1. Introduction
Ovarian cancer is the 5th most lethal malignancy for women; 13,980 women died from ovarian cancer last year in the United States [
1]. The US Surveillance, Epidemiology, and End Results (SEER) database reports that 5-year overall survival in patients with stage III and stage IV cancer is 25%. While the current standard treatment of ovarian cancer is cytoreductive surgery in conjunction with platinum plus taxane-based chemotherapy, the mortality of ovarian cancer has only slightly improved in the past decade [
2]. In general, ovarian cancers show an initial response to chemotherapy but gain anti-apoptotic ability by increasing the expression of anti-apoptotic molecules or inactivating pro-apoptotic cell death components, leading to tumor recurrence and drug resistance [
3]. In this regard, various attempts to restore the apoptotic mechanism of cancer cells are ongoing [
4].
Apoptosis is the most important regulated mechanism of cell death, led by two distinct pathways, the death receptor signal pathway (extrinsic apoptosis) and the mitochondrial pathway (intrinsic apoptosis), both result in the activation of caspases. To prevent inappropriate activation of caspases, apoptosis is strictly regulated by both pro-apoptotic and anti-apoptotic proteins. One such protein is receptor-interacting protein 1 (RIP1), which is ubiquitinated by two cellular inhibitor of apoptosis proteins (cIAPs), cIAP1 and cIAP2 [
5]. The ubiquitinated RIP1 complex leads to downstream signaling, resulting in the transcription of NF-kB gene targets. A second mitochondrial activator of caspases (SMAC) proteins released from mitochondria ubiquitinate XIAP to promote apoptosis [
6], but this mitochondrial pathway is blocked by high expression levels of IAP proteins in some chemotherapy-resistant recurrent cancers. In ovarian cancer patients, high cIAP1/2 expression correlated with significantly shorter survival compared to patients with low cIAP1/2 expressing cancers in multivariate analysis [
7].
Small molecules designed to mimic the IAP binding motif of SMAC, which binds to the BIR (baculoviral IAP repeat) 2 and BIR3 domains of cIAPs, are called SMAC mimetics (SMs) [
8]. After SMs remove cIAPs by auto-ubiquitination, de-ubiquitinated RIP1 can form distinct death complexes with tumor necrosis factor receptor type 1-associated death domain protein (TRADD), resulting in tumor necrosis factor-α (TNF-α) induced cell death through caspase-mediated apoptosis or RIP3/MLKL mediated necroptosis. SMs are intended to treat cancer with minimal toxicity to normal cells since they preferentially target cancer cells which depend on cIAPs more so than healthy cells. To date, SMs have entered clinical trials for hematological and solid cancers [
9].
We previously completed a phase 2 clinical study of the SMAC mimetic, birinapant, in women with relapsed platinum-resistant or platinum-refractory epithelial ovarian cancer. Birinapant showed consistent on-target suppression of cIAP1 in tumor biopsies and peripheral blood mononuclear cells, but single-agent anti-tumor activity did not meet the pre-specified primary endpoint of 20% response rate in order to justify proceeding with the development of the single agent [
10]. Because of the strong on-target effect but minimal efficacy, we undertook a drug screening to find rational combination therapies to enhance the anti-cancer activity of SMAC mimetics. Matrix drug screening using a library of 1912 compounds crossed with birinapant found that histone deacetylase (HDAC) inhibitors (entinostat, panobinostat, vorinostat, romidepsin, and Trichostatin A) are highly synergistic with SMs in killing ovarian cancer cells [
11]. Here, we tested the hypothesis that HDAC inhibitors sensitize cancer cells to SMs, suggesting that the combination of SM and HDAC inhibitors can be a novel anti-cancer therapy.
Histone deacetylases (HDACs) are important epigenetic regulators of gene expression, removing histone acetylation enzymatically. There are three main HDAC classes (I, II, and IV) comprised of at least 11 HDACs. Histone acetylation is generally associated with elevated gene transcription, but overexpression of HDACs in cancer is thought to repress tumor suppressor genes by histone deacetylation, thus resulting in tumor progression. With this goal, various HDAC inhibitors are in clinical trials as novel cancer therapeutics [
12].
In this study, we demonstrate a mechanism of synergy between SMs and HDAC inhibitors that involves TNF-α. Recently, a genome-wide siRNA screen identified that the regulation of TNF-α mRNA expression by transcription factor SP3 is a critical factor for SMs mediated cancer cell death [
13]. TNF-α is a trigger of the death receptor signal, which is necessary for immunity and anti-cancer effects through regulated cell death, especially in the presence of SM-facilitated apoptosis. We, therefore, hypothesized that HDAC inhibitors sensitize cancer cells to SMAC mimetics by enhancing TNF-α production via SP3. Here, we tested both birinapant, a dimeric peptidomimetic compound, and another SMAC mimetic, tolinapant, a non-peptidomimetic antagonist of cIAPs and XIAP, discovered using fragment-based drug design [
14]. These SMs were combined with different HDAC inhibitors, including the class I selective HDAC inhibitor, entinostat, reported to have an anti-tumor effect in an ovarian cancer model [
15], the broader-acting panobinostat, as well as romidepsin and vorinostat. We demonstrate that the synergy is not restricted to individual drugs but is a class effect of the SMs and HDAC inhibitors. By identifying the mechanism of this synergy, we hope to move this combination therapy forward clinically to present a novel treatment option for women with chemo-refractory ovarian cancer.
2. Materials and Methods
2.1. Cell Culture
Ovarian cancer cell lines were obtained from the NCI-Frederick Developmental Therapeutics Program tumor/cell line repository (Frederick, MD, USA). Human ovarian cancer cell lines (OVCAR3, OVCAR8, SKOV3, TOV21G, ES-2, A2780, and IGROV1) were grown in RPMI medium supplemented with 10% FBS, 1% penicillin/streptomycin. Mouse ovarian cancer cell line ID8 was grown in DMEM medium with 4% FBS, 1% penicillin/streptomycin, insulin (5 μg/mL), transferrin (5 μg/mL), sodium selenite (5 ng/mL) (ITX mix, Sigma -Aldrich #I-1884, St. Louis, MO, USA). All cells were maintained at 37 °C under a 5% CO
2 atmosphere. Cell lines were authenticated via Short Tandem Repeat at Frederick National Laboratory. Authenticity was confirmed against the ATCC database (
www.atcc.org/CulturesandProducts/CellBiology/STRProfileDatabase/tabid/174/Default.aspx (accessed on 1 May 2019)), CLIMA database (
http://bioinformatics.istge.it/clima/ (accessed on 1 May 2019)), and NCI-60 database published data.
2.2. XTT Viability Assay
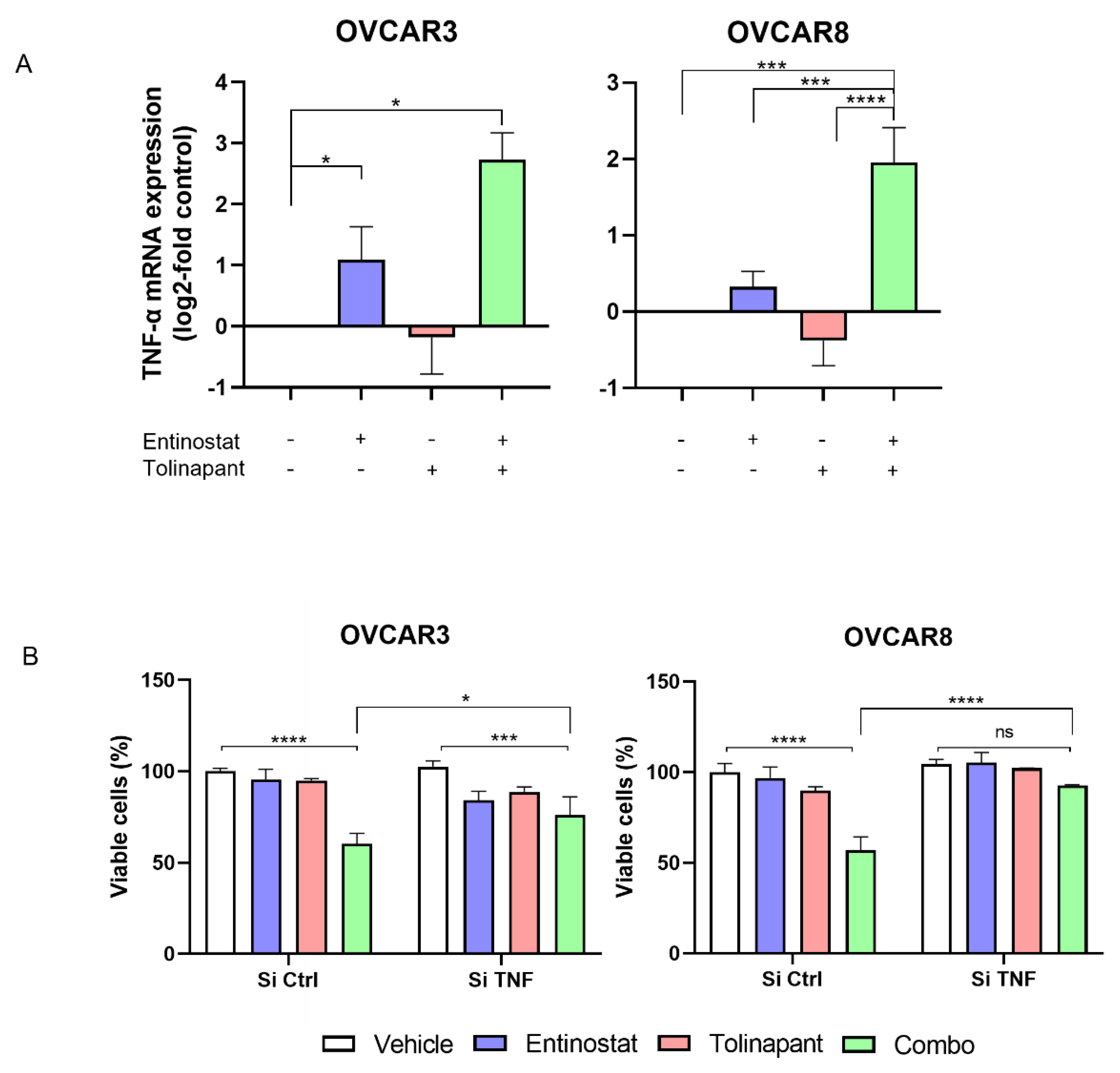
Further in vitro testing verified synergistic activity between tolinapant and entinostat, romidepsin, or vorinostat. OVCAR3, OVCAR8, SKOV3, TOV21G, ES-2, A2780, IGROV1, and ID8 cancer cell lines were seeded in 96-well plates at a density of 2–4 × 103 cells/well and incubated for 24 h prior to drug addition. In the case of dual drug treatment, both drugs were added simultaneously. Then, 72 h after drug application, XTT-PMS dye was added to each well, incubated for 1.5 h, and then OD 450 nm was measured to determine cell viability (Molecular Devices SpectraMax ID3 plate reader).
2.3. Western Blot
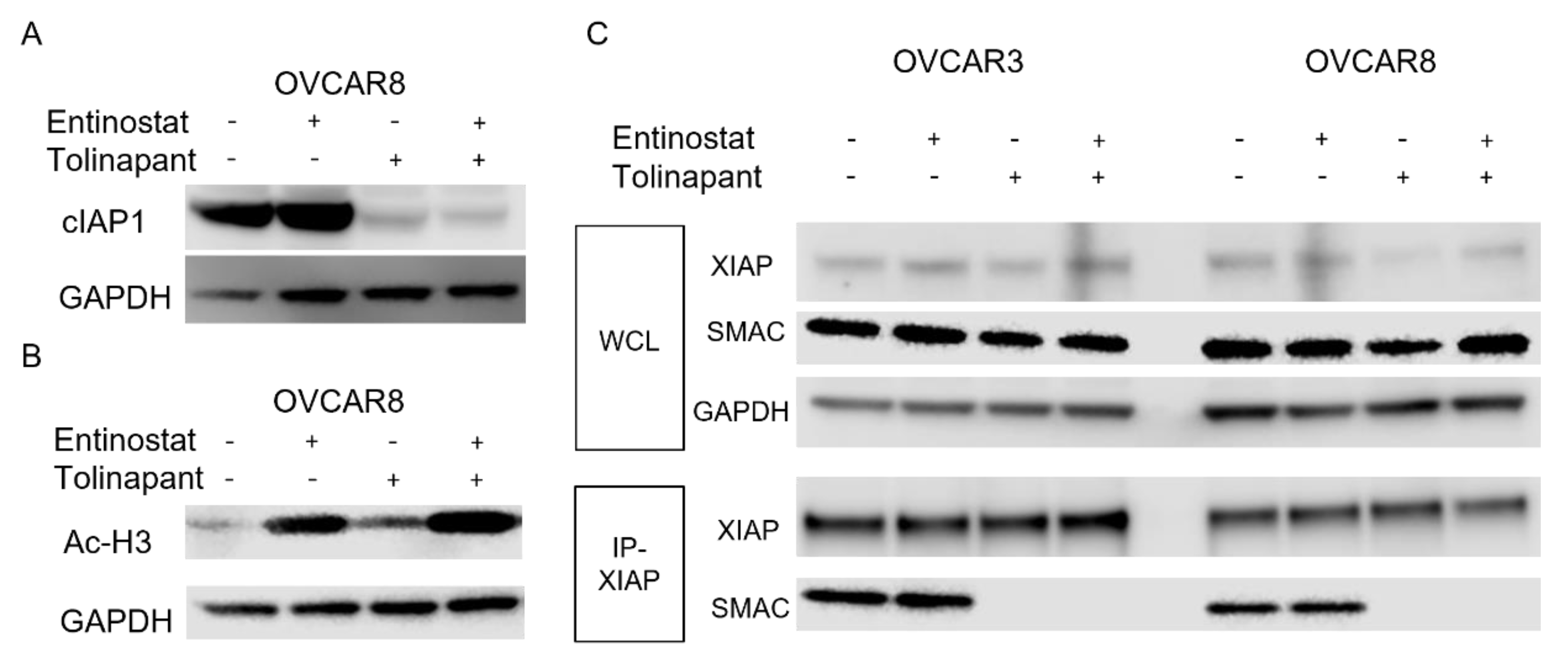
OVCAR3, OVCAR8, SKOV3, TOV21G, ES-2, A2780, IGROV1, and ID8 cells were plated in 6-well plates at a density of 1.0 × 10
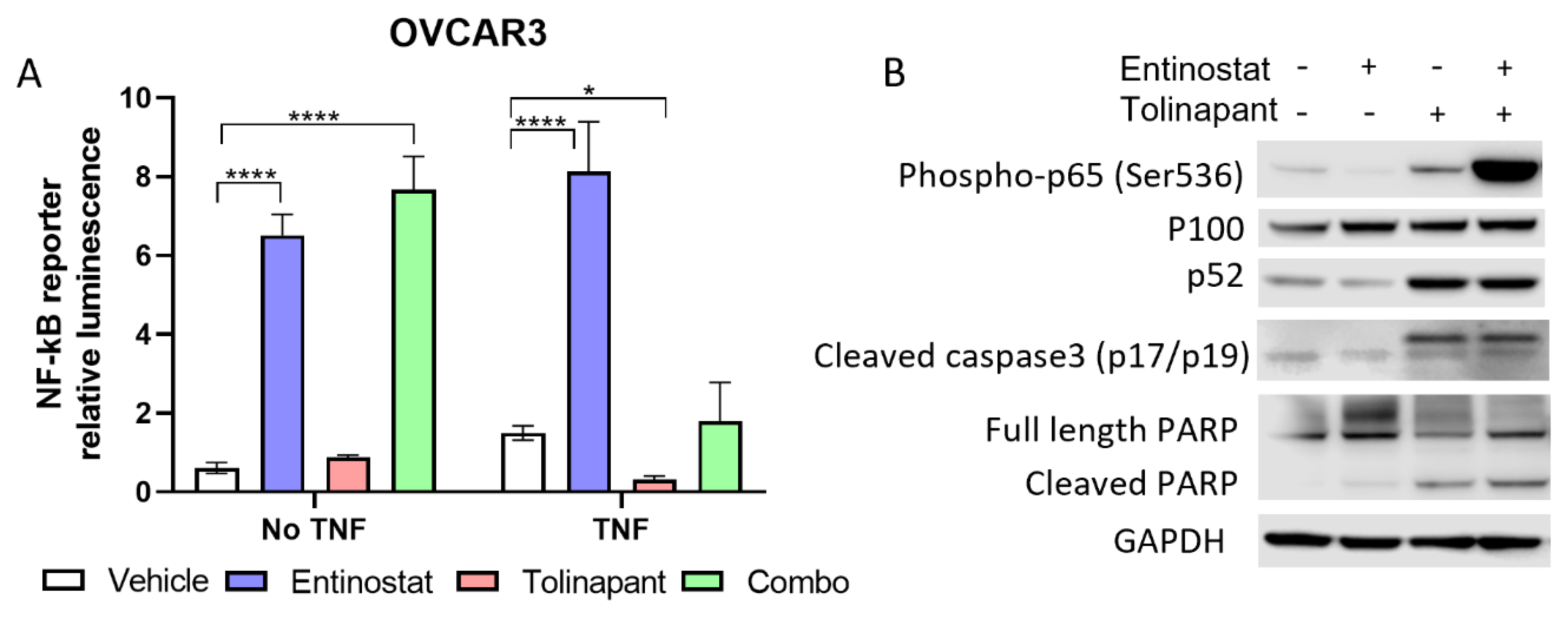
6 to allow for evaluation of protein expression following treatment with single agent tolinapant, entinostat alone, or combination therapy. Cells were treated for 24 h or 72 h, and then total protein was extracted using M-PER buffer (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Concentrations were estimated with the BCA Protein Assay Kit (Thermo Scientific). SDS-Page and Western analysis were performed using the NuPage system (Invitrogen, Waltham, MA, USA) and the Supersignal Chemiluminescent Substrate System (Thermo Scientific), respectively. The following primary antibodies were used: GAPDH (MAB374, Sigma-Aldrich), SMAC (#2954S CST), DR5 (#8074S CST), Acetyl-Histone H3 (K9/K14, #9677S CST), HDAC1 (#34589 CST), p52 (MAB05-361 Sigma-Aldrich), XIAP (#2042S CST), human cIAP1 (AF8181 R&D), mouse cIAP1 (ALX-803-335-C100 ENZO), SP3 (sc-365220 Santa Cruz), Phospho-NF-kB p65 (#3033 CST), Cleaved Caspase3 (#9664 CST). Protein signal quantitation used Image Studio Lite Ver 5.2 (LI-COR). Original blots can be found at
Supplementary File S1.
2.4. Co-Immunoprecipitation (Co-IP)
OVCAR3, OVCAR8, and TOV21G cells were treated with single-agent tolinapant, entinostat alone, or combination therapy for 24 h, and total protein was extracted using RIPA buffer (Thermo Scientific) according to the manufacturer’s protocol, followed by sonication (Fisherbrand™ Model 120 Sonic Dismembrator). Concentrations were estimated with the BCA Protein Assay Kit (Thermo Scientific). A total of 1 μg of total cellular protein and 1 μg of primary antibodies are incubated overnight at 4 degrees with protein A/G beads (sc-2003 Santa Cruz). Beads were washed and boiled in SDS sample buffer containing DTT before analysis of the eluted proteins by Western blotting. Western blots of the same lysate before immunoprecipitation were used for comparison.
2.5. Caspase Activity Assay
Caspase 3/7, Caspase 8, and Caspase 9 activity were measured in OVCAR3 and OVCAR8 cells lines using Caspase-Glo luminescence assays (Promega, G8091, G8201, G8211) according to the manufacturer’s specifications after exposing cells to panobinostat 10 nM alone and in combination with birinapant 20 μM for 24 h. All drug exposures occurred with or without 10 ng/mL of TNF-α. Activity data were normalized to viable cell number and measured in an identical plate by XTT assay as described.
2.6. NF-kB Reporter Assay
OVCAR3 and OVCAR8 cell lines were selected given their high canonical NF-kB activity. Cells were transduced with a luciferase reporter to measure NF-kB (p65) activation. Reporter cells were then plated at a density of 2000 cells/50 μL per well in a 96-well plate and then exposed to entinostat alone and in combination with tolinapant for 24 h. All drug exposures occurred with and without TNF-α (10 ng/mL). Quantitative luminescence was measured by Promega Luciferase Assay using a Molecular Devices SpectraMax ID3 plate reader and normalized to a cellular viability assay using XTT-PMS dye.
2.7. RNAi Experiments
OVCAR8 cells were transfected with siRNAs using DharmaFECT 1 Transfection Reagent per standard manufacture procedure (Horizon Discovery), and drugs were added 2 days later. The siRNAs used were as follows: ON-TARGET plus Non-targeting Pool (D-001810-10; Horizon Discovery, Waterbeach, UK), ON-TARGET plus siRNA against human TNF-α (L-010546; Horizon Discovery).
2.8. Cytokine Assay
Secreted cytokine levels of IL-6, IL-8, and TNF-α were measured in culture supernatants using the Mesoscale multiplex assay after treating OVCAR3 and OVCAR8 cells overnight with Panobinostat 20 μM alone and in combination with birinapant 10 nM per manufacturer’s protocol (MesoScale Discovery, Rockville, MD, USA).
2.9. RNA Extraction and Quantitative Real-Time PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany) per the manufacturer’s protocol. The final RNA concentration was determined with a NanoDrop spectrophotometer using the 260/280 absorbance ratio. Total purified RNA was reverse transcribed with random primers using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) per the manufacturer’s protocol. The resulting cDNA was used as a template for quantitative real-time PCR (qRT-PCR). Analysis of gene expression was performed on ViiA7 Real-time PCR System (Applied Biosystems) using TaqMan probe assays with GAPDH and ACTB as control. Quantitation and normalization of relative gene expression were accomplished using ddCT (Delta-Delta-cycle threshold) method. Catalog numbers for commercial primers are provided in
Supplementary Table S1.
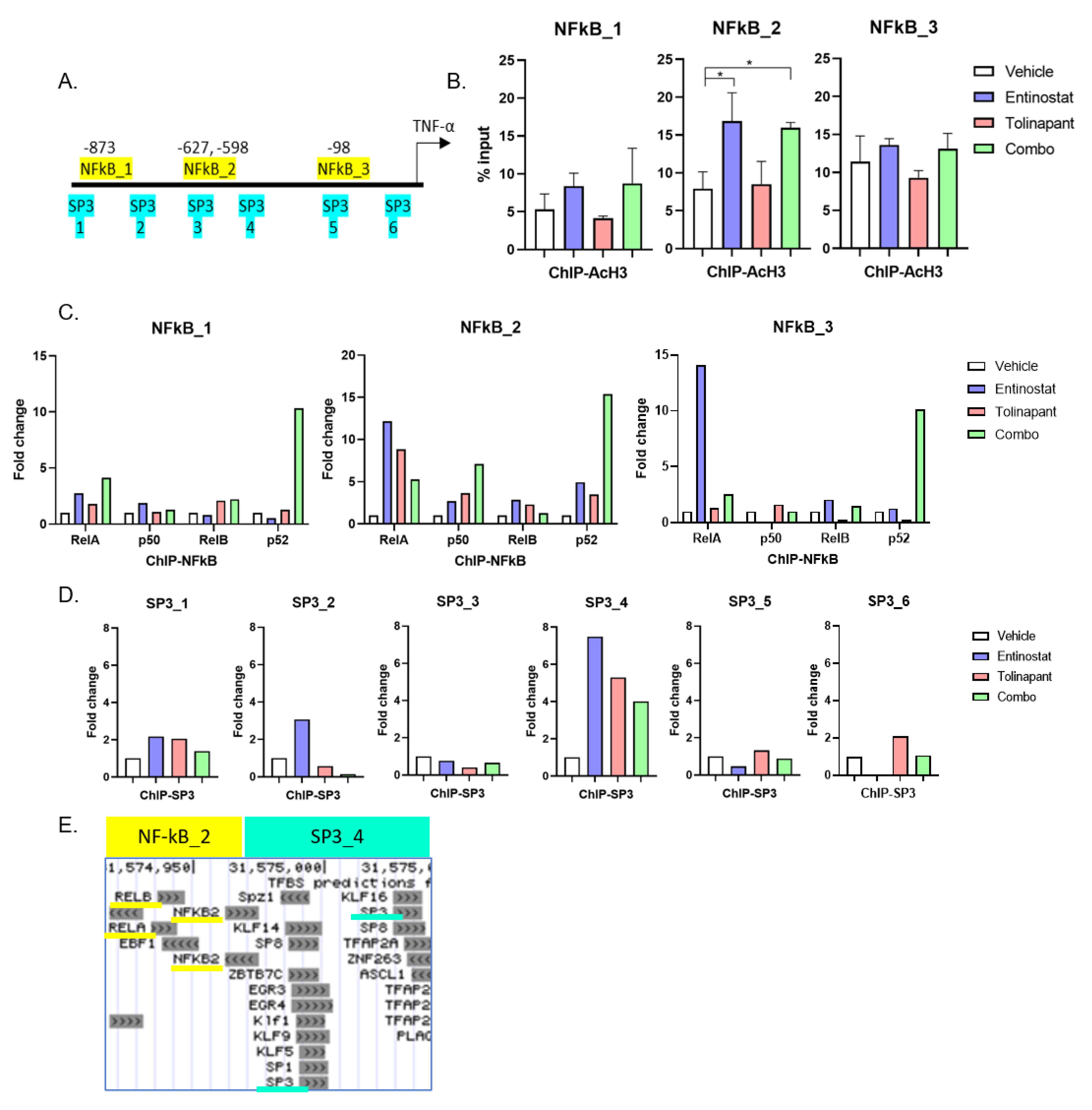
2.10. Chromatin Immunoprecipitation (ChIP)—qPCR Assay
The SimpleChIP Enzymatic Chromatin IP Kit (magnetic beads) was purchased from Cell Signaling Technology, and assays were performed according to the manufacturer’s instructions. Antibodies for RelA (NF-kB p65 #8242S CST), p50 (NF-kBp105/50 #13586S), RelB (#10544 CST), p52 (NF-kB p100/52 #37359S CST), Acetyl-Histone H3 (K27, #4353S CST), and SP3 (sc-365220 Santa Cruz). Genome browser (
https://genome.ucsc.edu/index.html (accessed on 1 August 2019)) and Jasper (
http://jaspar.genereg.net/ (accessed on 1 August 2019)) were used to evaluate DNA sequences for transcription factor binding sites. Analysis of ChIP was performed on a ViiA7 Real-time PCR System (Applied Biosystems) using QuantiTect SYBR Green PCR Kit (Qiagen). The quantification of transcription factors binding to target sites was calculated by measuring the ddCT (Delta-Delta-cycle threshold) ratio of chromatin immunoprecipitation (ChIP) to 2% Input, and the normal rabbit IgG antibody served as a negative control. All primers used for ChIP PCRs are listed in
Supplementary Table S2.
2.11. In Vivo Mouse Studies
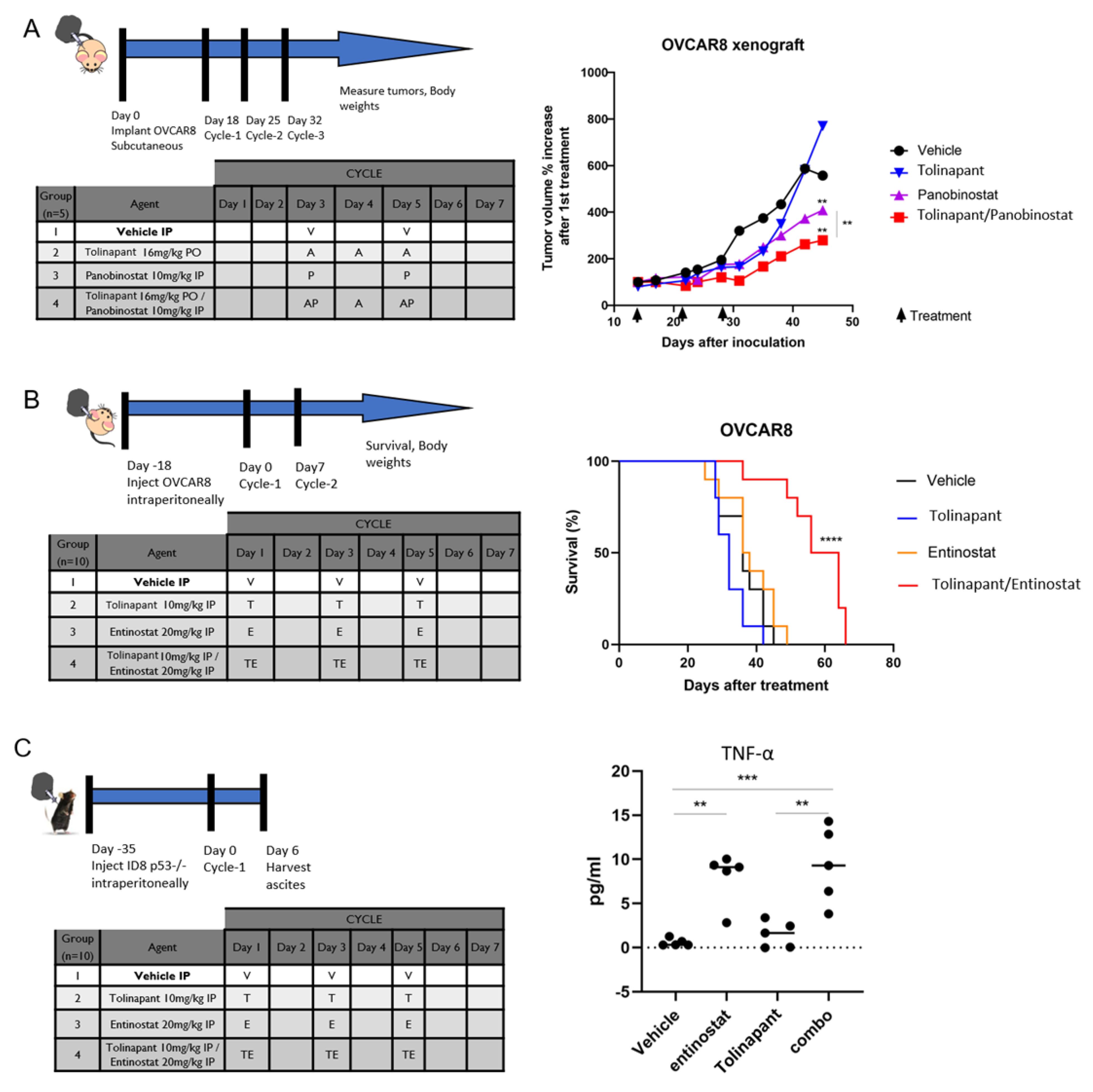
2.11.1. Xenograft Model
1–2 × 106 of OVCAR8 cells were counted and prepared as suspensions in 0.5 mL PBS for subcutaneous (flank) injections into 6–8 weeks old athymic nude female mice. Tumors were grown for two weeks before the mice were randomized into treatment groups. Mice then received intraperitoneal (IP) treatment with vehicle control (5% dextrose), Panobinostat 10 mg/kg, per oral (PO) treatment of tolinapant 16 mg/kg, or combination panobinostat plus tolinapant. Body weights and tumor measurements were taken twice weekly for 8–10 weeks or as required by humane endpoints. Subcutaneous tumor volumes were calculated according to the formula V = 1/2(length × width2).
2.11.2. Survival Study: Immune Deficient Model
1–2 × 106 OVCAR8 cells were injected IP into 6–8 weeks old athymic nude female mice and allowed to grow for 2 weeks before IP treatment with vehicle control (5% dextrose), entinostat 20 mg/kg, tolinapant 10 mg/kg, or combination entinostat plus tolinapant. Mice were followed until euthanasia endpoints and scored for overall survival. Animal care was provided in accordance with procedures in the Guide for the Care and Use of Laboratory Animals. Experiments were carried out according to a protocol approved by the NCI Animal Care and Use Committee.
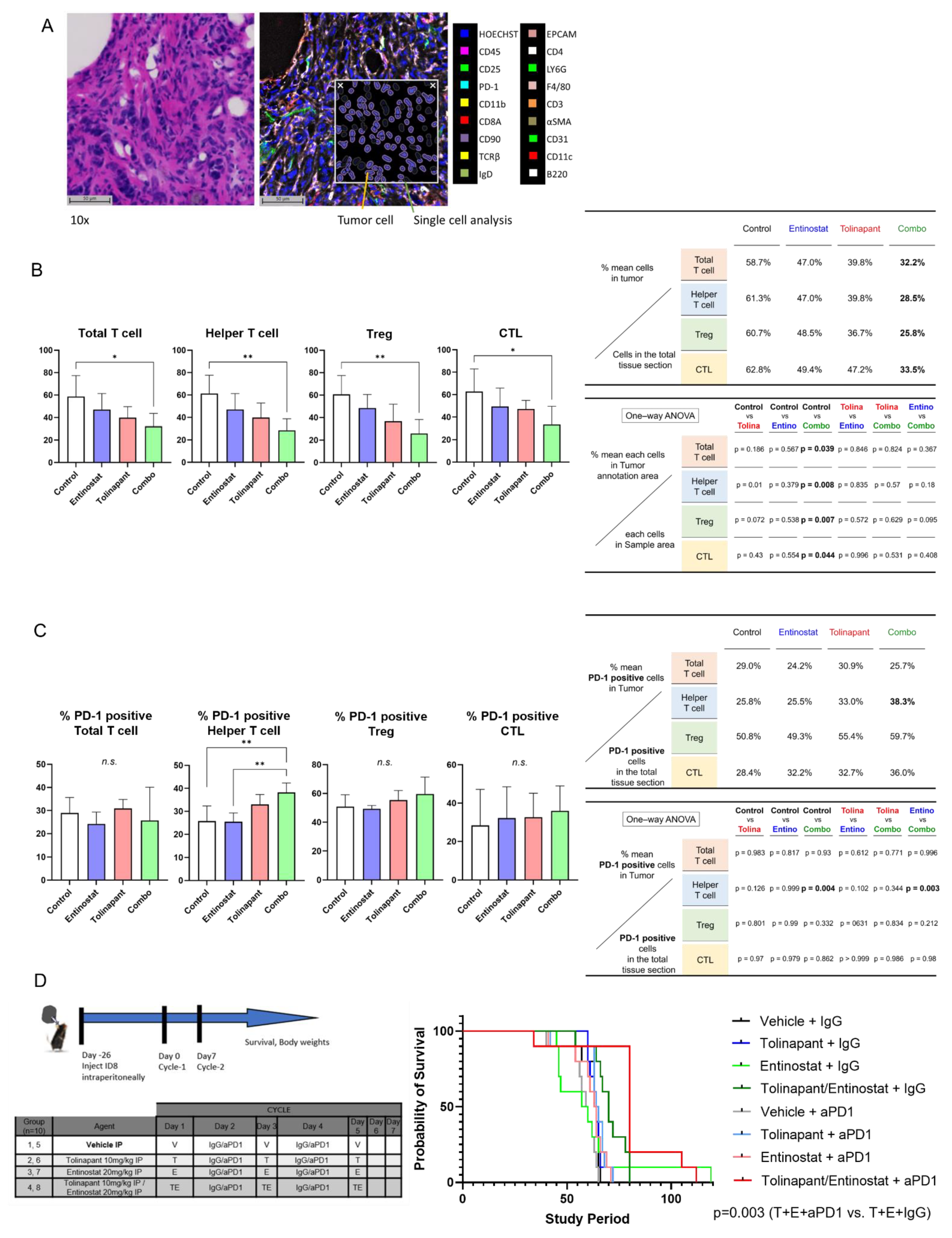
2.11.3. Survival Study: Immune Competent Model
2 × 10
6 ID8 p53−/− cells were injected IP into 6–8 weeks old C57/BL6 female mice and allowed to grow for 4 weeks before IP treatment with vehicle control (5% dextrose), entinostat 20 mg/kg, tolinapant 10 mg/kg, or combination entinostat plus tolinapant with 200 μg/mouse of control IgG or anti-mouse PD1 antibody (#29F.1A12, Bio X CellX, Lebanon, NH). ID8 p53−/− cells were kindly provided by Josephine Walton [
16]. Mice were followed until euthanasia endpoints and scored for overall survival. Animal care was provided in accordance with procedures in the Guide for the Care and Use of Laboratory Animals. Experiments were carried out according to a protocol approved by the NCI Animal Care and Use Committee.
2.12. Cytokine Assay in Mouse Ascites
The 2 × 106 ID8 p53−/− cells were injected IP into 6–8 weeks old C57/BL6 female mice and allowed to grow for 5 weeks before IP treatment with vehicle control (5% dextrose), entinostat 20 mg/kg, tolinapant 10 mg/kg, or combination entinostat plus tolinapant. Ascites were collected after 24 h from final treatment, and TNF-α were measured using Quantikine ELISA Mouse TNF-α Immunoassay kit per the manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA).
2.13. Statistical Analysis
All in vitro experiments were conducted in duplicate or triplicate for each experimental condition, as noted above. Results were analyzed for statistically significant differences using two-tailed
t-tests (2 groups) or ANOVA multiple comparison tests (3 or more groups) in GraphPad Prism version 8.0 for Windows (GraphPad Software, San Diego, CA, USA,
www.graphpad.com (accessed on 1 November 2019)). Overall survival was estimated using the Kaplan–Meier method, with differences between treatment groups evaluated using a long-rank test in GraphPad Prism version 8.0 for Windows;
p values < 0.05 were considered statistically significant. Synergistic effects on XTT cell viability assay between two drugs are analyzed with Combenefit (
https://www.cruk.cam.ac.uk/research-groups/jodrell-group/combenefit (accessed on 1 November 2019)) [
17]. Combination indexes were analyzed with CalcuSyn Version 2.0 (
http://www.biosoft.com/w/calcusyn.htm (accessed on 1 November 2019)). Quantification of Western blots was performed with densitometry using LI-COR Image Studio Lite version 5.2 (LI-COR Biotechnology, Lincoln, NE, USA).
2.14. Multiplexed Immuno-Fluorescence Imaging Using CODEX
2.14.1. Single Cell Analysis
CODEX platform [
18] was used to evaluate the impact of tolinapant, entinostat, and their combination on the immune tumor microenvironment (iTME) and to assess whether combination therapy may be a new treatment option in ovarian cancer. Multiplexed Immuno-Fluorescence Imaging was used as a powerful tool to evaluate iTME reproducibly. Multiplexed imaging technologies allow capturing many parameters of single cells while preserving their spatial location. We randomized a total of 20 C57B6 mice, each with tumor cells (ID8 p53KO) implanted in their ovary, into groups #1–5 as Control, #6–10 as the tolinapant group, #11–15 as the entinostat group, and #16–20 as the combination treatment group. After drug treatment, ovarian tumor resection was performed, and appropriate specimens were prepared for Multiplexed Immuno-Fluorescence Imaging. We acquired and processed CODEX images for optimal performance in HALO image analysis. With HALO algorithms and modules, CODEX multiplexed IF images from mice ovarian tumor tissue were generated using 18 barcoded antibodies. The major structures within ovarian cancer were imaged, such as the cortex, medulla, follicle, and fallopian tube. We evaluated tissue morphology based on Hoechst nuclear staining. The various settings for identifying Hoechst are as follows.: Nuclear Contrast Threshold 0.5, Minimum Nuclear Intensity 0.1, Maximum Image Brightness 1, and Nuclear Segmentation Aggressiveness 0.65. After that, we set the weak, moderate, and strong thresholds for all dyes other than Hoechst, respectively, and we counted the relative distributions of each cell in the ovarian tumor. The key tumor cell and primary T cell types could be visualized and compared in mouse ovarian tumor tissue. Specifically, the Helper T cell, cytotoxic T lymphocyte (CTL), regulatory T cell (Treg), and PD-1 positive T cell were identified.
2.14.2. Spatial Plot Analysis
After counting the number of each cell, the localization of each cell was identified by Spatial Plot. To evaluate the effects of various treatments on ovarian tumor tissue, we needed to distinguish tumor cells from the rest of the tumor within the ovarian tumor tissue. After identifying the localization of tumor cells, we created a Density Heatmap in Spatial Analysis to evaluate the density of tumor cells. A tumor annotation line (yellow line) was drawn for each sample using the Density Heatmap to determine the tumor site and other sites with a high degree of accuracy. The number of various cells was then counted in the tumor and other areas and analyzed using one-way ANOVA, showing that T cells (red) tend to cluster around the tumor cells (light blue) (
Supplementary Figure S9). A representative figure showing the relationship between the outer edge of the sample (black line), the tumor annotation line (yellow line), and the various T cells (blue: Helper T cells, green: Tregs, yellow: CTLs) is shown in
Supplementary Figure S10.
4. Discussion
IAP inhibition with SMs is a promising anti-cancer treatment option but has limited efficacy as a single agent. In this study, we demonstrated improved efficacy with the combination of SMs and HDAC inhibitors, a synergistic combination that was discovered through our global unbiased matrix drug screen that we previously reported [
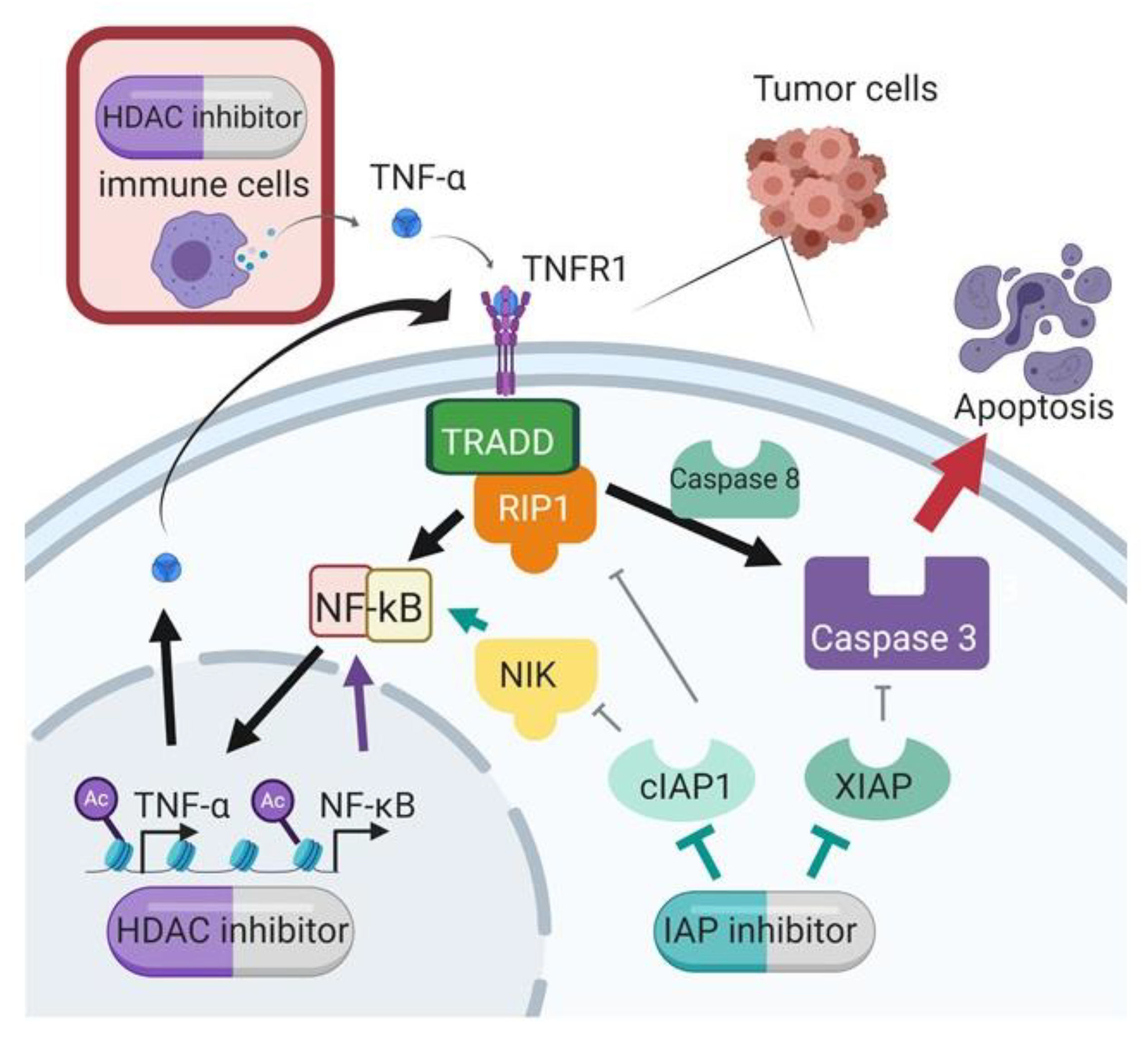
11]. Importantly, we demonstrated this synergy across multiple cell lines, and using different SMs and HDAC inhibitors, confirming that this is a class effect and not specific to individual targeted agents. Our data support a mechanism of synergy whereby the HDAC inhibition increases transcription of TNF-α, and the IAP inhibition by SMAC mimetic shunts TNF receptor signaling to activate caspase cleavage and subsequent apoptosis (
Figure 8).
Some ovarian cancer cell lines, such as SKOV3, TOV21G, IGROV1, and ES2, are susceptible to single-agent SM. These cell lines have some common features which might cause synergism with SMs, such as the secretion of TNF-α. In addition, SKOV3, TOV21G, and IGROV1 have
ARID1A mutation, and TOV21G and ES2 are clear cell subtypes [
23]. In contrast to most high-grade serous ovarian cancer, which has
TP53 mutation [
24], many (57–67%) of ovarian clear cell cancer (OCCC) have
ARID1A mutation [
25,
26].
ARID1A mutant ovarian cancers may be related to OCCC because
TP53 and
ARID1A are almost mutually exclusive in ovarian cancer. OCCC is the most refractory to standard chemotherapeutic regimens (platinum and taxane) among epithelial ovarian cancers, and the median survival of women with stage III/IV OCCC is significantly lower than those with HGS [
27]. Additionally, OCCC arises from endometriosis [
28]. Endometriosis is difficult to cure with current hormonal therapy, but IAP inhibition has demonstrated a positive effect in controlling endometriosis in a mouse model [
29,
30]. Endometriosis lesions highly express cIAPs [
28] which could contribute to the generation of endometriosis but also provide a target to prevent or treat future OCCC. Endometriosis-related ovarian cancer might have a higher sensitivity to SMAC mimetics because those cancers have a greater tendency to express TNF-α compared with high-grade serous ovarian cancer. Previous work showed that endometrioid ovarian cancers have a higher positive rate of TNF-α (83.3%, 10/12 cases) compared with high-grade serous ovarian cancer (40%, 20/50 cases) on immunohistochemistry staining [
31]. Therefore, SMs could be developed as a treatment option for endometriosis or OCCC.
In the presence of TNF-α, the SMs had a heightened anti-tumor effect. Mechanistically, the identified upregulation of TNF-α provides a stimulus for apoptosis that is augmented by the presence of the SMAC mimetic. Without the SM, the TNF-α in the presence of cIAP1 can stimulate classical NF-kB signaling and block apoptosis. The SM depletes cIAP1, and thus, TNF receptor signaling leads to caspase activation and apoptosis [
32]. Our results show that the HDAC inhibitor increases TNF-α mRNA and protein, and these are further increased with the combination. SMs-mediated cIAP1 depletion combined with caspases released from XIAP is essential for full activation of apoptosis downstream of the TNF-α signaling pathway. Cell lines OVCAR3 and OVCAR8 showed maximal activation of Caspase 3/7 and 9 with the combination of SMAC mimetic and HDAC inhibitor in the presence of TNF-a. OVCAR8 showed less reactivity to the addition of TNF-α, and this may be due to its decreased dependency on classical NF-kB signaling that we previously showed [
33,
34].
Sufficient levels of TNF-α in the environment may render tumors particularly susceptible to IAP antagonism [
35]. In our report, the HDAC inhibitor-induced TNF-α secretion corresponded to a synergistic anti-tumor effect with SMs. The synergy between birinapant and chemotherapeutic drugs, including docetaxel, gemcitabine, SN-38 (active metabolite of irinotecan), and 5-AC (5-azacytidine), has been previously reported [
11,
36]. Here, we describe a novel synergistic combination of SMs and HDAC inhibitors. Pretreatment with a neutralizing anti-TNF-α antibody [
30] or Si-RNA of TNF-α [
11] resulted in rescue from tumor cell death led by the birinapant and chemotherapeutic drugs. In the same way, we showed that depletion of TNF-α by RNA interference attenuated the synergistic effect between tolinapant and entinostat. This result suggests that HDAC inhibitor-induced TNF-α plays a critical role in the synergy.
A combination of tolinapant and entinostat was effective in vivo in several mouse models. Although we expected HDAC inhibitors to have some anti-tumor effect, neither panobinostat nor entinostat single agents showed anti-tumor activity in any of our models. A previous report indicated that the anti-tumor effect of entinostat single agent required adaptive immunity [
15]. Even so, we did not observe improvement in survival of the C57B6 immune-competent mouse treated with entinostat + anti-PD1 antibody compared to entinostat alone or vehicle-treated mice. Both SMs and HDAC inhibitors have limited efficacy when used as single agents in ovarian cancer models, but the synergistic anti-tumor effect was confirmed in all mouse models. Furthermore, mouse survival improved with a triplet combination of tolinapant, entinostat, and anti-PD1 antibody, indicating the importance of immune modulation.
The comprehensive, unbiased matrix drug screen that identified this combination treatment is a powerful discovery technique to identify synergistic drug strategies that may improve outcomes for patients with ovarian cancer, and a clinical trial is currently under development. An understanding of the mechanisms underlying the synergy will allow for the discovery of novel therapeutic options for women with recurrent ovarian cancers that acquire apoptosis resistance due to IAP upregulation.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







