
Association between Plasminogen Activator Inhibitor-1 and Osimertinib Tolerance in EGFR-Mutated Lung Cancer via Epithelial–Mesenchymal Transition

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Patients
2.3. Cell Culture
2.4. Reagents
2.5. Quantitative RealTime PCR (qRT-PCR)
2.6. Quantification of PAI-1 Protein
2.7. Immunohistochemical Staining
2.8. Generation of EGFR-TKI-Tolerant Cells
2.9. EGFR T790M, C797S Mutation, and Gene Copy Number Analyses
2.10. Validation of PAI-1-siRNA Knockdown Efficacy
2.11. Proliferation Assay
2.12. Analysis of PAI-1- or EMT-Related Gene Expression
2.13. Microarray
2.14. Animal Experimental Procedure
2.15. Statistical Analysis
3. Results
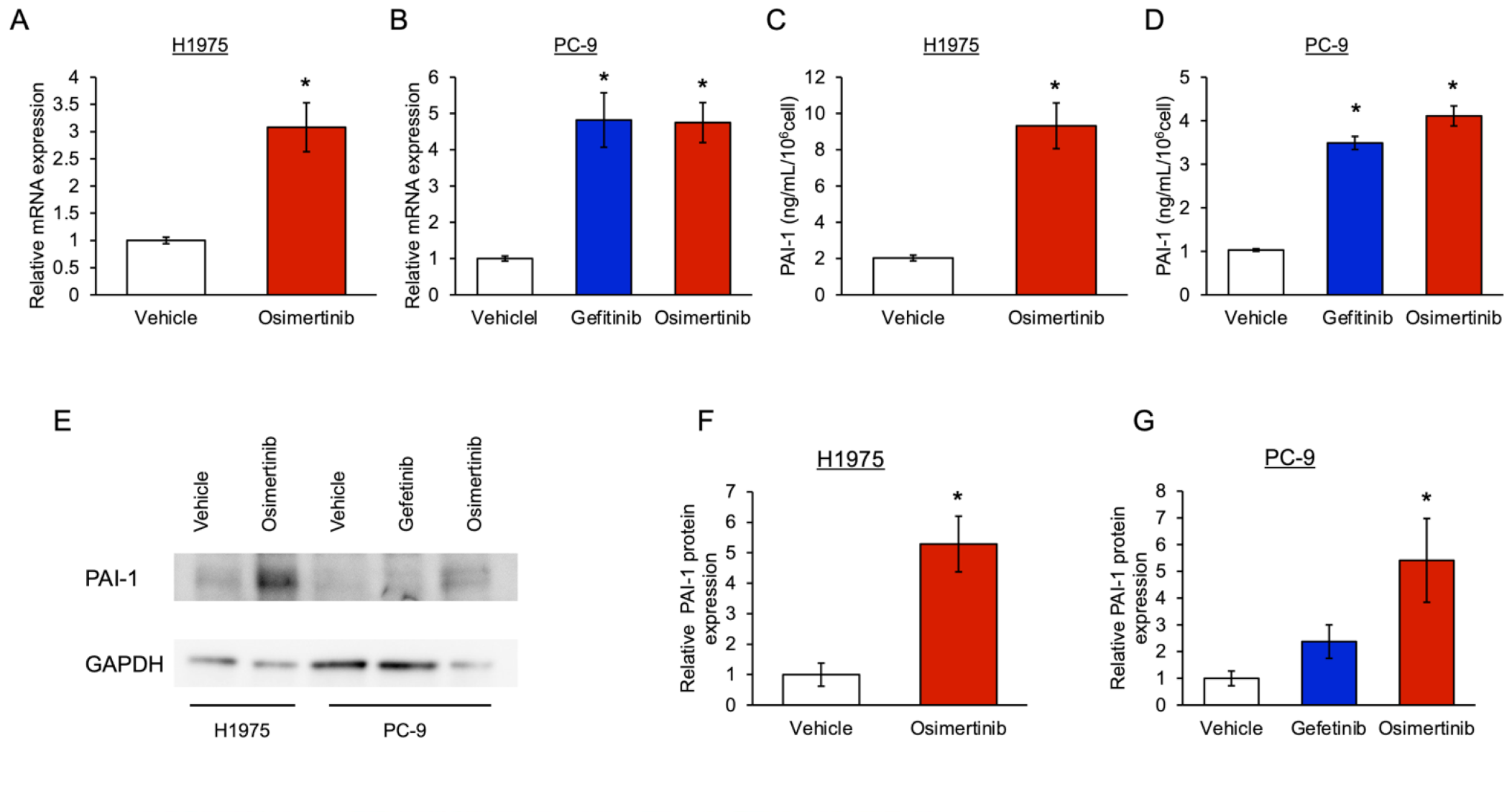
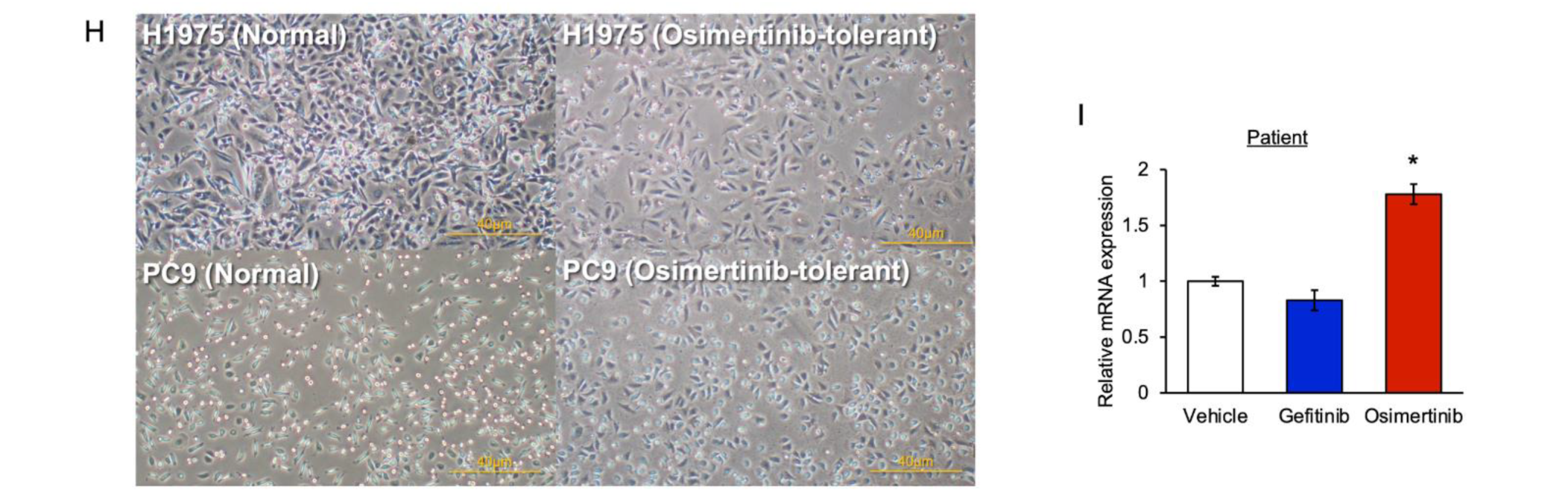
3.1. The Level of PAI-1 Increased in EGFR-Mutated Cancer Cells with Tolerance to EGFR-TKI
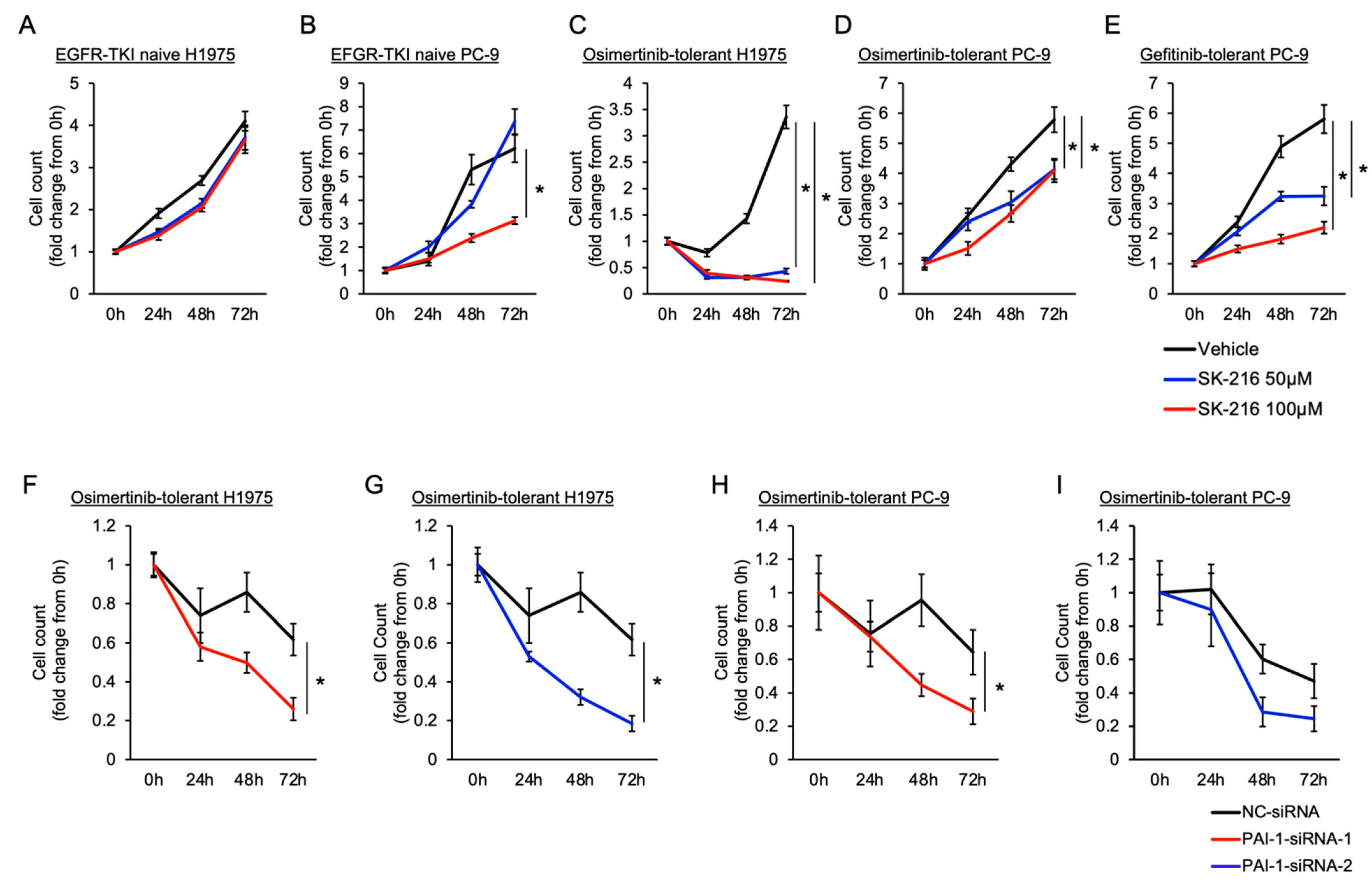
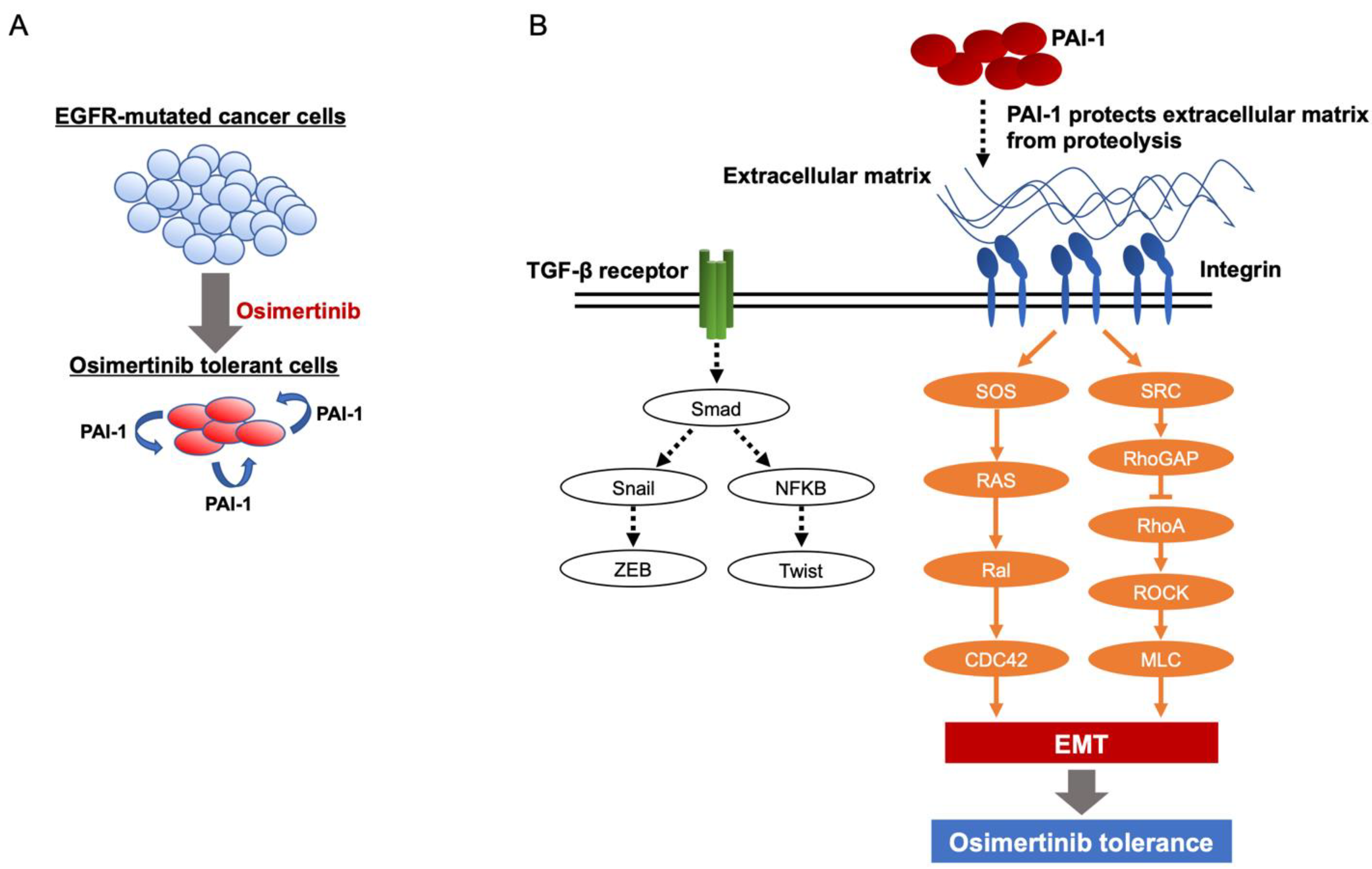
3.2. Involvement of PAI-1 in Tolerance to EGFR-TKI in EGFR-Mutated Lung Cancer Cells
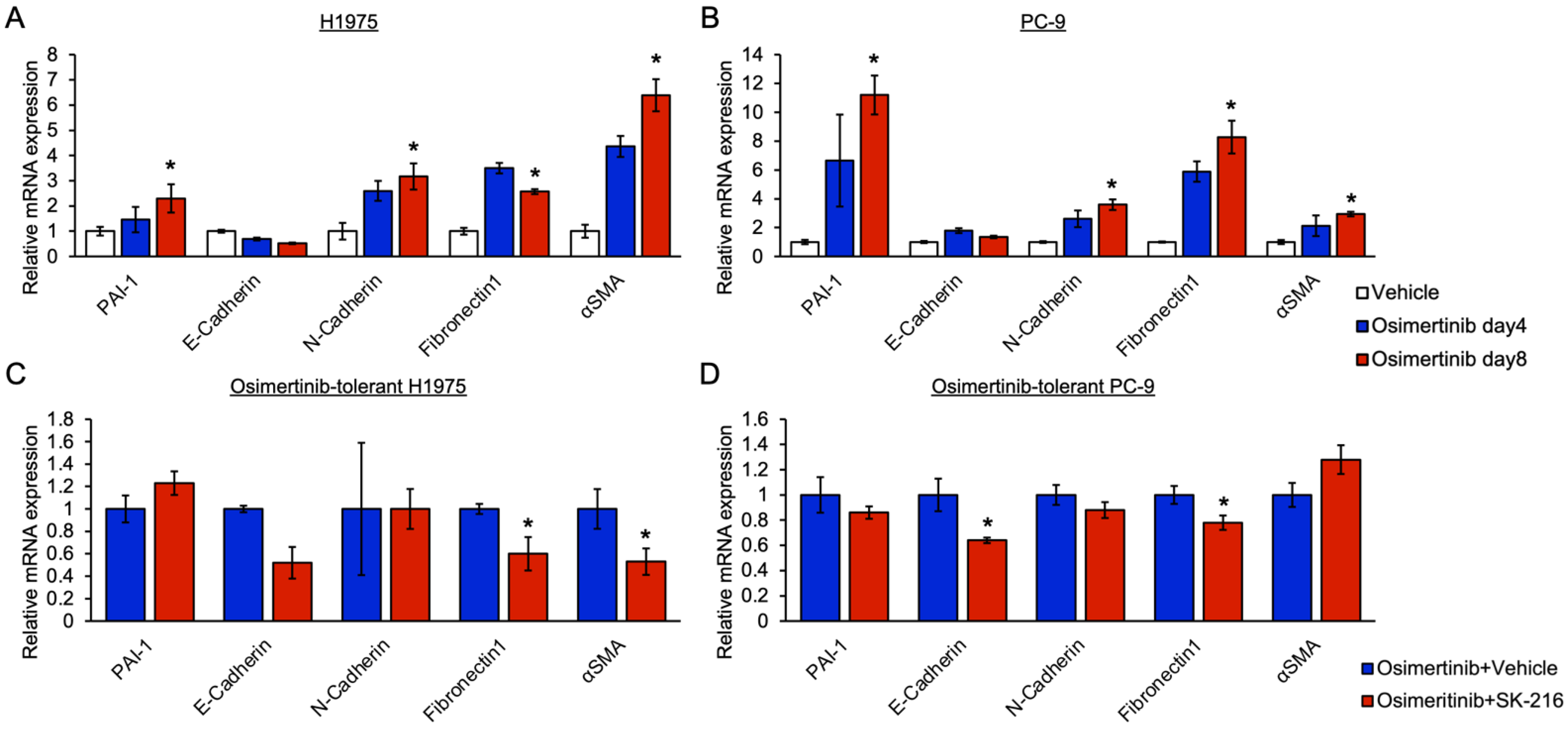
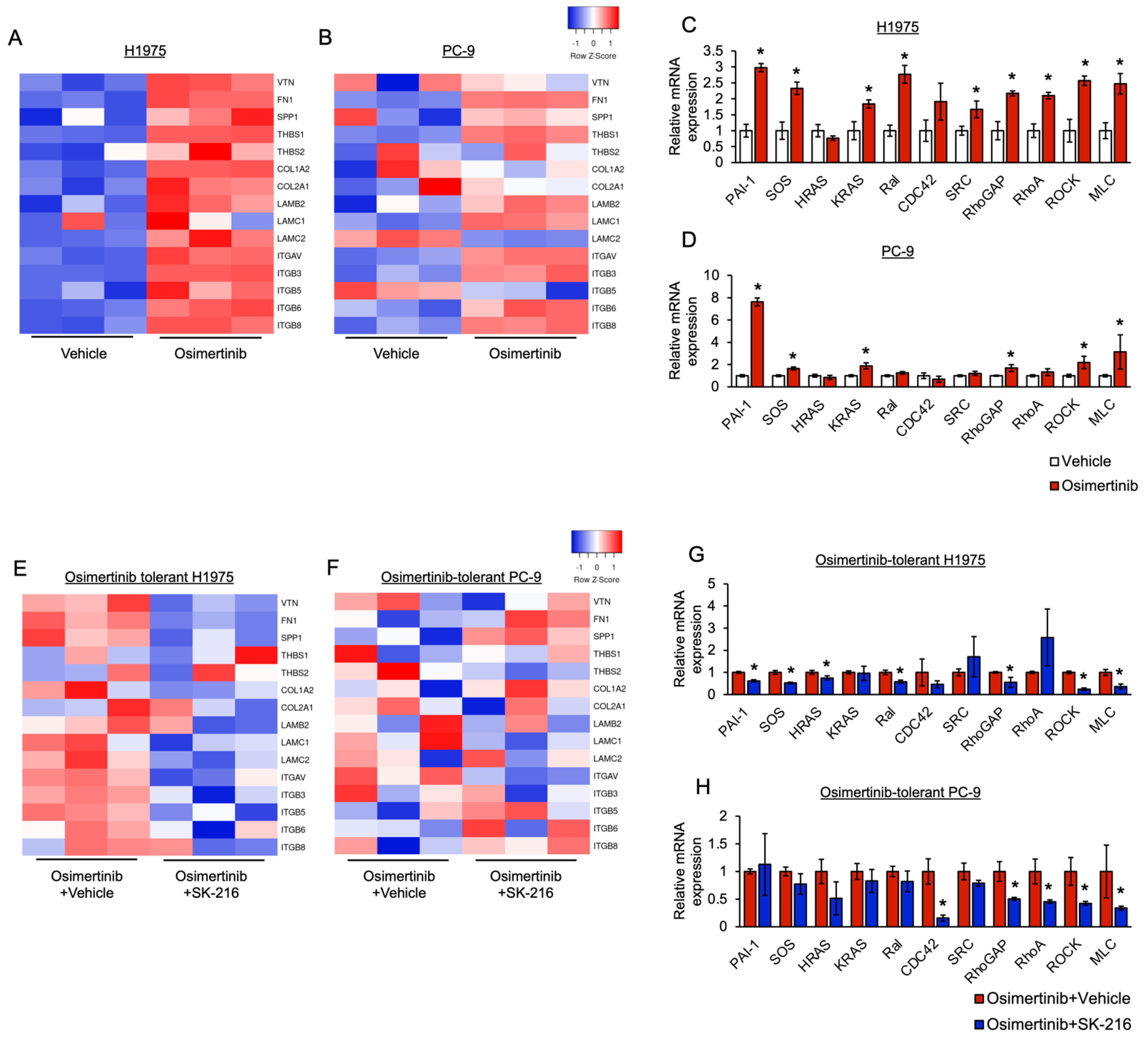
3.3. PAI-1 Was Found to Be Involved in Tolerance to Osimertinib in EGFR-Mutated Lung Cancer via Its Association with EMT
3.4. PAI-1 Was Found to Be Involved in Tolerance to Osimertinib via Its Association with Integrin-Initiated EMT
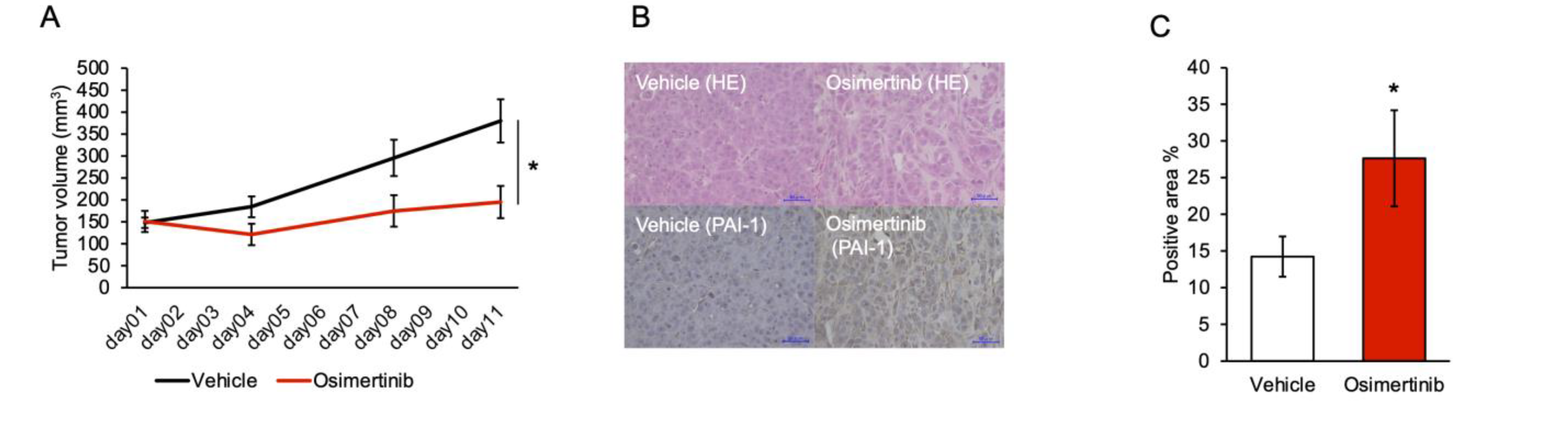
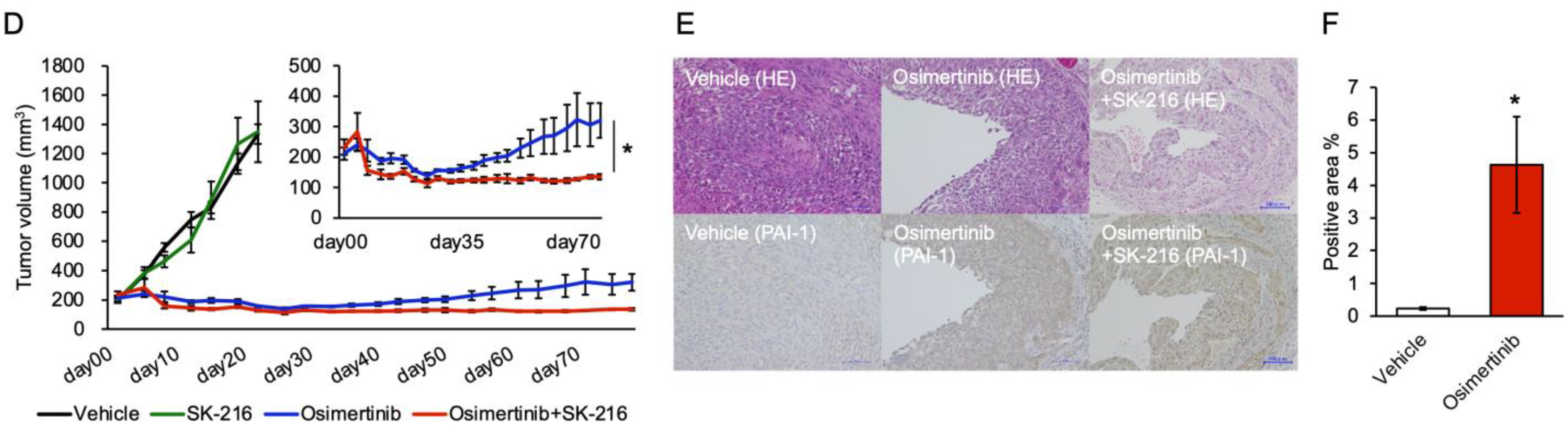
3.5. PAI-1 Inhibition Limited Regrowth of Osimertinib-Treated Tumors in the Subcutaneous Tumor Model
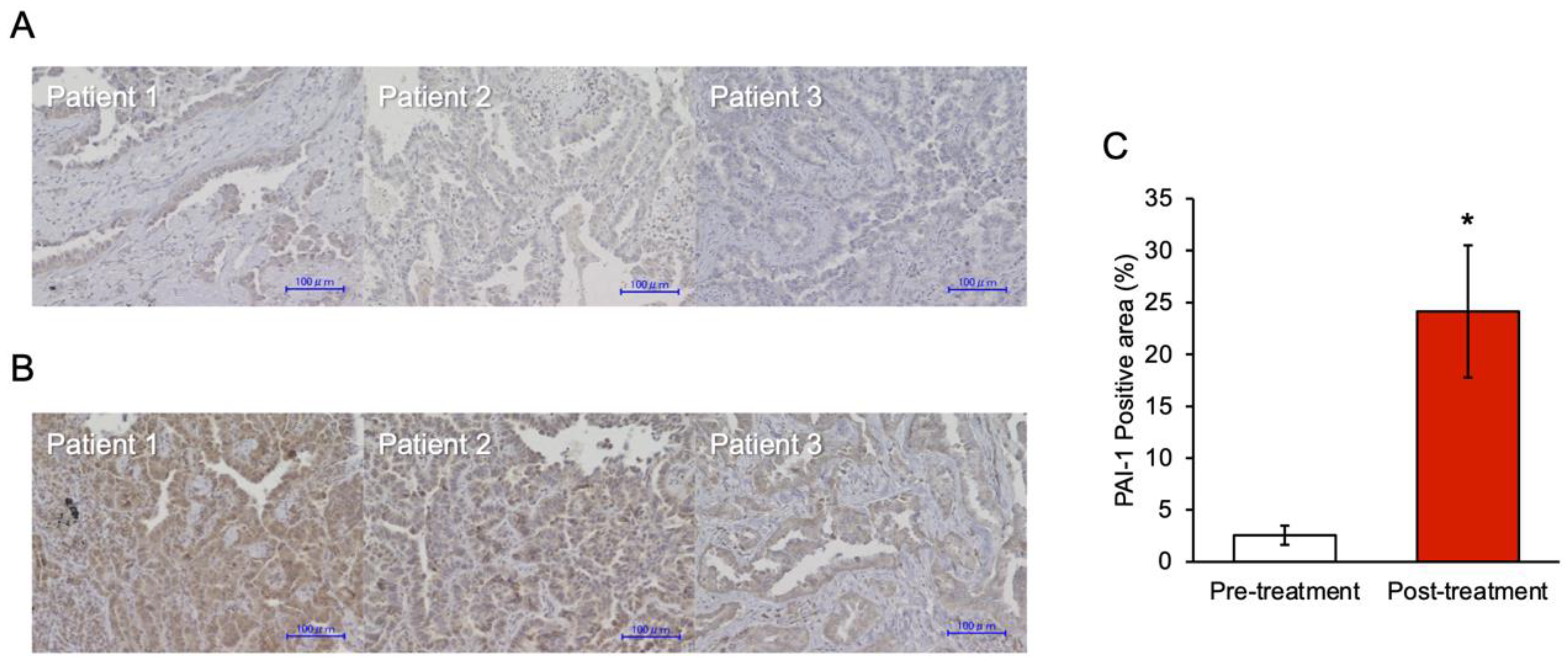
3.6. PAI-1 Expression in Tumor Specimens at the Time of Acquired Resistance to EGFR-TKIs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Nakayama, T.; Ioka, A.; Tsukuma, H. Trends in Lung Cancer Incidence by Histological Type in Osaka, Japan. Jpn. J. Clin. Oncol. 2008, 38, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Shiraishi, K.; Kunitoh, H.; Takenoshita, S.; Yokota, J.; Kohno, T. Gene Aberrations for Precision Medicine against Lung Adenocarcinoma. Cancer Sci. 2016, 107, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non-Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus Cisplatin plus Gemcitabine for First-Line Treatment of Asian Patients with Advanced Non-Small-Cell Lung Cancer Harbouring EGFR Mutations (LUX-Lung 6): An Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non-Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Li, X.-F.; Shen, W.-Z.; Jin, X.; Ren, P.; Zhang, J. Let-7c Regulated Epithelial-Mesenchymal Transition Leads to Osimertinib Resistance in NSCLC Cells with EGFR T790M Mutations. Sci. Rep. 2020, 10, 11236. [Google Scholar] [CrossRef]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. Targeting the EMT Transcription Factor Snail Overcomes Resistance to Osimertinib in EGFR-Mutant Non-Small Cell Lung Cancer. Thorac. Cancer 2021, 12, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhuo, X.; Wu, Y.; Fu, X.; Mao, C. PAR2 Blockade Reverses Osimertinib Resistance in Non-Small-Cell Lung Cancer Cells via Attenuating ERK-Mediated EMT and PD-L1 Expression. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119144. [Google Scholar] [CrossRef]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Kita, K.; Tanimoto, A.; Nishiyama, A.; Yano, S. Glycogen Synthase Kinase-3 Inhibition Overcomes Epithelial-Mesenchymal Transition-Associated Resistance to Osimertinib in EGFR-Mutant Lung Cancer. Cancer Sci. 2020, 111, 2374–2384. [Google Scholar] [CrossRef]
- Ji, W.; Choi, Y.J.; Kang, M.-H.; Sung, K.J.; Kim, D.H.; Jung, S.; Choi, C.-M.; Lee, J.C.; Rho, J.K. Efficacy of the CDK7 Inhibitor on EMT-Associated Resistance to 3rd Generation EGFR-TKIs in Non-Small Cell Lung Cancer Cell Lines. Cells 2020, 9, E2596. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.-B.; et al. Targeting the EMT Transcription Factor TWIST1 Overcomes Resistance to EGFR Inhibitors in EGFR-Mutant Non-Small-Cell Lung Cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- Jiang, X.-M.; Xu, Y.-L.; Yuan, L.-W.; Zhang, L.; Huang, M.-Y.; Ye, Z.-H.; Su, M.-X.; Chen, X.-P.; Zhu, H.; Ye, R.D.; et al. TGFβ2-Mediated Epithelial-Mesenchymal Transition and NF-ΚB Pathway Activation Contribute to Osimertinib Resistance. Acta Pharmacol. Sin. 2021, 42, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Mitsudomi, T. Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations. Cells 2021, 10, 1590. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR Overcomes EMT-Mediated Resistance in EGFR Mutant Non-Small Cell Lung Cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse Drug-Resistance Mechanisms Can Emerge from Drug-Tolerant Cancer Persister Cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Urano, T.; Umemura, K. PAI-1, Progress in Understanding the Clinical Problem and Its Aetiology. Br. J. Haematol. 2012, 157, 291–298. [Google Scholar] [CrossRef]
- Fang, H.; Placencio, V.R.; DeClerck, Y.A. Protumorigenic Activity of Plasminogen Activator Inhibitor-1 through an Antiapoptotic Function. J. Natl. Cancer Inst. 2012, 104, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- Rømer, M.U.; Larsen, L.; Offenberg, H.; Brünner, N.; Lademann, U.A. Plasminogen Activator Inhibitor 1 Protects Fibrosarcoma Cells from Etoposide-Induced Apoptosis through Activation of the PI3K/Akt Cell Survival Pathway. Neoplasia 2008, 10, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; Miyata, Y.; et al. Inhibition of PAI-1 Limits Chemotherapy Resistance in Lung Cancer through Suppressing Myofibroblast Characteristics of Cancer-Associated Fibroblasts. J. Cell Mol. Med. 2019, 23, 2984–2994. [Google Scholar] [CrossRef]
- Kang, J.; Kim, W.; Kwon, T.; Youn, H.; Kim, J.S.; Youn, B. Plasminogen Activator Inhibitor-1 Enhances Radioresistance and Aggressiveness of Non-Small Cell Lung Cancer Cells. Oncotarget 2016, 7, 23961–23974. [Google Scholar] [CrossRef]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An Open Source Plugin for the Quantitative Evaluation and Automated Scoring of Immunohistochemistry Images of Human Tissue Samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef]
- Murakami, A.; Takahashi, F.; Nurwidya, F.; Kobayashi, I.; Minakata, K.; Hashimoto, M.; Nara, T.; Kato, M.; Tajima, K.; Shimada, N.; et al. Hypoxia Increases Gefitinib-Resistant Lung Cancer Stem Cells through the Activation of Insulin-like Growth Factor 1 Receptor. PLoS ONE 2014, 9, e86459. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. IDEP: An Integrated Web Application for Differential Expression and Pathway Analysis of RNA-Seq Data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic. Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Hattori, N.; Hamada, H.; Masuda, T.; Omori, K.; Akita, S.; Iwamoto, H.; Fujitaka, K.; Kohno, N. Inhibition of PAI-1 Limits Tumor Angiogenesis Regardless of Angiogenic Stimuli in Malignant Pleural Mesothelioma. Cancer Res. 2016, 76, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Hattori, N.; Senoo, T.; Akita, S.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Kohno, N. SK-216, an Inhibitor of Plasminogen Activator Inhibitor-1, Limits Tumor Progression and Angiogenesis. Mol. Cancer Ther. 2013, 12, 2378–2388. [Google Scholar] [CrossRef]
- Pappot, H.; Pedersen, A.N.; Brünner, N.; Christensen, I.J. The Complex between Urokinase (UPA) and Its Type-1 Inhibitor (PAI-1) in Pulmonary Adenocarcinoma: Relation to Prognosis. Lung Cancer 2006, 51, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Foekens, J.A.; Peters, H.A.; Look, M.P.; Portengen, H.; Schmitt, M.; Kramer, M.D.; Brünner, N.; Jänicke, F.; Meijer-van Gelder, M.E.; Henzen-Logmans, S.C.; et al. The Urokinase System of Plasminogen Activation and Prognosis in 2780 Breast Cancer Patients. Cancer Res. 2000, 60, 636–643. [Google Scholar]
- Sakakibara, T.; Hibi, K.; Kodera, Y.; Ito, K.; Akiyama, S.; Nakao, A. Plasminogen Activator Inhibitor-1 as a Potential Marker for the Malignancy of Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2004, 10, 1375–1378. [Google Scholar] [CrossRef]
- Heiss, M.M.; Allgayer, H.; Gruetzner, K.U.; Babic, R.; Jauch, K.W.; Schildberg, F.W. Clinical Value of Extended Biologic Staging by Bone Marrow Micrometastases and Tumor-Associated Proteases in Gastric Cancer. Ann. Surg. 1997, 226, 736–744, discussion 744–745. [Google Scholar] [CrossRef]
- Ohba, K.; Miyata, Y.; Kanda, S.; Koga, S.; Hayashi, T.; Kanetake, H. Expression of Urokinase-Type Plasminogen Activator, Urokinase-Type Plasminogen Activator Receptor and Plasminogen Activator Inhibitors in Patients with Renal Cell Carcinoma: Correlation with Tumor Associated Macrophage and Prognosis. J. Urol. 2005, 174, 461–465. [Google Scholar] [CrossRef]
- Ren, J.; Chen, Y.; Song, H.; Chen, L.; Wang, R. Inhibition of ZEB1 Reverses EMT and Chemoresistance in Docetaxel-Resistant Human Lung Adenocarcinoma Cell Line. J. Cell Biochem. 2013, 114, 1395–1403. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Senoo, T.; Hattori, N.; Tanimoto, T.; Furonaka, M.; Ishikawa, N.; Fujitaka, K.; Haruta, Y.; Murai, H.; Yokoyama, A.; Kohno, N. Suppression of Plasminogen Activator Inhibitor-1 by RNA Interference Attenuates Pulmonary Fibrosis. Thorax 2010, 65, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Omori, K.; Hattori, N.; Senoo, T.; Takayama, Y.; Masuda, T.; Nakashima, T.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N. Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Transforming Growth Factor-β-Dependent Epithelial Mesenchymal Transition and Differentiation of Fibroblasts to Myofibroblasts. PLoS ONE 2016, 11, e0148969. [Google Scholar] [CrossRef]
- Yang, X.H.; Flores, L.M.; Li, Q.; Zhou, P.; Xu, F.; Krop, I.E.; Hemler, M.E. Disruption of Laminin-Integrin-CD151-Focal Adhesion Kinase Axis Sensitizes Breast Cancer Cells to ErbB2 Antagonists. Cancer Res. 2010, 70, 2256–2263. [Google Scholar] [CrossRef]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.-C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. Β1 Integrin Mediates an Alternative Survival Pathway in Breast Cancer Cells Resistant to Lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin Signalling during Tumour Progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Wang, C.; Wang, T.; Lv, D.; Li, L.; Yue, J.; Chen, H.-Z.; Xu, L. Acquired Resistance to EGFR TKIs Mediated by TGFβ1/Integrin Β3 Signaling in EGFR-Mutant Lung Cancer. Mol. Cancer Ther. 2019, 18, 2357–2367. [Google Scholar] [CrossRef]
- Ciambrone, G.J.; McKeown-Longo, P.J. Plasminogen Activator Inhibitor Type I Stabilizes Vitronectin-Dependent Adhesions in HT-1080 Cells. J. Cell Biol. 1990, 111, 2183–2195. [Google Scholar] [CrossRef]
- Burridge, K.; Fath, K.; Kelly, T.; Nuckolls, G.; Turner, C. Focal Adhesions: Transmembrane Junctions between the Extracellular Matrix and the Cytoskeleton. Annu. Rev. Cell Biol. 1988, 4, 487–525. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in Tissue Fibrosis. J. Cell Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef]
- Ge, X.; Zhang, Y.; Huang, F.; Wu, Y.; Pang, J.; Li, X.; Fan, F.; Liu, H.; Li, S. EGFR Tyrosine Kinase Inhibitor Almonertinib Induces Apoptosis and Autophagy Mediated by Reactive Oxygen Species in Non-Small Cell Lung Cancer Cells. Hum. Exp. Toxicol. 2021, 40, S49–S62. [Google Scholar] [CrossRef]
- Swiatkowska, M.; Szemraj, J.; Al-Nedawi, K.N.I.; Pawłowska, Z. Reactive Oxygen Species Upregulate Expression of PAI-1 in Endothelial Cells. Cell Mol. Biol. Lett. 2002, 7, 1065–1071. [Google Scholar]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-Dependent β-Catenin Signaling Mediates EGFR TKI Drug Persistence in EGFR Mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Sex | F | M | M | M | M |
| Age (years) | 66 | 65 | 52 | 66 | 63 |
| Histological classification | ad | ad | ad | ad | ad |
| Clinical stage at diagnosis | IIIA | IIA | IIB | I | IIIB |
| EGFR mutation category | Exon 19 del | Exon 19 del | Exon 19 del | Exon 21 L858R | Exon 18 D719C |
| Tissue collection site (at diagnosis) | Lung | Lung | Lung | Lung | Lung |
| Method of tissue collection (at diagnosis) | Surgery | Surgery | Surgery | Surgery | Surgery |
| Tissue collection site (second biopsy) | Lung | Lung | Lung | Bone | Lymph node |
| Method of tissue collection (second biopsy) | Surgery | Surgery | Surgery | CT | Surgery |
| Presence of T790M mutation (second biopsy) | Yes | Yes | Yes | No | No |
| EGFR-TKI used before the second biopsy | Gefitinib | Gefitinib | Erlotinib | Afatinib | Afatinib |
| EGFR-TKI used after the second biopsy | Osimertinib | Osimertinib | Osimertinib | None | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokumo, K.; Masuda, T.; Nakashima, T.; Namba, M.; Yamaguchi, K.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; Iwamoto, H.; Fujitaka, K.; et al. Association between Plasminogen Activator Inhibitor-1 and Osimertinib Tolerance in EGFR-Mutated Lung Cancer via Epithelial–Mesenchymal Transition. Cancers 2023, 15, 1092. https://doi.org/10.3390/cancers15041092
Tokumo K, Masuda T, Nakashima T, Namba M, Yamaguchi K, Sakamoto S, Horimasu Y, Miyamoto S, Iwamoto H, Fujitaka K, et al. Association between Plasminogen Activator Inhibitor-1 and Osimertinib Tolerance in EGFR-Mutated Lung Cancer via Epithelial–Mesenchymal Transition. Cancers. 2023; 15(4):1092. https://doi.org/10.3390/cancers15041092
Chicago/Turabian StyleTokumo, Kentaro, Takeshi Masuda, Taku Nakashima, Masashi Namba, Kakuhiro Yamaguchi, Shinjiro Sakamoto, Yasushi Horimasu, Shintaro Miyamoto, Hiroshi Iwamoto, Kazunori Fujitaka, and et al. 2023. "Association between Plasminogen Activator Inhibitor-1 and Osimertinib Tolerance in EGFR-Mutated Lung Cancer via Epithelial–Mesenchymal Transition" Cancers 15, no. 4: 1092. https://doi.org/10.3390/cancers15041092
APA StyleTokumo, K., Masuda, T., Nakashima, T., Namba, M., Yamaguchi, K., Sakamoto, S., Horimasu, Y., Miyamoto, S., Iwamoto, H., Fujitaka, K., Miyata, Y., Okada, M., Hamada, H., & Hattori, N. (2023). Association between Plasminogen Activator Inhibitor-1 and Osimertinib Tolerance in EGFR-Mutated Lung Cancer via Epithelial–Mesenchymal Transition. Cancers, 15(4), 1092. https://doi.org/10.3390/cancers15041092