

Lipocalin-2: A Nurturer of Tumor Progression and a Novel Candidate for Targeted Cancer Therapy

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Role of Iron in Cancer Progression
2.1. Lipocalin-2 as an Alternative Iron Regulator
2.2. Polarization of Tumor-Associated Macrophages by Iron
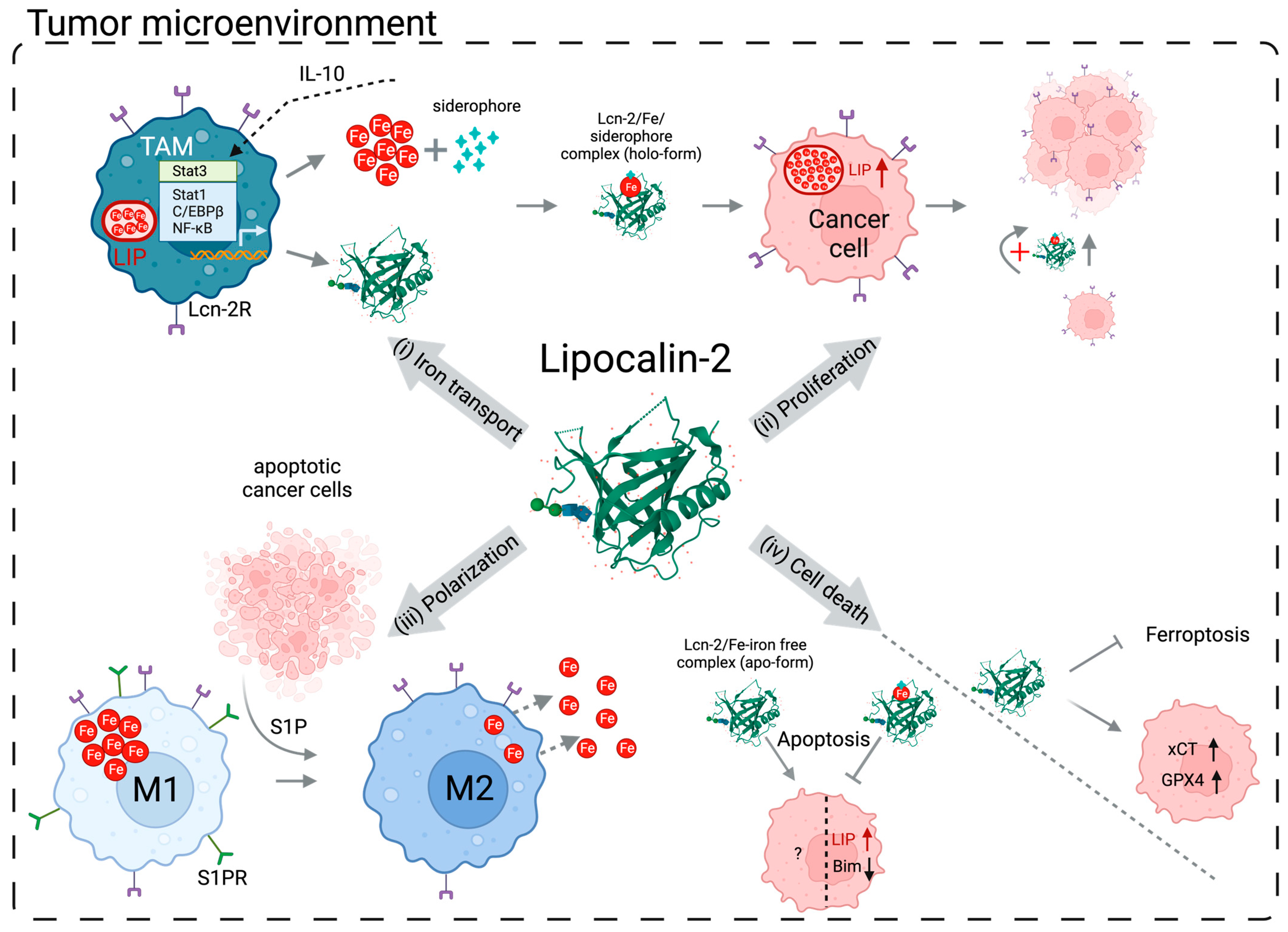
3. Lipocalin-2 in Cancer Progression
4. Cell Death and Lipocalin-2
4.1. Implication of Lipocalin-2 in Apoptosis
4.2. Implication of Lipocalin-2 in Ferroptosis
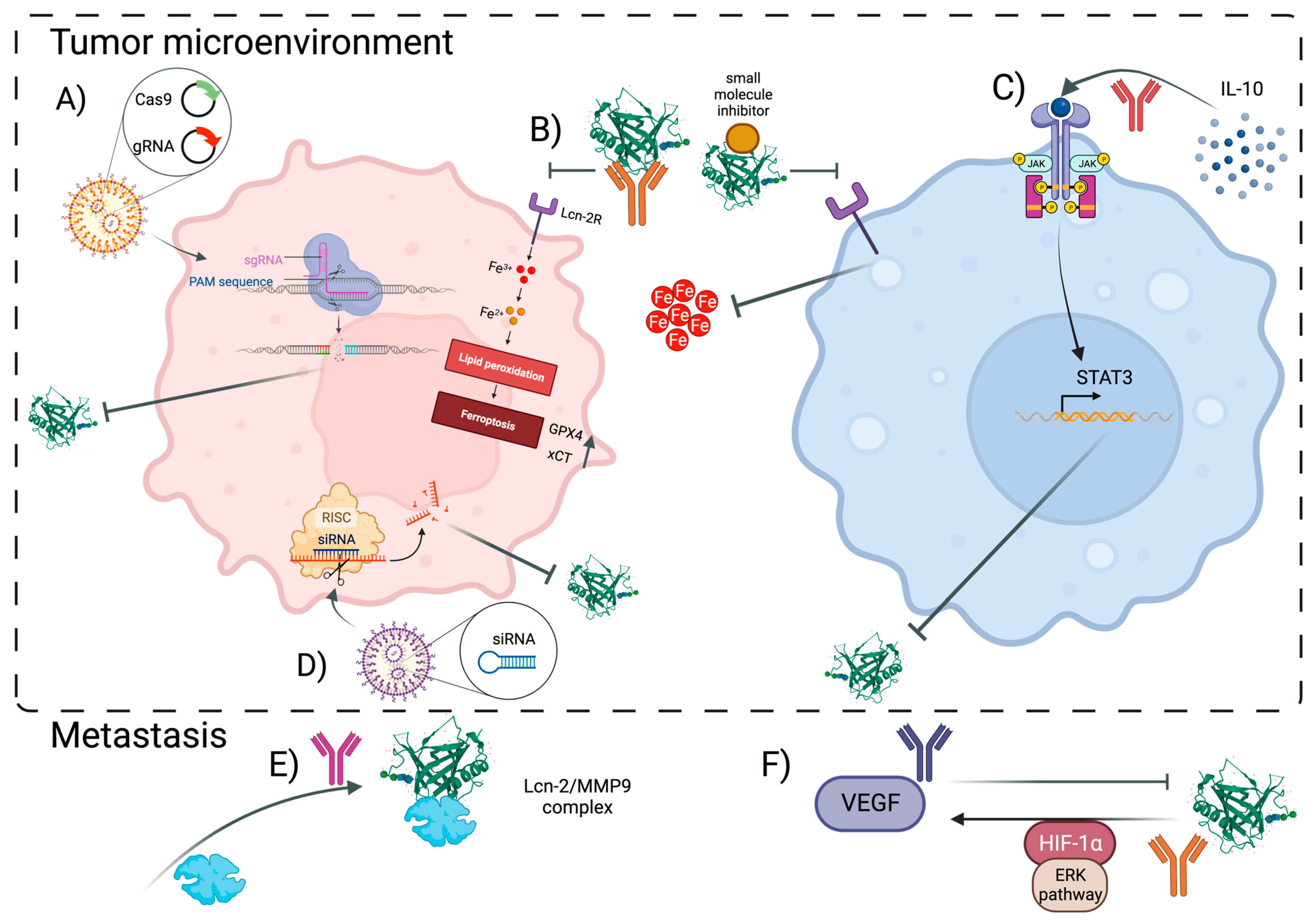
5. Lipocalin-2 as a Potential Therapeutic Target
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiang, P.; Sinha, S.; Aldape, K.; Hannenhalli, S.; Sahinalp, C.; Ruppin, E. Big data in basic and translational cancer research. Nat. Rev. Cancer 2022, 22, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef]
- Campbell, J.A. Effects of Precipitated Silica and of Iron Oxide on the Incidence of Primary Lung Tumours in Mice. Br. Med. J. 1940, 2, 275–280. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Duan, X.; He, K.; Li, J.; Cheng, M.; Song, H.; Liu, J.; Liu, P. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 105–114. [Google Scholar] [PubMed]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Clifton, M.; Hoette, T.M.; Mori, K.; Deng, S.X.; Qiu, A.; Viltard, M.; Williams, D.; Paragas, N.; Leete, T.; et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat. Chem. Biol. 2010, 6, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, L.R.; Hart, D.O.; Goetz, D.H.; Green, M.R. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 2010, 141, 1006–1017. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kashima, H.; Yamada, Y.; Kobara, H.; Asaka, R.; Ando, H.; Higuchi, S.; Ida, K.; Mvunta, D.H.; Shiozawa, T. Lipocalin 2 Enhances Migration and Resistance against Cisplatin in Endometrial Carcinoma Cells. PLoS ONE 2016, 11, e0155220. [Google Scholar] [CrossRef]
- Shiiba, M.; Saito, K.; Fushimi, K.; Ishigami, T.; Shinozuka, K.; Nakashima, D.; Kouzu, Y.; Koike, H.; Kasamatsu, A.; Sakamoto, Y.; et al. Lipocalin-2 is associated with radioresistance in oral cancer and lung cancer cells. Int. J. Oncol. 2013, 42, 1197–1204. [Google Scholar] [CrossRef]
- Mertens, C.; Mora, J.; Oren, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmstrom, P.; Forsare, C.; Ferno, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef]
- Baffy, G.; Miyashita, T.; Williamson, J.R.; Reed, J.C. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem. 1993, 268, 6511–6519. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Teodoro, J.G.; Richard, F.A.; Green, M.R. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science 2001, 293, 829–834. [Google Scholar] [CrossRef]
- Meier, J.K.; Schnetz, M.; Beck, S.; Schmid, T.; Dominguez, M.; Kalinovic, S.; Daiber, A.; Brune, B.; Jung, M. Iron-Bound Lipocalin-2 Protects Renal Cell Carcinoma from Ferroptosis. Metabolites 2021, 11, 329. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Jeong, E.A.; Lee, J.Y.; An, H.S.; Jang, H.M.; Ahn, Y.J.; Lee, J.; Kim, K.E.; Roh, G.S. Lipocalin-2 Deficiency Reduces Oxidative Stress and Neuroinflammation and Results in Attenuation of Kainic Acid-Induced Hippocampal Cell Death. Antioxidants 2021, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Sanchez, G.S.; Pita-Grisanti, V.; Quinones-Diaz, B.; Gumpper, K.; Cruz-Monserrate, Z.; Vivas-Mejia, P.E. Biological Functions and Therapeutic Potential of Lipocalin 2 in Cancer. Int. J. Mol. Sci. 2020, 21, 4365. [Google Scholar] [CrossRef]
- Wang, Y.P.; Yu, G.R.; Lee, M.J.; Lee, S.Y.; Chu, I.S.; Leem, S.H.; Kim, D.G. Lipocalin-2 negatively modulates the epithelial-to-mesenchymal transition in hepatocellular carcinoma through the epidermal growth factor (TGF-beta1)/Lcn2/Twist1 pathway. Hepatology 2013, 58, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Kubben, F.J.; Sier, C.F.; Hawinkels, L.J.; Tschesche, H.; van Duijn, W.; Zuidwijk, K.; van der Reijden, J.J.; Hanemaaijer, R.; Griffioen, G.; Lamers, C.B.; et al. Clinical evidence for a protective role of lipocalin-2 against MMP-9 autodegradation and the impact for gastric cancer. Eur. J. Cancer 2007, 43, 1869–1876. [Google Scholar] [CrossRef]
- Fernandez, C.A.; Yan, L.; Louis, G.; Yang, J.; Kutok, J.L.; Moses, M.A. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin. Cancer Res. 2005, 11, 5390–5395. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef]
- Inoue, S.; Kawanishi, S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987, 47, 6522–6527. [Google Scholar]
- Pfeifhofer-Obermair, C.; Tymoszuk, P.; Petzer, V.; Weiss, G.; Nairz, M. Iron in the Tumor Microenvironment-Connecting the Dots. Front. Oncol. 2018, 8, 549. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fischer-Fodor, E.; Miklasova, N.; Berindan-Neagoe, I.; Saha, B. Iron, inflammation and invasion of cancer cells. Clujul Med. 2015, 88, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Maccio, A.; Madeddu, C.; Gramignano, G.; Mulas, C.; Tanca, L.; Cherchi, M.C.; Floris, C.; Omoto, I.; Barracca, A.; Ganz, T. The role of inflammation, iron, and nutritional status in cancer-related anemia: Results of a large, prospective, observational study. Haematologica 2015, 100, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Gramignano, G.; Astara, G.; Demontis, R.; Sanna, E.; Atzeni, V.; Maccio, A. Pathogenesis and Treatment Options of Cancer Related Anemia: Perspective for a Targeted Mechanism-Based Approach. Front. Physiol. 2018, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Miharada, K.; Hiroyama, T.; Sudo, K.; Danjo, I.; Nagasawa, T.; Nakamura, Y. Lipocalin 2-mediated growth suppression is evident in human erythroid and monocyte/macrophage lineage cells. J. Cell Physiol. 2008, 215, 526–537. [Google Scholar] [CrossRef]
- Lim, D.; Jeong, J.H.; Song, J. Lipocalin 2 regulates iron homeostasis, neuroinflammation, and insulin resistance in the brains of patients with dementia: Evidence from the current literature. CNS Neurosci. Ther. 2021, 27, 883–894. [Google Scholar] [CrossRef]
- Nahm, C.H.; Lee, M.H.; Fujii, T.; Fujii, N.; Choi, J.W. Lipocalin-2, Soluble Transferrin Receptor, and Erythropoietin in Anemia During Mild Renal Dysfunction. Int. J. Gen. Med. 2023, 16, 3603–3612. [Google Scholar] [CrossRef]
- Bhurosy, T.; Jishan, A.; Boland, P.M.; Lee, Y.H.; Heckman, C.J. Underdiagnosis of iron deficiency anemia among patients with colorectal cancer: An examination of electronic medical records. BMC Cancer 2022, 22, 435. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Chen, D.; Jiang, Y.; Huang, W.; Ouyang, H.; Xing, W.; Zeng, M.; Xie, X.; Zeng, W. Impact of preoperative anemia on relapse and survival in breast cancer patients. BMC Cancer 2014, 14, 844. [Google Scholar] [CrossRef]
- Busti, F.; Marchi, G.; Ugolini, S.; Castagna, A.; Girelli, D. Anemia and Iron Deficiency in Cancer Patients: Role of Iron Replacement Therapy. Pharmaceuticals 2018, 11, 94. [Google Scholar] [CrossRef]
- Groopman, J.E.; Itri, L.M. Chemotherapy-induced anemia in adults: Incidence and treatment. J. Natl. Cancer Inst. 1999, 91, 1616–1634. [Google Scholar] [CrossRef]
- Kwok, J.C.; Richardson, D.R. The iron metabolism of neoplastic cells: Alterations that facilitate proliferation? Crit. Rev. Oncol. Hematol. 2002, 42, 65–78. [Google Scholar] [CrossRef]
- Jung, M.; Mertens, C.; Tomat, E.; Brune, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, S.; Wang, Y.; Zhu, Y.; Wang, J.; Guo, J. HMOX1 pathway signature predicts clinical benefit from immunotherapy plus tyrosine kinase inhibitor therapy in advanced renal cell carcinoma. Cancer Med. 2023, 12, 10512–10525. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.L.; Versini, A.; Khene, N.; Gaillet, C.; Caneque, T.; Muller, S.; Rodriguez, R. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chemistry 2020, 26, 7369–7373. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Yang, Q.; Huang, X. Src regulates Tyr(20) phosphorylation of transferrin receptor-1 and potentiates breast cancer cell survival. J. Biol. Chem. 2011, 286, 35708–35715. [Google Scholar] [CrossRef] [PubMed]
- Vela, D.; Vela-Gaxha, Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp. Mol. Med. 2018, 50, e436. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Hu, L.; Lu, K.; Wang, Y.; Xie, Y.; Gao, L.; Yang, G.; Xie, B.; He, W.; Chen, G.; et al. Ferroportin downregulation promotes cell proliferation by modulating the Nrf2-miR-17-5p axis in multiple myeloma. Cell Death Dis. 2019, 10, 624. [Google Scholar] [CrossRef]
- Yang, J.; Moses, M.A. Lipocalin 2: A multifaceted modulator of human cancer. Cell Cycle 2009, 8, 2347–2352. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef]
- Bauer, M.; Eickhoff, J.C.; Gould, M.N.; Mundhenke, C.; Maass, N.; Friedl, A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008, 108, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Schnetz, M.; Rehwald, C.; Grein, S.; Elwakeel, E.; Weigert, A.; Brune, B.; Jung, M. Iron-Bound Lipocalin-2 from Tumor-Associated Macrophages Drives Breast Cancer Progression Independent of Ferroportin. Metabolites 2021, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Tymoszuk, P.; Nairz, M.; Brigo, N.; Petzer, V.; Heeke, S.; Kircher, B.; Hermann-Kleiter, N.; Klepsch, V.; Theurl, I.; Weiss, G.; et al. Iron Supplementation Interferes With Immune Therapy of Murine Mammary Carcinoma by Inhibiting Anti-Tumor T Cell Function. Front. Oncol. 2020, 10, 584477. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Remsik, J.; Kiseliovas, V.; Derderian, C.; Sener, U.; Alghader, M.; Saadeh, F.; Nikishina, K.; Bale, T.; Iacobuzio-Donahue, C.; et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020, 369, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Rehwald, C.; Schnetz, M.; Urbschat, A.; Mertens, C.; Meier, J.K.; Bauer, R.; Baer, P.; Winslow, S.; Roos, F.C.; Zwicker, K.; et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br. J. Cancer 2020, 122, 421–433. [Google Scholar] [CrossRef]
- Ibrahim, O.; O’Sullivan, J. Iron chelators in cancer therapy. Biometals 2020, 33, 201–215. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef]
- Larrick, J.W.; Hyman, E.S. Acquired iron-deficiency anemia caused by an antibody against the transferrin receptor. N. Engl. J. Med. 1984, 311, 214–218. [Google Scholar] [CrossRef]
- Moura, I.C.; Lepelletier, Y.; Arnulf, B.; England, P.; Baude, C.; Beaumont, C.; Bazarbachi, A.; Benhamou, M.; Monteiro, R.C.; Hermine, O. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood 2004, 103, 1838–1845. [Google Scholar] [CrossRef]
- Paterson, J.; Webster, C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016, 20, 49–52. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Xu, S.; Bell, M.; Martling, C.R.; Venge, P. Immunoassays distinguishing between HNL/NGAL released in urine from kidney epithelial cells and neutrophils. Clin. Chim. Acta 2012, 413, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, S.; Roushandeh, A.M.; Ahmadzadeh, E.; Jahanian-Najafabadi, A.; Roudkenar, M.H. Implication and role of neutrophil gelatinase-associated lipocalin in cancer: Lipocalin-2 as a potential novel emerging comprehensive therapeutic target for a variety of cancer types. Mol. Biol. Rep. 2020, 47, 2327–2346. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Schroder, S.K.; Gasterich, N.; Weiskirchen, S.; Weiskirchen, R. Lipocalin 2 receptors: Facts, fictions, and myths. Front. Immunol. 2023, 14, 1229885. [Google Scholar] [CrossRef]
- Hvidberg, V.; Jacobsen, C.; Strong, R.K.; Cowland, J.B.; Moestrup, S.K.; Borregaard, N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005, 579, 773–777. [Google Scholar] [CrossRef]
- Cabedo Martinez, A.I.; Weinhaupl, K.; Lee, W.K.; Wolff, N.A.; Storch, B.; Zerko, S.; Konrat, R.; Kozminski, W.; Breuker, K.; Thevenod, F.; et al. Biochemical and Structural Characterization of the Interaction between the Siderocalin NGAL/LCN2 (Neutrophil Gelatinase-associated Lipocalin/Lipocalin 2) and the N-terminal Domain of Its Endocytic Receptor SLC22A17. J. Biol. Chem. 2016, 291, 2917–2930. [Google Scholar] [CrossRef]
- Jung, M.; Oren, B.; Mora, J.; Mertens, C.; Dziumbla, S.; Popp, R.; Weigert, A.; Grossmann, N.; Fleming, I.; Brune, B. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci. Signal. 2016, 9, ra64. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef]
- Cruz, D.N.; Gaiao, S.; Maisel, A.; Ronco, C.; Devarajan, P. Neutrophil gelatinase-associated lipocalin as a biomarker of cardiovascular disease: A systematic review. Clin. Chem. Lab. Med. 2012, 50, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Lee, H.T.; Rapoport, D.; Drexler, I.R.; Foster, K.; Yang, J.; Schmidt-Ott, K.M.; Chen, X.; Li, J.Y.; Weiss, S.; et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Investig. 2005, 115, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.W.; Yang, Q.; Mody, N.; Graham, T.E.; Hsu, C.H.; Xu, Z.; Houstis, N.E.; Kahn, B.B.; Rosen, E.D. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 2007, 56, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Weng, Y.C.; Chiang, I.C.; Huang, Y.T.; Liao, Y.C.; Chen, Y.C.; Kao, C.Y.; Liu, Y.L.; Lee, T.H.; Chou, W.H. Neutralization of Lipocalin-2 Diminishes Stroke-Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 6253. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Xiao, C.; Fu, X.; Wang, Y.; Liu, H.; Jiang, Y.; Zhao, Z.; You, F. Transferrin receptor regulates malignancies and the stemness of hepatocellular carcinoma-derived cancer stem-like cells by affecting iron accumulation. PLoS ONE 2020, 15, e0243812. [Google Scholar] [CrossRef]
- Leseigneur, C.; Le-Bury, P.; Pizarro-Cerda, J.; Dussurget, O. Emerging Evasion Mechanisms of Macrophage Defenses by Pathogenic Bacteria. Front. Cell Infect. Microbiol. 2020, 10, 577559. [Google Scholar] [CrossRef]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B., Jr.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the maintenance of homeostasis. Cell Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Tang, X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013, 332, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Van Overmeire, E.; Laoui, D.; Keirsse, J.; Van Ginderachter, J.A.; Sarukhan, A. Mechanisms driving macrophage diversity and specialization in distinct tumor microenvironments and parallelisms with other tissues. Front. Immunol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Galan, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, 709–725. [Google Scholar] [CrossRef]
- Zhang, N.; Kim, S.H.; Gainullina, A.; Erlich, E.C.; Onufer, E.J.; Kim, J.; Czepielewski, R.S.; Helmink, B.A.; Dominguez, J.R.; Saunders, B.T.; et al. LYVE1+ macrophages of murine peritoneal mesothelium promote omentum-independent ovarian tumor growth. J. Exp. Med. 2021, 218, e20210924. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef]
- Torroella-Kouri, M.; Silvera, R.; Rodriguez, D.; Caso, R.; Shatry, A.; Opiela, S.; Ilkovitch, D.; Schwendener, R.A.; Iragavarapu-Charyulu, V.; Cardentey, Y.; et al. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009, 69, 4800–4809. [Google Scholar] [CrossRef]
- Lahmar, Q.; Keirsse, J.; Laoui, D.; Movahedi, K.; Van Overmeire, E.; Van Ginderachter, J.A. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim. Biophys. Acta 2016, 1865, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Urbschat, A.; Thiemens, A.K.; Mertens, C.; Rehwald, C.; Meier, J.K.; Baer, P.C.; Jung, M. Macrophage-secreted Lipocalin-2 Promotes Regeneration of Injured Primary Murine Renal Tubular Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 2038. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.N.; Jung, M.; Weigert, A.; Brune, B. S1P Provokes Tumor Lymphangiogenesis via Macrophage-Derived Mediators Such as IL-1beta or Lipocalin-2. Mediators Inflamm. 2017, 2017, 7510496. [Google Scholar] [CrossRef]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef]
- Marques, O.; Porto, G.; Rema, A.; Faria, F.; Cruz Paula, A.; Gomez-Lazaro, M.; Silva, P.; Martins da Silva, B.; Lopes, C. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016, 16, 187. [Google Scholar] [CrossRef]
- Leftin, A.; Ben-Chetrit, N.; Joyce, J.A.; Koutcher, J.A. Imaging endogenous macrophage iron deposits reveals a metabolic biomarker of polarized tumor macrophage infiltration and response to CSF1R breast cancer immunotherapy. Sci. Rep. 2019, 9, 857. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, J.W.; Ylaya, K.; Chung, E.J.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.Y.; Hewitt, S.M. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef]
- Provatopoulou, X.; Gounaris, A.; Kalogera, E.; Zagouri, F.; Flessas, I.; Goussetis, E.; Nonni, A.; Papassotiriou, I.; Zografos, G. Circulating levels of matrix metalloproteinase-9 (MMP-9), neutrophil gelatinase-associated lipocalin (NGAL) and their complex MMP-9/NGAL in breast cancer disease. BMC Cancer 2009, 9, 390. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Chang, C.J.; Cheng, J.S. Survival, treatment regimens and medical costs of women newly diagnosed with metastatic triple-negative breast cancer. Sci. Rep. 2022, 12, 729. [Google Scholar] [CrossRef] [PubMed]
- Stoesz, S.P.; Friedl, A.; Haag, J.D.; Lindstrom, M.J.; Clark, G.M.; Gould, M.N. Heterogeneous expression of the lipocalin NGAL in primary breast cancers. Int. J. Cancer 1998, 79, 565–572. [Google Scholar] [CrossRef]
- Wenners, A.S.; Mehta, K.; Loibl, S.; Park, H.; Mueller, B.; Arnold, N.; Hamann, S.; Weimer, J.; Ataseven, B.; Darb-Esfahani, S.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) predicts response to neoadjuvant chemotherapy and clinical outcome in primary human breast cancer. PLoS ONE 2012, 7, e45826. [Google Scholar] [CrossRef]
- Yang, J.; Bielenberg, D.R.; Rodig, S.J.; Doiron, R.; Clifton, M.C.; Kung, A.L.; Strong, R.K.; Zurakowski, D.; Moses, M.A. Lipocalin 2 promotes breast cancer progression. Proc. Natl. Acad. Sci. USA 2009, 106, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Villodre, E.S.; Hu, X.; Larson, R.; Finetti, P.; Gomez, K.; Balema, W.; Stecklein, S.R.; Santiago-Sanchez, G.; Krishnamurthy, S.; Song, J.; et al. Lipocalin 2 promotes inflammatory breast cancer tumorigenesis and skin invasion. Mol. Oncol. 2021, 15, 2752–2765. [Google Scholar] [CrossRef] [PubMed]
- Malone, M.K.; Smrekar, K.; Park, S.; Blakely, B.; Walter, A.; Nasta, N.; Park, J.; Considine, M.; Danilova, L.V.; Pandey, N.B.; et al. Cytokines secreted by stromal cells in TNBC microenvironment as potential targets for cancer therapy. Cancer Biol. Ther. 2020, 21, 560–569. [Google Scholar] [CrossRef]
- Morales-Valencia, J.; Lau, L.; Marti-Nin, T.; Ozerdem, U.; David, G. Therapy-induced senescence promotes breast cancer cells plasticity by inducing Lipocalin-2 expression. Oncogene 2022, 41, 4361–4370. [Google Scholar] [CrossRef]
- Pitteri, S.J.; Faca, V.M.; Kelly-Spratt, K.S.; Kasarda, A.E.; Wang, H.; Zhang, Q.; Newcomb, L.; Krasnoselsky, A.; Paczesny, S.; Choi, G.; et al. Plasma proteome profiling of a mouse model of breast cancer identifies a set of up-regulated proteins in common with human breast cancer cells. J. Proteome Res. 2008, 7, 1481–1489. [Google Scholar] [CrossRef]
- Berger, T.; Cheung, C.C.; Elia, A.J.; Mak, T.W. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 2995–3000. [Google Scholar] [CrossRef]
- Oren, B.; Urosevic, J.; Mertens, C.; Mora, J.; Guiu, M.; Gomis, R.R.; Weigert, A.; Schmid, T.; Grein, S.; Brune, B.; et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J. Pathol. 2016, 239, 274–285. [Google Scholar] [CrossRef]
- Chiang, K.C.; Yeh, T.S.; Wu, R.C.; Pang, J.S.; Cheng, C.T.; Wang, S.Y.; Juang, H.H.; Yeh, C.N. Lipocalin 2 (LCN2) is a promising target for cholangiocarcinoma treatment and bile LCN2 level is a potential cholangiocarcinoma diagnostic marker. Sci. Rep. 2016, 6, 36138. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.; Bell, D.; Tao, J.; Preziosi, M.; Pradhan-Sundd, T.; Singh, S.; Poddar, M.; Luo, J.; Ranganathan, S.; Chikina, M.; et al. Hepatocyte-Derived Lipocalin 2 Is a Potential Serum Biomarker Reflecting Tumor Burden in Hepatoblastoma. Am. J. Pathol. 2018, 188, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mertens, C.; Bauer, R.; Rehwald, C.; Brune, B. Lipocalin-2 and iron trafficking in the tumor microenvironment. Pharmacol. Res. 2017, 120, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Tomasello, B.; Lavoro, A.; Falzone, L.; Gattuso, G.; Russo, A.; Paratore, S.; McCubrey, J.A.; Libra, M. Bioinformatic analysis of the LCN2-SLC22A17-MMP9 network in cancer: The role of DNA methylation in the modulation of tumor microenvironment. Front. Cell Dev. Biol. 2022, 10, 945586. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.L.; Lee, S.T.; Min, I.S.; Park, Y.R.; Lee, J.H.; Kim, D.G.; Kim, S.W. Lipocalin 2 negatively regulates cell proliferation and epithelial to mesenchymal transition through changing metabolic gene expression in colorectal cancer. Cancer Sci. 2017, 108, 2176–2186. [Google Scholar] [CrossRef]
- Tong, Z.; Kunnumakkara, A.B.; Wang, H.; Matsuo, Y.; Diagaradjane, P.; Harikumar, K.B.; Ramachandran, V.; Sung, B.; Chakraborty, A.; Bresalier, R.S.; et al. Neutrophil gelatinase-associated lipocalin: A novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 2008, 68, 6100–6108. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, E.K.; Lee, K.J.; Hong, S.W.; Yoon, Y.; Kim, J.S. Ectopic expression of neutrophil gelatinase-associated lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int. J. Cancer 2006, 118, 2490–2497. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2017, 8, 992. [Google Scholar] [CrossRef]
- Richardson, D.R. 24p3 and its receptor: Dawn of a new iron age? Cell 2005, 123, 1175–1177. [Google Scholar] [CrossRef]
- Rahimi, S.; Roushandeh, A.M.; Ebrahimi, A.; Samadani, A.A.; Kuwahara, Y.; Roudkenar, M.H. CRISPR/Cas9-mediated knockout of Lcn2 effectively enhanced CDDP-induced apoptosis and reduced cell migration capacity of PC3 cells. Life Sci. 2019, 231, 116586. [Google Scholar] [CrossRef] [PubMed]
- Han, M.Y.; Nie, J.W.; Li, Y.Y.; Zhu, Y.Z.; Wu, G. Downregulation of NGAL is Required for the Inhibition of Proliferation and the Promotion of Apoptosis of Human Gastric Cancer MGC-803 Cells. Cell Physiol. Biochem. 2018, 50, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, X.; Han, Y.; Wang, Y. The effect of lipocalin-2 (LCN2) on apoptosis: A proteomics analysis study in an LCN2 deficient mouse model. BMC Genomics 2021, 22, 892. [Google Scholar] [CrossRef]
- Lin, H.H.; Liao, C.J.; Lee, Y.C.; Hu, K.H.; Meng, H.W.; Chu, S.T. Lipocalin-2-induced cytokine production enhances endometrial carcinoma cell survival and migration. Int. J. Biol. Sci. 2011, 7, 74–86. [Google Scholar] [CrossRef]
- Xu, G.; Ahn, J.; Chang, S.; Eguchi, M.; Ogier, A.; Han, S.; Park, Y.; Shim, C.; Jang, Y.; Yang, B.; et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J. Biol. Chem. 2012, 287, 4808–4817. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Kim, S.; Park, J.Y.; Lee, W.H.; Mori, K.; Kim, S.H.; Kim, I.K.; Suk, K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J. Immunol. 2007, 179, 3231–3241. [Google Scholar] [CrossRef]
- Dong, S.; Li, X.; Jiang, W.; Chen, Z.; Zhou, W. Current understanding of ferroptosis in the progression and treatment of pancreatic cancer. Cancer Cell Int. 2021, 21, 480. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Schreiber, S.L.; Conrad, M. Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol. Cell 2022, 82, 728–740. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Choudhary, B.S.; Shah, S.G.; Khapare, N.; Dwivedi, N.; Gaikwad, A.; Joshi, N.; Raichanna, J.; Basu, S.; Gurjar, M.; et al. Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int. J. Cancer 2021, 149, 1495–1511. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- De Bundel, D.; Schallier, A.; Loyens, E.; Fernando, R.; Miyashita, H.; Van Liefferinge, J.; Vermoesen, K.; Bannai, S.; Sato, H.; Michotte, Y.; et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci. 2011, 31, 5792–5803. [Google Scholar] [CrossRef]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A targetable LIFR-NF-kappaB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef]
- Guo, P.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel. Proc. Natl. Acad. Sci. USA 2019, 116, 18295–18303. [Google Scholar] [CrossRef]
- Leng, X.; Ding, T.; Lin, H.; Wang, Y.; Hu, L.; Hu, J.; Feig, B.; Zhang, W.; Pusztai, L.; Symmans, W.F.; et al. Inhibition of lipocalin 2 impairs breast tumorigenesis and metastasis. Cancer Res. 2009, 69, 8579–8584. [Google Scholar] [CrossRef] [PubMed]
- Fougere, M.; Gaudineau, B.; Barbier, J.; Guaddachi, F.; Feugeas, J.P.; Auboeuf, D.; Jauliac, S. NFAT3 transcription factor inhibits breast cancer cell motility by targeting the Lipocalin 2 gene. Oncogene 2010, 29, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Gwira, J.A.; Wei, F.; Ishibe, S.; Ueland, J.M.; Barasch, J.; Cantley, L.G. Expression of neutrophil gelatinase-associated lipocalin regulates epithelial morphogenesis in vitro. J. Biol. Chem. 2005, 280, 7875–7882. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Wang, J.; Gao, W.; Lin, Y.; Jin, W.; Chang, G.; Wang, R.; Li, Q.; Ma, L.; et al. C/EBP zeta targets to neutrophil gelatinase-associated lipocalin (NGAL) as a repressor for metastasis of MDA-MB-231 cells. Biochim. Biophys. Acta 2011, 1813, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Sanchez, G.S.; Noriega-Rivera, R.; Hernandez-O’Farrill, E.; Valiyeva, F.; Quinones-Diaz, B.; Villodre, E.S.; Debeb, B.G.; Rosado-Albacarys, A.; Vivas-Mejia, P.E. Targeting Lipocalin-2 in Inflammatory Breast Cancer Cells with Small Interference RNA and Small Molecule Inhibitors. Int. J. Mol. Sci. 2021, 22, 8581. [Google Scholar] [CrossRef]
- Jung, M.; Weigert, A.; Tausendschon, M.; Mora, J.; Oren, B.; Sola, A.; Hotter, G.; Muta, T.; Brune, B. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol. Cell Biol. 2012, 32, 3938–3948. [Google Scholar] [CrossRef]
- Warszawska, J.M.; Gawish, R.; Sharif, O.; Sigel, S.; Doninger, B.; Lakovits, K.; Mesteri, I.; Nairz, M.; Boon, L.; Spiel, A.; et al. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J. Clin. Investig. 2013, 123, 3363–3372. [Google Scholar] [CrossRef]
- Yang, J.; McNeish, B.; Butterfield, C.; Moses, M.A. Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB J. 2013, 27, 45–50. [Google Scholar] [CrossRef]
- Hu, C.; Yang, K.; Li, M.; Huang, W.; Zhang, F.; Wang, H. Lipocalin 2: A potential therapeutic target for breast cancer metastasis. Onco Targets Ther. 2018, 11, 8099–8106. [Google Scholar] [CrossRef]
- Nuntagowat, C.; Leelawat, K.; Tohtong, R. NGAL knockdown by siRNA in human cholangiocarcinoma cells suppressed invasion by reducing NGAL/MMP-9 complex formation. Clin. Exp. Metastasis 2010, 27, 295–305. [Google Scholar] [CrossRef]
- Guo, P.; You, J.O.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. Inhibiting metastatic breast cancer cell migration via the synergy of targeted, pH-triggered siRNA delivery and chemokine axis blockade. Mol. Pharm. 2014, 11, 755–765. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Živalj, M.; Van Ginderachter, J.A.; Stijlemans, B. Lipocalin-2: A Nurturer of Tumor Progression and a Novel Candidate for Targeted Cancer Therapy. Cancers 2023, 15, 5159. https://doi.org/10.3390/cancers15215159
Živalj M, Van Ginderachter JA, Stijlemans B. Lipocalin-2: A Nurturer of Tumor Progression and a Novel Candidate for Targeted Cancer Therapy. Cancers. 2023; 15(21):5159. https://doi.org/10.3390/cancers15215159
Chicago/Turabian StyleŽivalj, Maida, Jo A. Van Ginderachter, and Benoit Stijlemans. 2023. "Lipocalin-2: A Nurturer of Tumor Progression and a Novel Candidate for Targeted Cancer Therapy" Cancers 15, no. 21: 5159. https://doi.org/10.3390/cancers15215159
APA StyleŽivalj, M., Van Ginderachter, J. A., & Stijlemans, B. (2023). Lipocalin-2: A Nurturer of Tumor Progression and a Novel Candidate for Targeted Cancer Therapy. Cancers, 15(21), 5159. https://doi.org/10.3390/cancers15215159