
The Prospect of Harnessing the Microbiome to Improve Immunotherapeutic Response in Pancreatic Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Prospect of Immunotherapy in Pancreatic Cancer Treatment

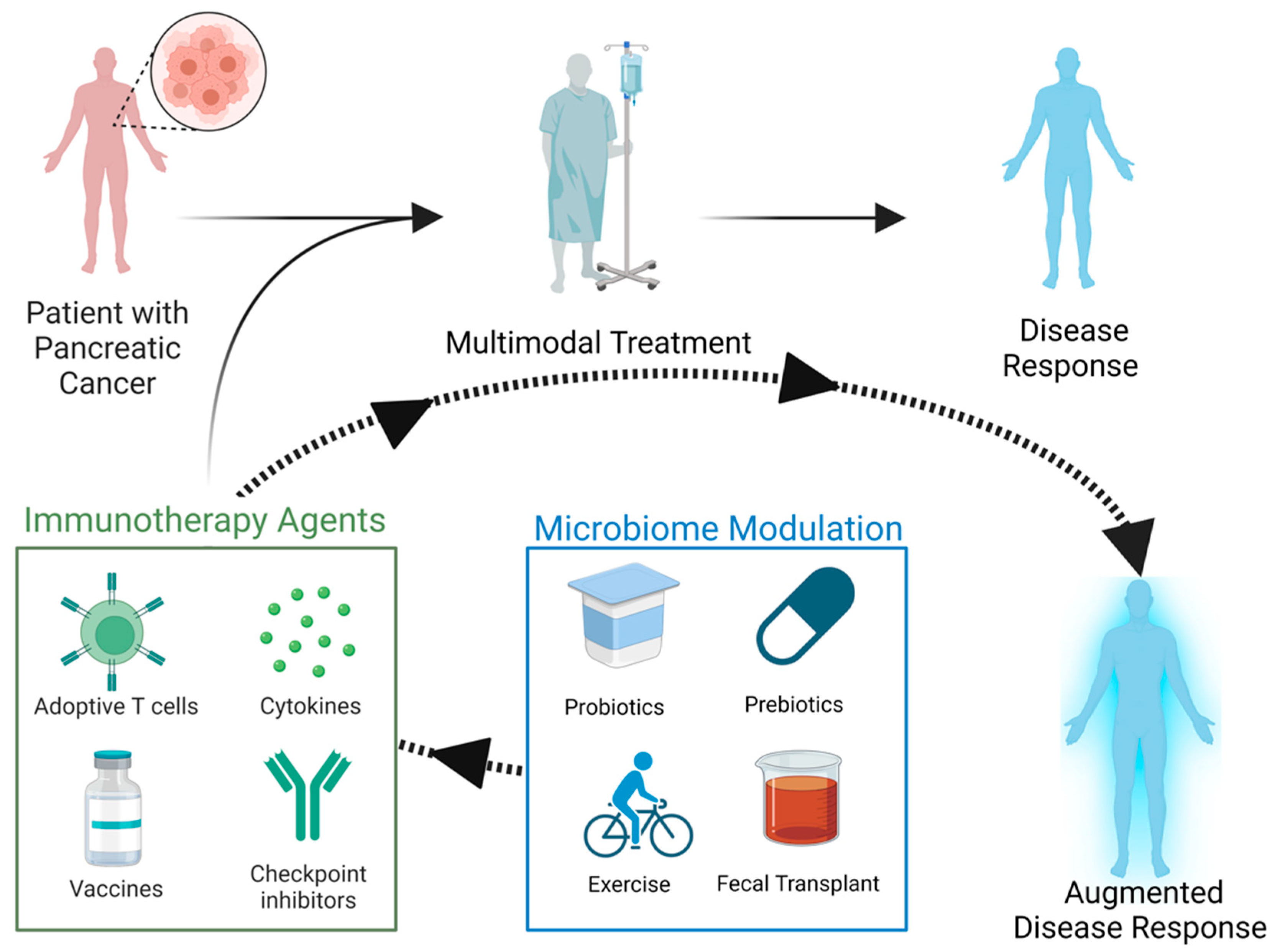{kind=link}
{kind=link}
| Author | Immunotherapy Category | Result |
|---|---|---|
| Zhao [22] | ICI | Enhanced immunotherapy response after IL2-inducible T-cell kinase inhibition. |
| Hung [23] | CAR-T | Protease-activated receptor 1 CAR-T cells enhanced PDAC xenograft killing and decreased matrix metalloproteinase 1 levels in the TME. |
| Xu [24] | ICI BiTE | Conversion of immunosuppressive Tregs to immune-enhancing Tregs. |
| Beelen [25] | ICI | Increased NK-cell-induced PDAC organoid cell death with anti-PD-L1 or anti-HER2 therapy. |
| Qiang [26] | Hu-mAb | TGF-β blockade in orthotopic PDAC xenografts enhances sensitivity to chemotherapy. |
| Koh [27] | Immunostimulatory | Ex vivo activated NK cells inhibit PDAC xenograft growth in combination with gemcitabine. |
| Li [28] | CAR-T | CAR-T cells targeting glypican-1 resulted in PDAC xenograft regression and increased T-cell signaling. |
| Peng [29] | ICI Vaccine | ICI reverses T-cell dysfunction and enhances neoantigen vaccine-induced T-cell responses and PDAC xenograft regression. |
| Mirji [30] | ICI | Enhanced immune activation and PDAC xenograft response to ICI with the microbial metabolite trimethylamine N-oxide. |
| Pushalkar [10] | ICI | Depletion of microbiota increases PD-1 expression and efficacy of anti-PD-1 ICI. |
| Winograd [31] | ICI | ICI resistance can be overcome with the induction of T-cell immunity. |
| Luu [32] | CAR-T | Microbial-derived short-chain fatty acids enhance antitumor immunity against PDAC xenografts. |
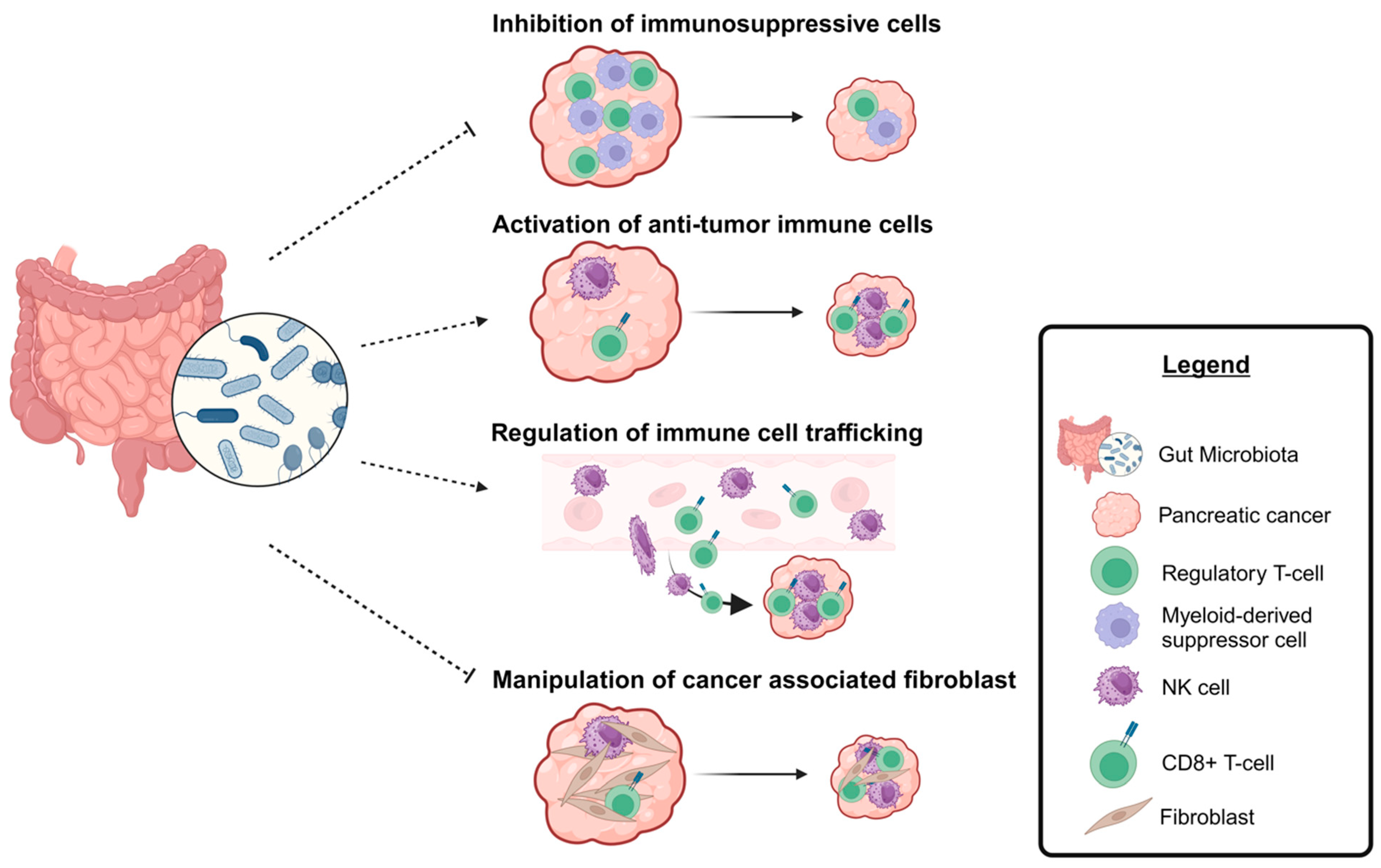
3. Overview of Microbiome–PDAC Research
4. The Relationship of the Microbiome with Conventional Chemotherapy in PDAC
5. Overcoming Challenges in PDAC Immunotherapy with the Microbiome
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Cancer Facts & Figures 2023. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2023/2023-cancer-facts-and-figures.pdf (accessed on 30 March 2023).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase ib/ii study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Thomas, R.M.; Jobin, C. The microbiome and cancer: Is the “oncobiome” mirage real? Trends Cancer 2015, 1, 24–35. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019, 178, 795–806.e12. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Nishijima, S.; Kojima, Y.; Hisada, Y.; Imbe, K.; Miyoshi-Akiyama, T.; Suda, W.; Kimura, M.; Aoki, R.; Sekine, K.; et al. Metagenomic identification of microbial signatures predicting pancreatic cancer from a multinational study. Gastroenterology 2022, 163, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Kartal, E.; Schmidt, T.S.B.; Molina-Montes, E.; Rodríguez-Perales, S.; Wirbel, J.; Maistrenko, O.M.; Akanni, W.A.; Alashkar Alhamwe, B.; Alves, R.J.; Carrato, A.; et al. A faecal microbiota signature with high specificity for pancreatic cancer. Gut 2022, 71, 1359–1372. [Google Scholar] [CrossRef]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yang, G.; Yang, J.; Ren, J.; You, L.; Zhao, Y. Human microbiota colonization and pancreatic ductal carcinoma. Crit. Rev. Microbiol. 2023, 49, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.J.; Zhang, L.; Zhou, H.; Chia, D.; Elashoff, D.; Akin, D.; Paster, B.J.; Joshipura, K.; Wong, D.T.W. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 2012, 61, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Finck, A.V.; Blanchard, T.; Roselle, C.P.; Golinelli, G.; June, C.H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 2022, 28, 678–689. [Google Scholar] [CrossRef]
- Sparano, J.A.; Fisher, R.I.; Sunderland, M.; Margolin, K.; Ernest, M.L.; Sznol, M.; Atkins, M.B.; Dutcher, J.P.; Micetich, K.C.; Weiss, G.R. Randomized phase III trial of treatment with high-dose interleukin-2 either alone or in combination with interferon alfa-2a in patients with advanced melanoma. J. Clin. Oncol. 1993, 11, 1969–1977. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef]
- Zhao, M.; Li, L.; Kiernan, C.H.; Castro Eiro, M.D.; Dammeijer, F.; van Meurs, M.; Brouwers-Haspels, I.; Wilmsen, M.E.P.; Grashof, D.G.B.; van de Werken, H.J.G.; et al. Overcoming immune checkpoint blockade resistance in solid tumors with intermittent ITK inhibition. Sci. Rep. 2023, 13, 15678. [Google Scholar] [CrossRef]
- Hung, H.-C.; Fan, M.-H.; Wang, D.; Miao, C.H.; Su, P.; Liu, C.-L. Effect of chimeric antigen receptor T cells against protease-activated receptor 1 for treating pancreatic cancer. BMC Med. 2023, 21, 338. [Google Scholar] [CrossRef]
- Xu, Y.; Fu, J.; Henderson, M.; Lee, F.; Jurcak, N.; Henn, A.; Wahl, J.; Shao, Y.; Wang, J.; Lyman, M.; et al. CLDN18.2 bite engages effector and regulatory T cells for antitumor immune response in preclinical models of pancreatic cancer. Gastroenterology 2023, 165, 1219–1232. [Google Scholar] [CrossRef]
- Beelen, N.A.; Aberle, M.R.; Bruno, V.; Olde Damink, S.W.M.; Bos, G.M.J.; Rensen, S.S.; Wieten, L. Antibody-dependent cellular cytotoxicity-inducing antibodies enhance the natural killer cell anti-cancer response against patient-derived pancreatic cancer organoids. Front. Immunol. 2023, 14, 1133796. [Google Scholar] [CrossRef]
- Qiang, L.; Hoffman, M.T.; Ali, L.R.; Castillo, J.I.; Kageler, L.; Temesgen, A.; Lenehan, P.; Wang, S.J.; Bello, E.; Cardot-Ruffino, V.; et al. Transforming Growth Factor-β Blockade in Pancreatic Cancer Enhances Sensitivity to Combination Chemotherapy. In Gastroenterology; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Koh, E.-K.; Lee, H.-R.; Son, W.-C.; Park, G.-Y.; Kim, J.; Bae, J.-H.; Park, Y.-S. Combinatorial immunotherapy with gemcitabine and ex vivo-expanded NK cells induces anti-tumor effects in pancreatic cancer. Sci. Rep. 2023, 13, 7656. [Google Scholar] [CrossRef]
- Li, N.; Quan, A.; Li, D.; Pan, J.; Ren, H.; Hoeltzel, G.; de Val, N.; Ashworth, D.; Ni, W.; Zhou, J.; et al. The IgG4 hinge with CD28 transmembrane domain improves VHH-based CAR T cells targeting a membrane-distal epitope of GPC1 in pancreatic cancer. Nat. Commun. 2023, 14, 1986. [Google Scholar] [CrossRef]
- Peng, H.; Li, L.; Zuo, C.; Chen, M.Y.; Zhang, X.; Myers, N.B.; Hogg, G.D.; DeNardo, D.G.; Goedegebuure, S.P.; Hawkins, W.G.; et al. Combination TIGIT/PD-1 blockade enhances the efficacy of neoantigen vaccines in a model of pancreatic cancer. Front. Immunol. 2022, 13, 1039226. [Google Scholar] [CrossRef]
- Mirji, G.; Worth, A.; Bhat, S.A.; El Sayed, M.; Kannan, T.; Goldman, A.R.; Tang, H.-Y.; Liu, Q.; Auslander, N.; Dang, C.V.; et al. The microbiome-derived metabolite TMAO drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci. Immunol. 2022, 7, eabn0704. [Google Scholar] [CrossRef]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.L.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. IMPACT Study Investigators Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Pettenati, C.; Ingersoll, M.A. Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat. Rev. Urol. 2018, 15, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Lobo, N.; Brooks, N.A.; Zlotta, A.R.; Cirillo, J.D.; Boorjian, S.; Black, P.C.; Meeks, J.J.; Bivalacqua, T.J.; Gontero, P.; Steinberg, G.D.; et al. 100 years of Bacillus Calmette-Guérin immunotherapy: From cattle to COVID-19. Nat. Rev. Urol. 2021, 18, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, G.; Tang, T.-Y.; Gao, X.; Liang, T.-B. Personalized pancreatic cancer therapy: From the perspective of mRNA vaccine. Mil. Med. Res. 2022, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic cancer and immunotherapy: A clinical overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; Vonderheide, R.H. Immune vulnerabilities of mutant KRAS in pancreatic cancer. In Trends in Cancer; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Hippe, D.S.; Srinivasan, S.; Proll, S.C.; Miltiadous, O.; Li, N.; Zhang, P.; Ensbey, K.S.; Hoffman, N.G.; Schmidt, C.R.; et al. Intestinal microbiota controls graft-versus-host disease independent of donor-host genetic disparity. Immunity 2023, 56, 1876–1893.e8. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune checkpoint inhibitors in cancer therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Physiology or Medicine 2018. NobelPrize.org. Available online: https://www.nobelprize.org/prizes/medicine/2018/summary (accessed on 2 May 2023).
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. KEYNOTE-024 Investigators Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Stein, A.; Paschold, L.; Tintelnot, J.; Goekkurt, E.; Henkes, S.-S.; Simnica, D.; Schultheiss, C.; Willscher, E.; Bauer, M.; Wickenhauser, C.; et al. Efficacy of Ipilimumab vs FOLFOX in Combination With Nivolumab and Trastuzumab in Patients With Previously Untreated ERBB2-Positive Esophagogastric Adenocarcinoma: The AIO INTEGA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1150–1158. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Boland, A.J.; O’Kane, A.A.; Buick, R.; Longley, D.B.; Scott, C.J. Antibody therapy in pancreatic cancer: mAb-ye we’re onto something? Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188557. [Google Scholar] [CrossRef]
- Sorbara, M.; Cordelier, P.; Bery, N. Antibody-Based Approaches to Target Pancreatic Tumours. Antibodies 2022, 11, 47. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Lee, S.T.; Gan, H.K.; Scott, A.M. Radiolabeled antibodies for cancer imaging and therapy. Cancers 2022, 14, 1454. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Michelakos, T.; Cai, L.; Villani, V.; Sabbatino, F.; Kontos, F.; Fernández-Del Castillo, C.; Yamada, T.; Neyaz, A.; Taylor, M.S.; Deshpande, V.; et al. Tumor microenvironment immune response in pancreatic ductal adenocarcinoma patients treated with neoadjuvant therapy. J. Natl. Cancer Inst. 2021, 113, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Fernandez-Zapico, M.E. Gemcitabine activates natural killer cells to attenuate pancreatic cancer recurrence. Gastroenterology 2016, 151, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R. Epithelial and stromal co-evolution and complicity in pancreatic cancer. Nat. Rev. Cancer 2023, 23, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Gartrell, R.D.; Enzler, T.; Kim, P.S.; Fullerton, B.T.; Fazlollahi, L.; Chen, A.X.; Minns, H.E.; Perni, S.; Weisberg, S.P.; Rizk, E.M.; et al. Neoadjuvant chemoradiation alters the immune microenvironment in pancreatic ductal adenocarcinoma. Oncoimmunology 2022, 11, 2066767. [Google Scholar] [CrossRef]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109-301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 2017, 14, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, L.B.; White, J.R.; Jones, C.B.; Slottke, R.E.; Ernst, S.E.; Moran, A.E.; Graff, J.N.; Sfanos, K.S. Composition of gastrointestinal microbiota in association with treatment response in individuals with metastatic castrate resistant prostate cancer progressing on enzalutamide and initiating treatment with anti-PD-1 (pembrolizumab). Neoplasia 2022, 32, 100822. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Izard, J. Microbiota, oral microbiome, and pancreatic cancer. Cancer J. 2014, 20, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Jiang, J.; Xie, H.; Li, A.; Lu, H.; Xu, S.; Zhou, L.; Zhang, H.; Cui, G.; Chen, X.; et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 2017, 8, 95176–95191. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018, 39, 1068–1078. [Google Scholar] [CrossRef]
- Sethi, V.; Kurtom, S.; Tarique, M.; Lavania, S.; Malchiodi, Z.; Hellmund, L.; Zhang, L.; Sharma, U.; Giri, B.; Garg, B.; et al. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology 2018, 155, 33–37.e6. [Google Scholar] [CrossRef]
- Yu, Q.; Newsome, R.C.; Beveridge, M.; Hernandez, M.C.; Gharaibeh, R.Z.; Jobin, C.; Thomas, R.M. Intestinal microbiota modulates pancreatic carcinogenesis through intratumoral natural killer cells. Gut Microbes 2022, 14, 2112881. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. European Study Group for Pancreatic Cancer A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.A.; Moertel, C.G.; Fleming, T.R.; Rubin, J.R.; Krook, J.E.; Everson, L.K.; Windschitl, H.E.; Twito, D.I.; Marschke, R.F.; Foley, J.F. A comparison of three chemotherapeutic regimens in the treatment of advanced pancreatic and gastric carcinoma. Fluorouracil vs fluorouracil and doxorubicin vs fluorouracil, doxorubicin, and mitomycin. JAMA 1985, 253, 2061–2067. [Google Scholar] [PubMed]
- Duschinsky, R.; Pleven, E.; Heidelberger, C. The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Vitiello, G.A.; Cohen, D.J.; Miller, G. Harnessing the microbiome for pancreatic cancer immunotherapy. Trends Cancer 2019, 5, 670–676. [Google Scholar] [CrossRef]
- Huang, X.; Li, M.; Hou, S.; Tian, B. Role of the microbiome in systemic therapy for pancreatic ductal adenocarcinoma (Review). Int. J. Oncol. 2021, 59. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Q.; Liao, Q.; Zhao, Y. Pancreatic cancer, gut microbiota, and therapeutic efficacy. J. Cancer 2020, 11, 2749–2758. [Google Scholar] [CrossRef]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.R.; Nicholson, J.K.; Kinross, J.M. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356–365. [Google Scholar] [CrossRef]
- Huang, J.; Liu, W.; Kang, W.; He, Y.; Yang, R.; Mou, X.; Zhao, W. Effects of microbiota on anticancer drugs: Current knowledge and potential applications. eBioMedicine 2022, 83, 104197. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.-A.; Mani, S.; et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Mundry, C.S.; Eberle, K.C.; Singh, P.K.; Hollingsworth, M.A.; Mehla, K. Local and systemic immunosuppression in pancreatic cancer: Targeting the stalwarts in tumor’s arsenal. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, C.; Duan, Y.; Heinrich, B.; Rosato, U.; Diggs, L.P.; Ma, L.; Roy, S.; Fu, Q.; Brown, Z.J.; et al. Gut microbiome directs hepatocytes to recruit mdscs and promote cholangiocarcinoma. Cancer Discov. 2021, 11, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Roles of differently polarized macrophages in the initiation and progressionof pancreatic cancer. Front. Immunol. 2023, 14, 1237711. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e8. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Herranz, M.; Bittinger, K.; Rafail, S.; Guedan, S.; Pierini, S.; Tanes, C.; Ganetsky, A.; Morgan, M.A.; Gill, S.; Tanyi, J.L.; et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight 2018, 3, e94952. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Su, H.; Johnson, C.H.; Khan, S.A.; Kluger, H.; Lu, L. Intratumour microbiome associated with the infiltration of cytotoxic CD8+ T cells and patient survival in cutaneous melanoma. Eur. J. Cancer 2021, 151, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ghaddar, B.; Biswas, A.; Harris, C.; Omary, M.B.; Carpizo, D.R.; Blaser, M.J.; De, S. Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell 2022, 40, 1240–1253.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.-J.; Sugimoto, H.; Chang, C.-C.; et al. Oncogenic collagen I homotrimers from cancer cells bind to α3β1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell 2022, 40, 818–834.e9. [Google Scholar] [CrossRef]
- Sethi, V.; Vitiello, G.A.; Saxena, D.; Miller, G.; Dudeja, V. The role of the microbiome in immunologic development and its implication for pancreatic cancer immunotherapy. Gastroenterology 2019, 156, 2097–2115.e2. [Google Scholar] [CrossRef]
- Nelson, M.H.; Diven, M.A.; Huff, L.W.; Paulos, C.M. Harnessing the microbiome to enhance cancer immunotherapy. J. Immunol. Res. 2015, 2015, 368736. [Google Scholar] [CrossRef]
- McCulloch, J.A.; Davar, D.; Rodrigues, R.R.; Badger, J.H.; Fang, J.R.; Cole, A.M.; Balaji, A.K.; Vetizou, M.; Prescott, S.M.; Fernandes, M.R.; et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 2022, 28, 545–556. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 75. [Google Scholar] [CrossRef]
- Yuan, X.; Duan, Y.; Xiao, Y.; Sun, K.; Qi, Y.; Zhang, Y.; Ahmed, Z.; Moiani, D.; Yao, J.; Li, H.; et al. Vitamin E enhances cancer immunotherapy by reinvigorating dendritic cells via targeting checkpoint SHP1. Cancer Discov. 2022, 12, 1742–1759. [Google Scholar] [CrossRef]
- Cortellino, S.; Raveane, A.; Chiodoni, C.; Delfanti, G.; Pisati, F.; Spagnolo, V.; Visco, E.; Fragale, G.; Ferrante, F.; Magni, S.; et al. Fasting renders immunotherapy effective against low-immunogenic breast cancer while reducing side effects. Cell Rep. 2022, 40, 111256. [Google Scholar] [CrossRef]
- Greathouse, K.L.; Wyatt, M.; Johnson, A.J.; Toy, E.P.; Khan, J.M.; Dunn, K.; Clegg, D.J.; Reddy, S. Diet-microbiome interactions in cancer treatment: Opportunities and challenges for precision nutrition in cancer. Neoplasia 2022, 29, 100800. [Google Scholar] [CrossRef]
- Mills, S.; Stanton, C.; Lane, J.A.; Smith, G.J.; Ross, R.P. Precision nutrition and the microbiome, part I: Current state of the science. Nutrients 2019, 11, 923. [Google Scholar] [CrossRef]
- O’Keefe, S.J.D. Editorial: Food, nutrition and the microbiome in health and disease. Curr. Opin. Gastroenterol. 2022, 38, 144–145. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Dalton, A.; Mermier, C.; Zuhl, M. Exercise influence on the microbiome-gut-brain axis. Gut Microbes 2019, 10, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.J.; McPherson, A.C.; Phelps, C.M.; Pandey, S.P.; Laughlin, C.R.; Shapira, J.H.; Medina Sanchez, L.; Rana, M.; Richie, T.G.; Mims, T.S.; et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 2023, 186, 1846–1862.e26. [Google Scholar] [CrossRef]


| Study Title | Malignancy | Immunotherapy Category | Phase | ClinicalTrials.gov or EudraCT Study ID |
|---|---|---|---|---|
| Immunotherapy and Irreversible Electroporation in the Treatment of Advanced Pancreatic Adenocarcinoma | PDAC | ICI | II | NCT03080974 |
| GVAX Pancreas Vaccine (With CY) in Combination with Nivolumab and SBRT for Patients with Borderline Resectable Pancreatic Cancer | PDAC | ICI Vaccine | II | NCT03161379 |
| SD- 101, Nivolumab, and Radiation Therapy in Treating Patients with Chemotherapy Refractory Metastatic Pancreatic Cancer | mPDAC | ICI | I | NCT04050085 |
| Pilot Study With CY, Pembrolizumab, GVAX, and IMC-CS4 (LY3022855) in Patients with Borderline Resectable Adenocarcinoma of the Pancreas | PDAC | ICI Vaccine | I | NCT03153410 |
| Vaccine Therapy and Sargramostim in Treating Patients with Pancreas Cancer That Cannot Be Removed by Surgery | PDAC Pancreatic ACC | Vaccine | I | NCT00669734 |
| Epacadostat, Pembrolizumab, and CRS-207, With or Without CY/GVAX Pancreas in Patients with Metastatic Pancreas Cancer | mPDAC | ICI Vaccine | II | NCT03006302 |
| Testing the Combination of Anetumab Ravtansine with Either Nivolumab, Nivolumab and Ipilimumab, or Gemcitabine and Nivolumab in Advanced Pancreatic Cancer | PDAC | ICI | I/II | NCT03816358 |
| Study of Pembrolizumab with or without Defactinib following Chemotherapy as a Neoadjuvant and Adjuvant Treatment for Resectable Pancreatic Ductal Adenocarcinoma | PDAC | ICI | II | NCT03727880 |
| VX15/2503 and Immunotherapy in Resectable Pancreatic and Colon Cancer | CRC PDAC | ICI | I | NCT03373188 |
| Neoadjuvant CAN-2409 in Combination with Chemoradiation or SBRT for Borderline Resectable Pancreatic Adenocarcinoma | PDAC | Viral immunotherapy | II | NCT02446093 |
| Pancreatic Tumor Cell Vaccine (GVAX), Low Dose Cyclophosphamide, Fractionated SBRT, and FOLFIRINOX Chemotherapy in Patients with Resected Adenocarcinoma of the Pancreas | PDAC | Vaccine | I | NCT01595321 |
| Study of CRS-207, Nivolumab, and Ipilimumab with or without GVAX Pancreas Vaccine (With CY) in Patients with Pancreatic Cancer | PDAC | ICI Vaccine | II | NCT03190265 |
| A study of ELI-002 in Subjects with KRAS Mutated Pancreatic Ductal Adenocarcinoma (PDAC) and Other Solid Tumors | PDAC CRC NSCLC Ovarian cancer Cholangiocarcinoma Gallbladder carcinoma | Immunotherapy targeting KRAS mutants | I | NCT04853017 |
| CAR T Cell Immunotherapy for Pancreatic Cancer | PDAC | CAR-T | I | NCT03323944 |
| Study of Autologous T-cells in Patients with Metastatic Pancreatic Cancer | mPDAC | CAR-T | I | NCT03638193 |
| Th-1 Dendritic Cell Immunotherapy Plus Standard Chemotherapy for Pancreatic Adenocarcinoma (DECIST) | PDAC | Vaccine | I | NCT04157127 |
| Nivolumab and Ipilimumab and Radiation Therapy in MSS and MSI High Colorectal and Pancreatic Cancer | CRC PDAC | ICI | II | NCT03104439 |
| Plerixafor and Cemiplimab in Metastatic Pancreatic Cancer | mPDAC | ICI | II | NCT04177810 |
| An Open Label, Dose Escalation Followed by Dose Expansion, Safety and Tolerability Trial of CAN04, a Fully Humanized Monoclonal Antibody Against IL1RAP, in Subjects with Solid Malignant Tumors | NSCLC PDAC | Hu-mAb | I | 2017-001111-36 |
| A Phase Ib/II, Open-Label, Multicenter, Randomized Umbrella Study Evaluating the Efficacy and Safety of Multiple Immunotheraby-Based Treatment Combinations in Patients with Metastastic Pancreatic Adenocarcinoma (Morpheus-Pancreatic Cancer) | mPDAC | ICI Cytokine Inhibitor | Ib/II | 2016-004126-42 |
| Safety and Efficacy of IMM-101 Combined with Stereotactic Radiotherapy in Patients with Limited MEtastatic PANcreatic Cancer (MEPANC-1) | mPDAC | Immune Stimulatory | II | 2020-003945-13 |
| First-in-Human Study of ICT01 in Patients with Advanced Cancer (EVICTION) | PDAC Various other solid tumors | Hu-mAb Immune Stimulatory ICI | I/II | NCT04243499 2019-003847-31 |
| Trial Of Hypofractionated Radiotherapy in Combination with MEDI4736 and Tremelimumab for Patients with Metastatic Melanoma and Lung, Breast and Pancreatic Cancers | PDAC NSCLC Melanoma | ICI | I | NCT02639026 |
| Radiation Therapy in Combination with Durvalumab for People with Pancreatic Cancer | PDAC | ICI | I/II | NCT03245541 |
| Study Title | Malignancy | Immunotherapy Category | Phase | ClinicalTrials.gov Study ID |
|---|---|---|---|---|
| Modulation of the Gut Microbiome with Pembrolizumab Following Chemotherapy in Resectable Pancreatic Cancer | PDAC | ICI | II | NCT05462496 |
| ARGONAUT: Development and Analysis of a Blood and Stool Sample Bank for Cancer Patients, Enabling the Systemic Study of the Effect of Gut Microbiomes on Response to Treatment | PDAC CRC Triple Negative Breast Cancer NSCLC | ICI | Prospective Cohort Study | NCT04638751 |
| The Mechanism of Enhancing the Antitumor Effects of CAR-T on PC by Gut Microbiota Regulation | PDAC | CAR-T | Case Control | NCT04203459 (China) |
| Feasibility Study of Microbial Ecosystem Therapeutics (MET-4) to Evaluate Effects of Fecal Microbiome in Patients on ImmunOtherapy (MET4-IO) | Solid Tumors | ICI | II/III | NCT03686202 (Canada) |
| Gut Microbiome Modulation to Enable Efficacy of Checkpoint-based Immunotherapy in Pancreatic Adenocarcinoma | PDAC | ICI | IV | NCT03891979 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogers, S.; Charles, A.; Thomas, R.M. The Prospect of Harnessing the Microbiome to Improve Immunotherapeutic Response in Pancreatic Cancer. Cancers 2023, 15, 5708. https://doi.org/10.3390/cancers15245708
Rogers S, Charles A, Thomas RM. The Prospect of Harnessing the Microbiome to Improve Immunotherapeutic Response in Pancreatic Cancer. Cancers. 2023; 15(24):5708. https://doi.org/10.3390/cancers15245708
Chicago/Turabian StyleRogers, Sherise, Angel Charles, and Ryan M. Thomas. 2023. "The Prospect of Harnessing the Microbiome to Improve Immunotherapeutic Response in Pancreatic Cancer" Cancers 15, no. 24: 5708. https://doi.org/10.3390/cancers15245708
APA StyleRogers, S., Charles, A., & Thomas, R. M. (2023). The Prospect of Harnessing the Microbiome to Improve Immunotherapeutic Response in Pancreatic Cancer. Cancers, 15(24), 5708. https://doi.org/10.3390/cancers15245708