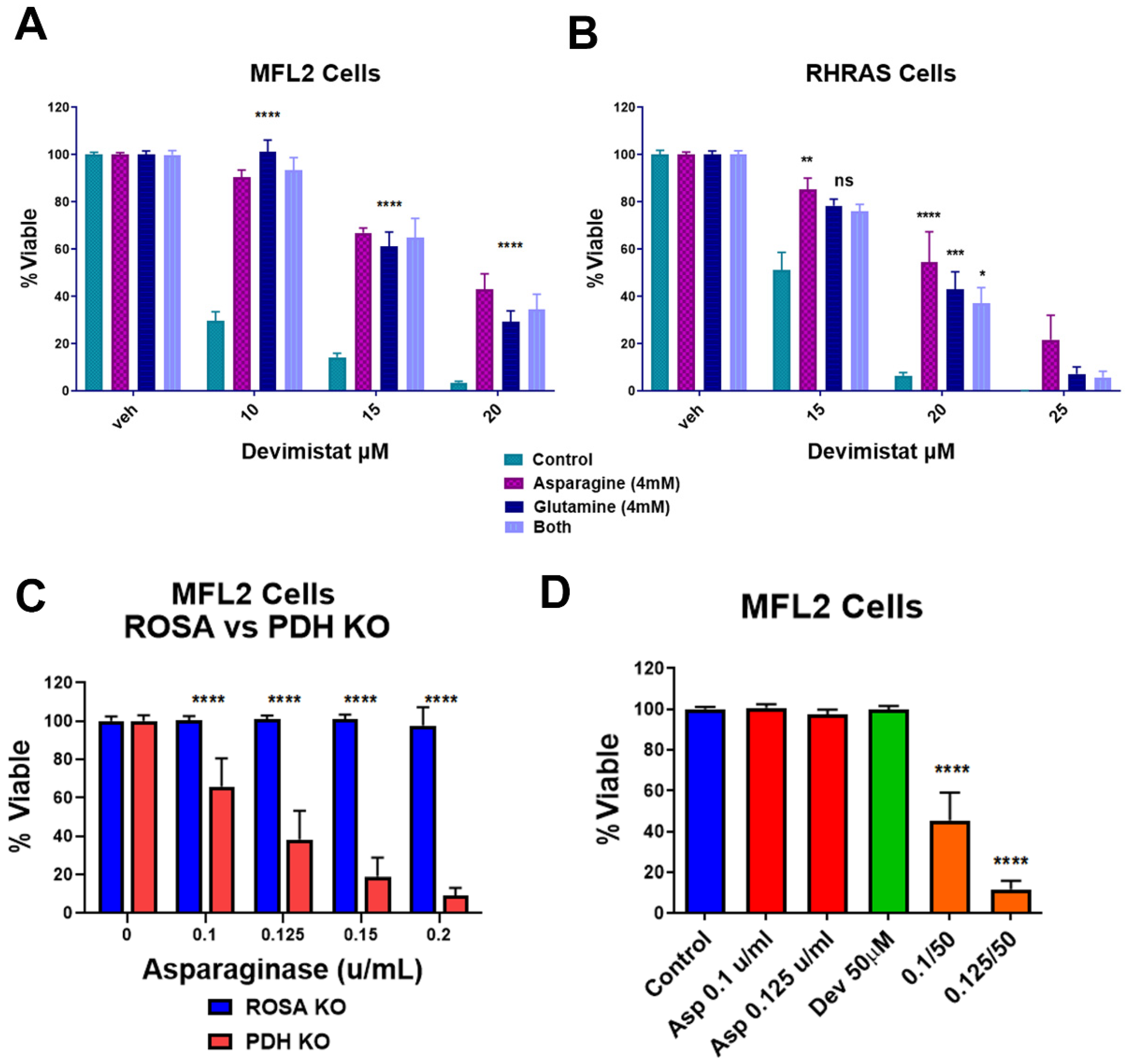
3.3. TCA Cycle Inhibition Induces Increased Glutamine and Asparagine Dependence
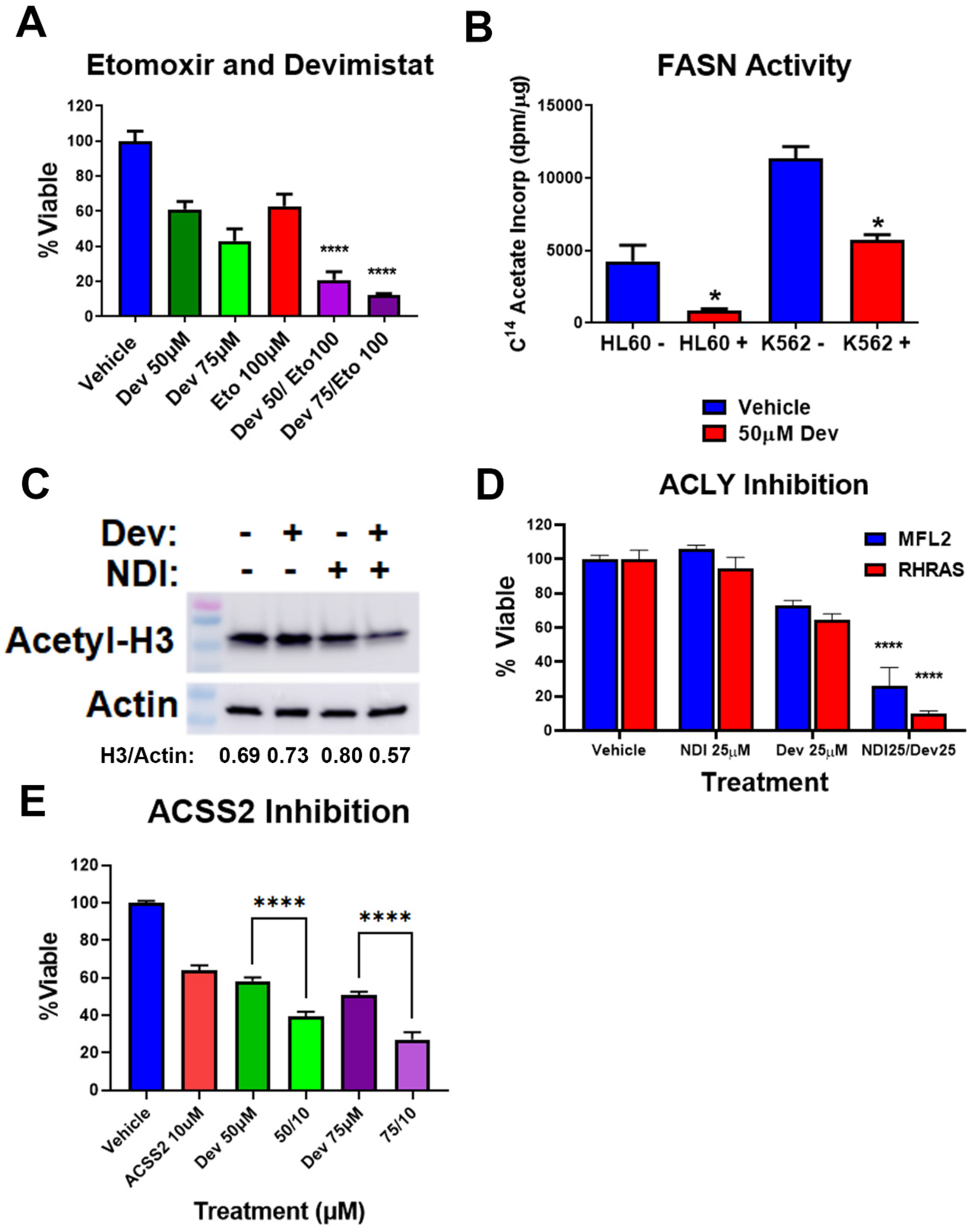
3.4. Acetyl-CoA Metabolism Is Altered by PDH Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Klepin, H.D.; Rao, A.V.; Pardee, T.S. Acute Myeloid Leukemia and Myelodysplastic Syndromes in Older Adults. J. Clin. Oncol. 2014, 32, 2541–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Zhu, D.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med. Chem. 2013, 5, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Anderson, R.; Miller, L.D.; Isom, S.; Chou, J.W.; Pladna, K.M.; Schramm, N.J.; Ellis, L.R.; Howard, D.S.; Bhave, R.R.; Manuel, M.; et al. Phase II trial of cytarabine and mitoxantrone with devimistat in acute myeloid leukemia. Nat. Commun. 2022, 13, 167. [Google Scholar] [CrossRef]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Feng, X.; Chen, Y.; Selfridge, J.E.; Gorityala, S.; Du, Z.; Wang, J.M.; Hao, Y.; Cioffi, G.; Conlon, R.A.; et al. 5-Fluorouracil Enhances the Antitumor Activity of the Glutaminase Inhibitor CB-839 against PIK3CA-Mutant Colorectal Cancers. Cancer Res 2020, 80, 4815–4827. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardaci, S.; Zheng, L.; Mackay, G.; van den Broek, N.J.F.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Bowlby, S.C.; Thomas, M.J.; D’Agostino, R.B., Jr.; Kridel, S.J. Nicotinamide Phosphoribosyl Transferase (Nampt) Is Required for De Novo Lipogenesis in Tumor Cells. PLoS ONE 2012, 7, e40195. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Tajeddine, N.; Kroemer, G. Disruption of the hexokinase–VDAC complex for tumor therapy. Oncogene 2008, 27, 4633–4635. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, C. Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Icard, P.; Wu, Z.; Fournel, L.; Coquerel, A.; Lincet, H.; Alifano, M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett. 2020, 471, 125–134. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.-H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9. [Google Scholar] [CrossRef] [Green Version]
- Leithner, K.; Hrzenjak, A.; Trötzmüller, M.; Moustafa, T.; Köfeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene 2015, 34, 1044–1050. [Google Scholar] [CrossRef]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.-C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choinière, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.e6. [Google Scholar] [CrossRef]
- Zavorka Thomas, M.E.; Lu, X.; Talebi, Z.; Jeon, J.Y.; Buelow, D.R.; Gibson, A.A.; Uddin, M.E.; Brinton, L.T.; Nguyen, J.; Collins, M.; et al. Gilteritinib Inhibits Glutamine Uptake and Utilization in FLT3-ITD–Positive AML. Mol. Cancer Ther. 2021, 20, 2207–2217. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Dorai, T.; Pinto, J.T.; Denton, T.T. The metabolic importance of the overlooked asparaginase II pathway. Anal. Biochem. 2020, 644, 114084. [Google Scholar] [CrossRef]
- Chan, W.-K.; Horvath, T.D.; Tan, L.; Link, T.; Harutyunyan, K.G.; Pontikos, M.A.; Anishkin, A.; Du, D.; Martin, L.A.; Yin, E.; et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity against Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2019, 18, 1587–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A Phase I Study of the First-in-Class Antimitochondrial Metabolism Agent, CPI-613, in Patients with Advanced Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, R.; Pladna, K.M.; Schramm, N.J.; Wheeler, F.B.; Kridel, S.; Pardee, T.S. Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia. Cancers 2023, 15, 484. https://doi.org/10.3390/cancers15020484
Anderson R, Pladna KM, Schramm NJ, Wheeler FB, Kridel S, Pardee TS. Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia. Cancers. 2023; 15(2):484. https://doi.org/10.3390/cancers15020484
Chicago/Turabian StyleAnderson, Rebecca, Kristin M. Pladna, Nathaniel J. Schramm, Frances B. Wheeler, Steven Kridel, and Timothy S. Pardee. 2023. "Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia" Cancers 15, no. 2: 484. https://doi.org/10.3390/cancers15020484
APA StyleAnderson, R., Pladna, K. M., Schramm, N. J., Wheeler, F. B., Kridel, S., & Pardee, T. S. (2023). Pyruvate Dehydrogenase Inhibition Leads to Decreased Glycolysis, Increased Reliance on Gluconeogenesis and Alternative Sources of Acetyl-CoA in Acute Myeloid Leukemia. Cancers, 15(2), 484. https://doi.org/10.3390/cancers15020484