
Transcriptomic Deconvolution of Neuroendocrine Neoplasms Predicts Clinically Relevant Characteristics
 , ,
, ,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
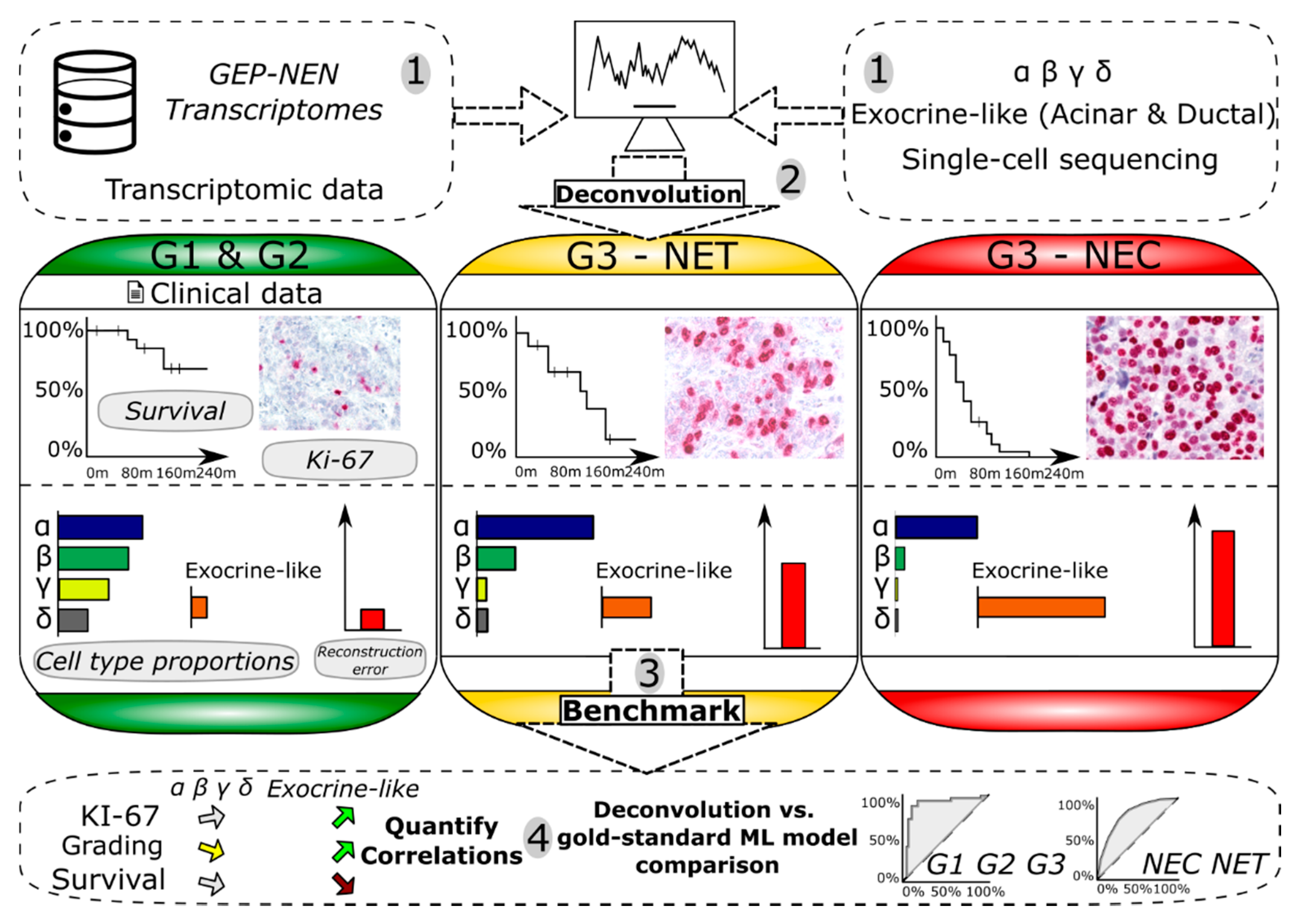
2.1. Overview of the Developed Framework
2.2. Datasets
2.3. Bioinformatics Processing
2.4. Deconvolution Algorithms
2.5. Machine Learning and Survival Time Prediction Test
2.6. Data Availability
2.7. Code Availability
3. Results
3.1. Creation of a Deconvolution Machine Learning Model in the Absence of Neoplastic Training Data
3.2. Deconvolution Algorithms, Cell Type Models, and Evaluation Datasets
3.3. Deconvolution of panNEN and Non-Pancreatic GEP-NEN Transcriptomes into Endocrine and Exocrine-Like Cell Type Proportions
3.4. Cell Type Proportion Predictions Differ by Grading, Study, and Deconvolution Model
3.5. Biological Contextualization of the Deconvolution Model Effectiveness and Cell Type Proportions
3.6. Correlation of Predicted Cell Type Proportions with Prognostic and Clinical Characteristics
3.7. Machine Learning-Based Prediction of Grading, NEC, or NET Status and Patient Survival Time
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Sanzo, M.; Cipolloni, L.; Borro, M.; La Russa, R.; Santurro, A.; Scopetti, M.; Simmaco, M.; Frati, P. Clinical Applications of Personalized Medicine: A New Paradigm and Challenge. Curr. Pharm. Biotechnol. 2017, 18, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.J.; Javed, Z.; Sadia, H.; Qureshi, I.A.; Irshad, A.; Ahmed, R.; Malik, K.; Raza, S.; Abbas, A.; Pezzani, R.; et al. Clinical Applications of Artificial Intelligence and Machine Learning in Cancer Diagnosis: Looking into the Future. Cancer Cell Int. 2021, 21, 270. [Google Scholar] [CrossRef]
- Rindi, G.; Wiedenmann, B. Neuroendocrine Neoplasia of the Gastrointestinal Tract Revisited: Towards Precision Medicine. Nat. Rev. Endocrinol. 2020, 16, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and Prognostic Factors for Treatment and Survival in 305 Patients with Advanced Gastrointestinal Neuroendocrine Carcinoma (WHO G3): The NORDIC NEC Study. Ann. Oncol. 2013, 24, 152–160. [Google Scholar] [CrossRef]
- Dasari, A.; Mehta, K.; Byers, L.A.; Sorbye, H.; Yao, J.C. Comparative Study of Lung and Extrapulmonary Poorly Differentiated Neuroendocrine Carcinomas: A SEER Database Analysis of 162,983 Cases. Cancer 2018, 124, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Basturk, O.; Yang, Z.; Tang, L.H.; Hruban, R.H.; Adsay, V.; McCall, C.M.; Krasinskas, A.M.; Jang, K.-T.; Frankel, W.L.; Balci, S.; et al. The High-Grade (WHO G3) Pancreatic Neuroendocrine Tumor Category Is Morphologically and Biologically Heterogenous and Includes Both Well Differentiated and Poorly Differentiated Neoplasms. Am. J. Surg. Pathol. 2015, 39, 683–690. [Google Scholar] [CrossRef]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E.; Dhall, D.; Jih, L.; Basturk, O.; Allen, P.J.; Klimstra, D.S. Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Differentiated Neuroendocrine Carcinomas. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small Cell and Large Cell Neuroendocrine Carcinomas of the Pancreas Are Genetically Similar and Distinct from Well-Differentiated Pancreatic Neuroendocrine Tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Elvebakken, H.; Perren, A.; Scoazec, J.-Y.; Tang, L.H.; Federspiel, B.; Klimstra, D.S.; Vestermark, L.W.; Ali, A.S.; Zlobec, I.; Myklebust, T.Å.; et al. A Consensus Developed Morphological Re-Evaluation of 196 High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms and Its Clinical Correlations. Neuroendocrinology 2020, 9, 883–894. [Google Scholar] [CrossRef]
- Tang, L.H.; Basturk, O.; Sue, J.J.; Klimstra, D.S. A Practical Approach to the Classification of WHO Grade 3 (G3) Well-Differentiated Neuroendocrine Tumor (WD-NET) and Poorly Differentiated Neuroendocrine Carcinoma (PD-NEC) of the Pancreas. Am. J. Surg. Pathol. 2016, 40, 1192–1202. [Google Scholar] [CrossRef]
- Tang, L.H. Pancreatic Neuroendocrine Neoplasms: Landscape and Horizon. Arch. Pathol. Lab. Med. 2020, 144, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Bilotta, M.; Mafficini, A.; Luchini, C.; Furlan, D.; Inzani, F.; Petrone, G.; Bonvissuto, D.; La Rosa, S.; Schinzari, G.; et al. Gene Expression Profiling of Pancreas Neuroendocrine Tumors with Different Ki67-Based Grades. Cancers 2021, 13, 2054. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; Drier, Y.; Dreijerink, K.M.A.; Brosens, L.A.A.; Deshpande, V.; Epstein, C.B.; Conemans, E.B.; Morsink, F.H.M.; Graham, M.K.; Valk, G.D.; et al. Enhancer Signatures Stratify and Predict Outcomes of Non-Functional Pancreatic Neuroendocrine Tumors. Nat. Med. 2019, 25, 1260–1265. [Google Scholar] [CrossRef]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grötzinger, C.; Barbi, S.; Bersani, S.; Körner, J.; Wafy, I.; Mafficini, A.; Lawlor, R.T.; et al. A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-Genome Landscape of Pancreatic Neuroendocrine Tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Diedisheim, M.; Dermine, S.; Jouinot, A.; Septier, A.; Gaujoux, S.; Dousset, B.; Cadiot, G.; Larger, E.; Bertherat, J.; Scharfmann, R.; et al. Prognostic Transcriptome Classes of Duodenopancreatic Neuroendocrine Tumors. Endocr. Relat. Cancer 2021, 28, 563–571. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A Precision Oncology Approach to the Pharmacological Targeting of Mechanistic Dependencies in Neuroendocrine Tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep Learning: New Computational Modelling Techniques for Genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Schaefer, J.; Lehne, M.; Schepers, J.; Prasser, F.; Thun, S. The Use of Machine Learning in Rare Diseases: A Scoping Review. Orphanet J. Rare Dis. 2020, 15, 145. [Google Scholar] [CrossRef]
- Rashid, H.; Tanveer, M.A.; Aqeel Khan, H. Skin Lesion Classification Using GAN Based Data Augmentation. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2019, 2019, 916–919. [Google Scholar]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-Cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-Cell Transcriptomes Identify Human Islet Cell Signatures and Reveal Cell-Type-Specific Expression Changes in Type 2 Diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef]
- Tosti, L.; Hang, Y.; Debnath, O.; Tiesmeyer, S.; Trefzer, T.; Steiger, K.; Ten, F.W.; Lukassen, S.; Ballke, S.; Kühl, A.A.; et al. Single-Nucleus and In Situ RNA–Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology 2021, 160, 1330–1344.e11. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A Single-Cell Survey of the Small Intestinal Epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global Genomic and Transcriptomic Analysis of Human Pancreatic Islets Reveals Novel Genes Influencing Glucose Metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef]
- Horak, P.; Heining, C.; Kreutzfeldt, S.; Hutter, B.; Mock, A.; Hüllein, J.; Fröhlich, M.; Uhrig, S.; Jahn, A.; Rump, A.; et al. Comprehensive Genomic and Transcriptomic Analysis for Guiding Therapeutic Decisions in Patients with Rare Cancers. Cancer Discov. 2021, 11, 2780–2795. [Google Scholar] [CrossRef]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; et al. Pancreatic Endocrine Tumors: Expression Profiling Evidences a Role for AKT-mTOR Pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef]
- Kawasaki, K.; Toshimitsu, K.; Matano, M.; Fujita, M.; Fujii, M.; Togasaki, K.; Ebisudani, T.; Shimokawa, M.; Takano, A.; Takahashi, S.; et al. An Organoid Biobank of Neuroendocrine Neoplasms Enables Genotype-Phenotype Mapping. Cell 2020, 183, 1420–1435.e21. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de Novo Haploid Genome Assemblies Demonstrates the Enduring Quality of the Reference Assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Anders, S.; Kim, V.; Huber, W. RNA-Seq Workflow: Gene-Level Exploratory Analysis and Differential Expression. F1000 Res. 2015, 4, 1070. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Ballester, B.; Smedley, D.; Zhang, J.; Rice, P.; Kasprzyk, A. BioMart Central Portal—Unified Access to Biological Data. Nucleic Acids Res. 2009, 37, W23–W27. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing Classifier Performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Tibshirani, R.; Khatri, P.; Bodian, D.L.; Staedtler, F.; Perry, N.M.; Hastie, T.; Sarwal, M.M.; Davis, M.M.; Butte, A.J. Cell Type-Specific Gene Expression Differences in Complex Tissues. Nat. Methods 2010, 7, 287–289. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Dong, M.; Thennavan, A.; Urrutia, E.; Li, Y.; Perou, C.M.; Zou, F.; Jiang, Y. SCDC: Bulk Gene Expression Deconvolution by Multiple Single-Cell RNA Sequencing References. Brief. Bioinform. 2021, 22, 416–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Gong, T.; Clarke, R.; Chen, L.; Shih, I.-M.; Zhang, Z.; Levine, D.A.; Xuan, J.; Wang, Y. UNDO: A Bioconductor R Package for Unsupervised Deconvolution of Mixed Gene Expressions in Tumor Samples. Bioinformatics 2015, 31, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk Tissue Cell Type Deconvolution with Multi-Subject Single-Cell Expression Reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust Enumeration of Cell Subsets from Tissue Expression Profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring Tumour Purity and Stromal and Immune Cell Admixture from Expression Data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Kuhn, M.; Johnson, K. Feature Engineering and Selection: A Practical Approach for Predictive Models; CRC Press: Boca Raton, FL, USA, 2019; ISBN 9781351609470. [Google Scholar]
- Tritschler, S.; Theis, F.J.; Lickert, H.; Böttcher, A. Systematic Single-Cell Analysis Provides New Insights into Heterogeneity and Plasticity of the Pancreas. Mol. Metab. 2017, 6, 974–990. [Google Scholar] [CrossRef]
- Bonner-Weir, S.; Inada, A.; Yatoh, S.; Li, W.-C.; Aye, T.; Toschi, E.; Sharma, A. Transdifferentiation of Pancreatic Ductal Cells to Endocrine Beta-Cells. Biochem. Soc. Trans. 2008, 36, 353–356. [Google Scholar]
- Puri, S.; Folias, A.E.; Hebrok, M. Plasticity and Dedifferentiation within the Pancreas: Development, Homeostasis, and Disease. Cell Stem. Cell 2015, 16, 18–31. [Google Scholar] [CrossRef]
- Yachida, S.; Totoki, Y.; Noe, M.; Nakatani, Y.; Horie, M.; Kawasaki, K.; Nakamura, H.; Saito-Adachi, M.; Suzuki, M.; Takai, E.; et al. Comprehensive Genomic Profiling of Neuroendocrine Carcinomas of the Gastrointestinal System. Cancer Discov. 2022, 12, 692–711. [Google Scholar] [CrossRef]
- Konukiewitz, B.; Jesinghaus, M.; Steiger, K.; Schlitter, A.M.; Kasajima, A.; Sipos, B.; Zamboni, G.; Weichert, W.; Pfarr, N.; Klöppel, G. Pancreatic Neuroendocrine Carcinomas Reveal a Closer Relationship to Ductal Adenocarcinomas than to Neuroendocrine Tumors G3. Hum. Pathol. 2018, 77, 70–79. [Google Scholar] [CrossRef]
- Venizelos, A.; Elvebakken, H.; Perren, A.; Nikolaienko, O.; Deng, W.; Lothe, I.M.B.; Couvelard, A.; Hjortland, G.O.; Sundlöv, A.; Svensson, J.; et al. The Molecular Characteristics of High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr. Relat. Cancer 2021, 29, 1–14. [Google Scholar] [CrossRef]
- Simon, T.; Riemer, P.; Detjen, K.; Di Domenico, A.; Bormann, F.; Menne, A.; Khouja, S.; Monjé, N.; Childs, L.H.; Lenze, D.; et al. DNA Methylation Reveals Distinct Cells of Origin for Pancreatic Neuroendocrine Carcinomas (PanNECs) and Pancreatic Neuroendocrine Tumors (PanNETs). bioRxiv 2021. passed peer-review, in production. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Klein, D.; Lanzoni, G.; García-Santana, C.; Montalvo, A.; Pláceres-Uray, F.; Mazza, E.M.C.; Ricordi, C.; Inverardi, L.A.; et al. P2RY1/ALK3-Expressing Cells within the Adult Human Exocrine Pancreas Are BMP-7 Expandable and Exhibit Progenitor-like Characteristics. Cell Rep. 2018, 22, 2408–2420. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Klein, D.; van Dijk, J.; Muñiz-Anquela, R.; Moreno-Hernández, Y.B.; Lanzoni, G.; Sadiq, S.; Navarro-Rubio, B.; García, M.T.; et al. Single-Cell Resolution Analysis of the Human Pancreatic Ductal Progenitor Cell Niche. Proc. Natl. Acad. Sci. USA 2020, 117, 10876–10887. [Google Scholar] [CrossRef]
- Grün, D.; Muraro, M.J.; Boisset, J.-C.; Wiebrands, K.; Lyubimova, A.; Dharmadhikari, G.; van den Born, M.; van Es, J.; Jansen, E.; Clevers, H.; et al. De Novo Prediction of Stem Cell Identity Using Single-Cell Transcriptome Data. Cell Stem Cell 2016, 19, 266–277. [Google Scholar] [CrossRef]
- Gopalan, V.; Singh, A.; Rashidi Mehrabadi, F.; Wang, L.; Ruppin, E.; Arda, H.E.; Hannenhalli, S. A Transcriptionally Distinct Subpopulation of Healthy Acinar Cells Exhibit Features of Pancreatic Progenitors and PDAC. Cancer Res. 2021, 81, 3958–3970. [Google Scholar] [CrossRef] [PubMed]
- Baldan, J.; Houbracken, I.; Rooman, I.; Bouwens, L. Adult Human Pancreatic Acinar Cells Dedifferentiate into an Embryonic Progenitor-like State in 3D Suspension Culture. Sci. Rep. 2019, 9, 4040. [Google Scholar] [CrossRef]
- De Lisle, R.C.; Logsdon, C.D. Pancreatic Acinar Cells in Culture: Expression of Acinar and Ductal Antigens in a Growth-Related Manner. Eur. J. Cell Biol. 1990, 51, 64–75. [Google Scholar]
- Storz, P. Acinar Cell Plasticity and Development of Pancreatic Ductal Adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue Injury Adaptation and a Precursor to the Dysplasia-Cancer Sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Yosefov-Levi, O.; Kolodkin-Gal, D.; Granit, R.Z.; Peters, L.; Kalifa, R.; Xia, L.; Nasereddin, A.; Shiff, I.; Amran, O.; et al. Single-Cell Transcriptomes of Pancreatic Preinvasive Lesions and Cancer Reveal Acinar Metaplastic Cells’ Heterogeneity. Nat. Commun. 2020, 11, 4516. [Google Scholar] [CrossRef]
- Chan, C.S.; Laddha, S.V.; Lewis, P.; Koletsky, M.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.; Bose, P.; Chan, T.A.; et al. ATRX, DAXX or MEN1 Mutant Pancreatic Neuroendocrine Tumors Are a Distinct Alpha-Cell Signature Subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef]
- Di Domenico, A.; Pipinikas, C.P.; Maire, R.S.; Bräutigam, K.; Simillion, C.; Dettmer, M.S.; Vassella, E.; Thirlwell, C.; Perren, A.; Marinoni, I. Epigenetic Landscape of Pancreatic Neuroendocrine Tumours Reveals Distinct Cells of Origin and Means of Tumour Progression. Commun Biol. 2020, 3, 740. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Yoon, S.-J.; Park, J.; Shin, Y.; Choi, Y.; Park, S.W.; Kang, S.-G.; Son, H.Y.; Huh, Y.-M. Deconvolution of Diffuse Gastric Cancer and the Suppression of CD34 on the BALB/c Nude Mice Model. BMC Cancer 2020, 20, 314. [Google Scholar] [CrossRef]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial Maps of Prostate Cancer Transcriptomes Reveal an Unexplored Landscape of Heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Peng, X.L.; Moffitt, R.A.; Torphy, R.J.; Volmar, K.E.; Yeh, J.J. De Novo Compartment Deconvolution and Weight Estimation of Tumor Samples Using DECODER. Nat. Commun. 2019, 10, 4729. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Ma, Z.; Lytle, N.K.; Chen, B.; Jyotsana, N.; Novak, S.W.; Cho, C.J.; Caplan, L.; Ben-Levy, O.; Neininger, A.C.; Burnette, D.T.; et al. Single-Cell Transcriptomics Reveals a Conserved Metaplasia Program in Pancreatic Injury. Gastroenterology 2022, 162, 604–620.E20. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.C.; Bankaitis, E.D.; Boyer, D.; Xu, X.; Van de Casteele, M.; Magnuson, M.A.; Heimberg, H.; Wright, C.V.E. Spatiotemporal Patterns of Multipotentiality in Ptf1a-Expressing Cells during Pancreas Organogenesis and Injury-Induced Facultative Restoration. Development 2013, 140, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, A.G.; Hollister-Lock, J.; Sullivan, B.A.; Tsuchida, R.; Bonner-Weir, S.; Weir, G.C. Beta Cell Identity Changes with Mild Hyperglycemia: Implications for Function, Growth, and Vulnerability. Mol. Metab. 2020, 35, 100959. [Google Scholar] [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal--a One-Stop Shop for Cancer Genomics Data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, D.E.; Yu, R.; Won, K.-J.; Stoffers, D.A. Single Cell Transcriptomic Profiling of Mouse Pancreatic Progenitors. Physiol. Genom. 2017, 49, 105–114. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-Cell RNA-Seq Profiling of Human Preimplantation Embryos and Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Terry, M.T.; Patricia, M. Grambsch Modeling Survival Data: Extending the Cox Model. Stat. Med. 2001, 20, 2053–2054. [Google Scholar]
- Gaujoux, R.; Seoighe, C. A Flexible R Package for Nonnegative Matrix Factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef]
- Scholkopf, B.; Smola, A.J.; Williamson, R.C.; Bartlett, P.L. New Support Vector Algorithms. Neural Comput. 2000, 12, 1207–1245. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Purpose | ID—Source | Reference |
|---|---|---|---|---|
| Baron | scRNAseq | Training | GSE84133, GEO | [22] |
| Califano | bulk RNAseq | Benchmark | GSE98894, GEO | [18] |
| Diedisheim | bulk RNAseq | Benchmark | DOI: 10.1530/ERC-21-0051 | [17] |
| Fadista | bulk RNAseq | Out-group test | GSE50244, GEO | [27] |
| Haber | scRNAseq | HISC Training | GSE92332, GEO | [26] |
| Lawlor | scRNAseq | Training | GSE86473, GEO | [23] |
| Fröhling | bulk RNAseq | Benchmark | EGAS00001004813 | [28] |
| Missiaglia | microarray | Benchmark | GSE73338, GEO | [29] |
| Riemer | bulk RNAseq | Benchmark | EGAD00001006657 | unpublished |
| Sadanandam | microarray | Benchmark | GSE73339, GEO | [15] |
| Sato | bulk RNAseq | Benchmark | JGAS000237, NBDC | [30] |
| Scarpa | bulk RNAseq | Benchmark | EGAS00001001732, ICGC | [16] |
| Segerstolpe | scRNAseq | Training | E-MTAB-5061, Array Express | [24] |
| Tosti | snRNAseq | Training | EGAD00001006396, EGA | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otto, R.; Detjen, K.M.; Riemer, P.; Fattohi, M.; Grötzinger, C.; Rindi, G.; Wiedenmann, B.; Sers, C.; Leser, U. Transcriptomic Deconvolution of Neuroendocrine Neoplasms Predicts Clinically Relevant Characteristics. Cancers 2023, 15, 936. https://doi.org/10.3390/cancers15030936
Otto R, Detjen KM, Riemer P, Fattohi M, Grötzinger C, Rindi G, Wiedenmann B, Sers C, Leser U. Transcriptomic Deconvolution of Neuroendocrine Neoplasms Predicts Clinically Relevant Characteristics. Cancers. 2023; 15(3):936. https://doi.org/10.3390/cancers15030936
Chicago/Turabian StyleOtto, Raik, Katharina M. Detjen, Pamela Riemer, Melanie Fattohi, Carsten Grötzinger, Guido Rindi, Bertram Wiedenmann, Christine Sers, and Ulf Leser. 2023. "Transcriptomic Deconvolution of Neuroendocrine Neoplasms Predicts Clinically Relevant Characteristics" Cancers 15, no. 3: 936. https://doi.org/10.3390/cancers15030936
APA StyleOtto, R., Detjen, K. M., Riemer, P., Fattohi, M., Grötzinger, C., Rindi, G., Wiedenmann, B., Sers, C., & Leser, U. (2023). Transcriptomic Deconvolution of Neuroendocrine Neoplasms Predicts Clinically Relevant Characteristics. Cancers, 15(3), 936. https://doi.org/10.3390/cancers15030936