
Deepening Our Understanding of the Factors Affecting Landscape of Myeloproliferative Neoplasms: What Do We Know about Them?


Abstract
:Simple Summary
Abstract
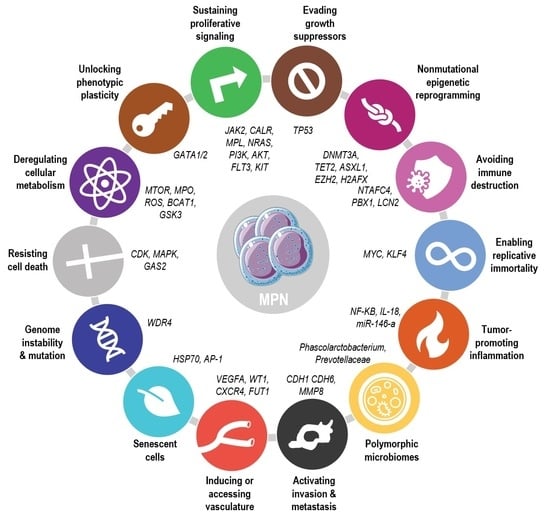
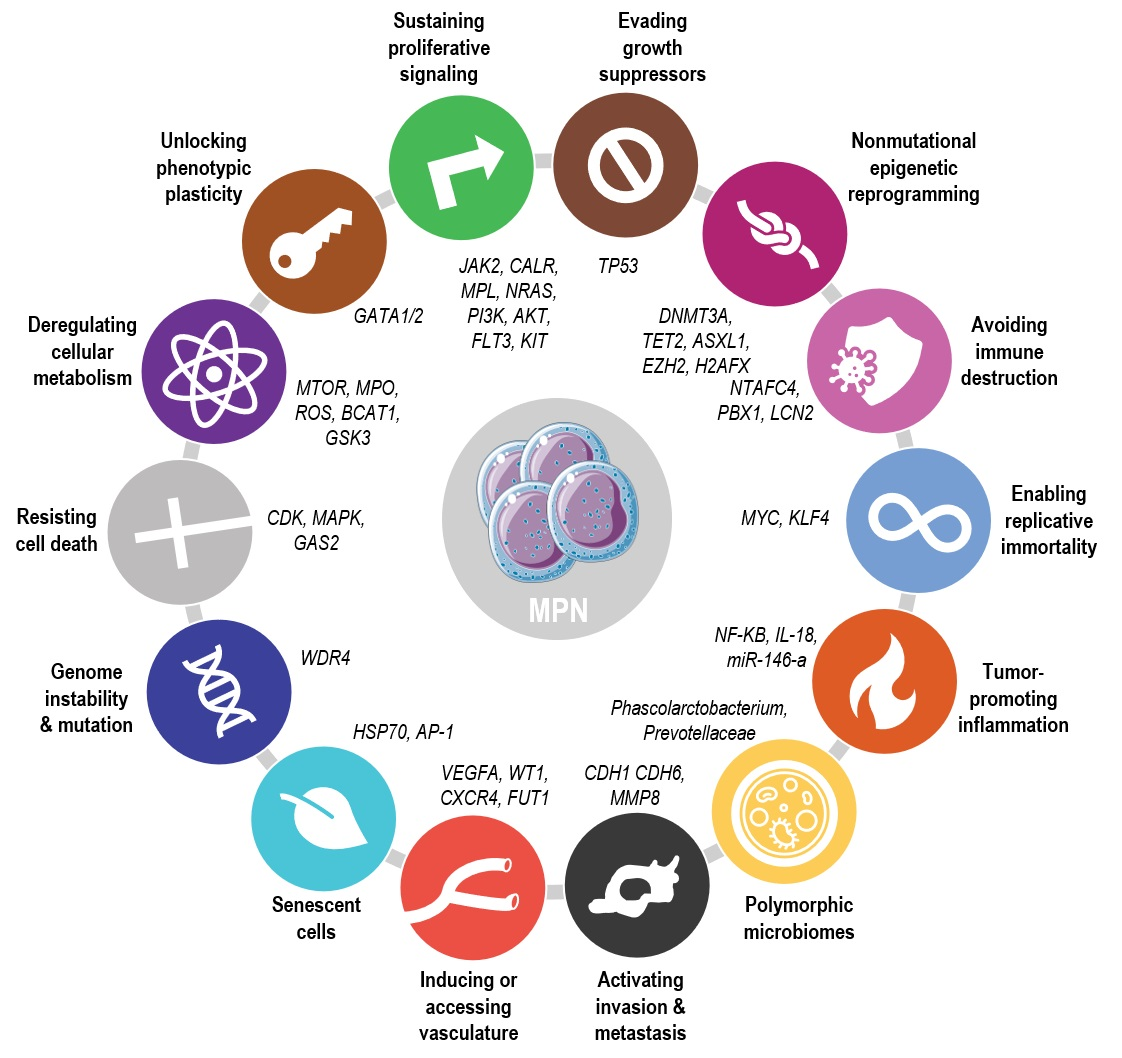{kind=link}
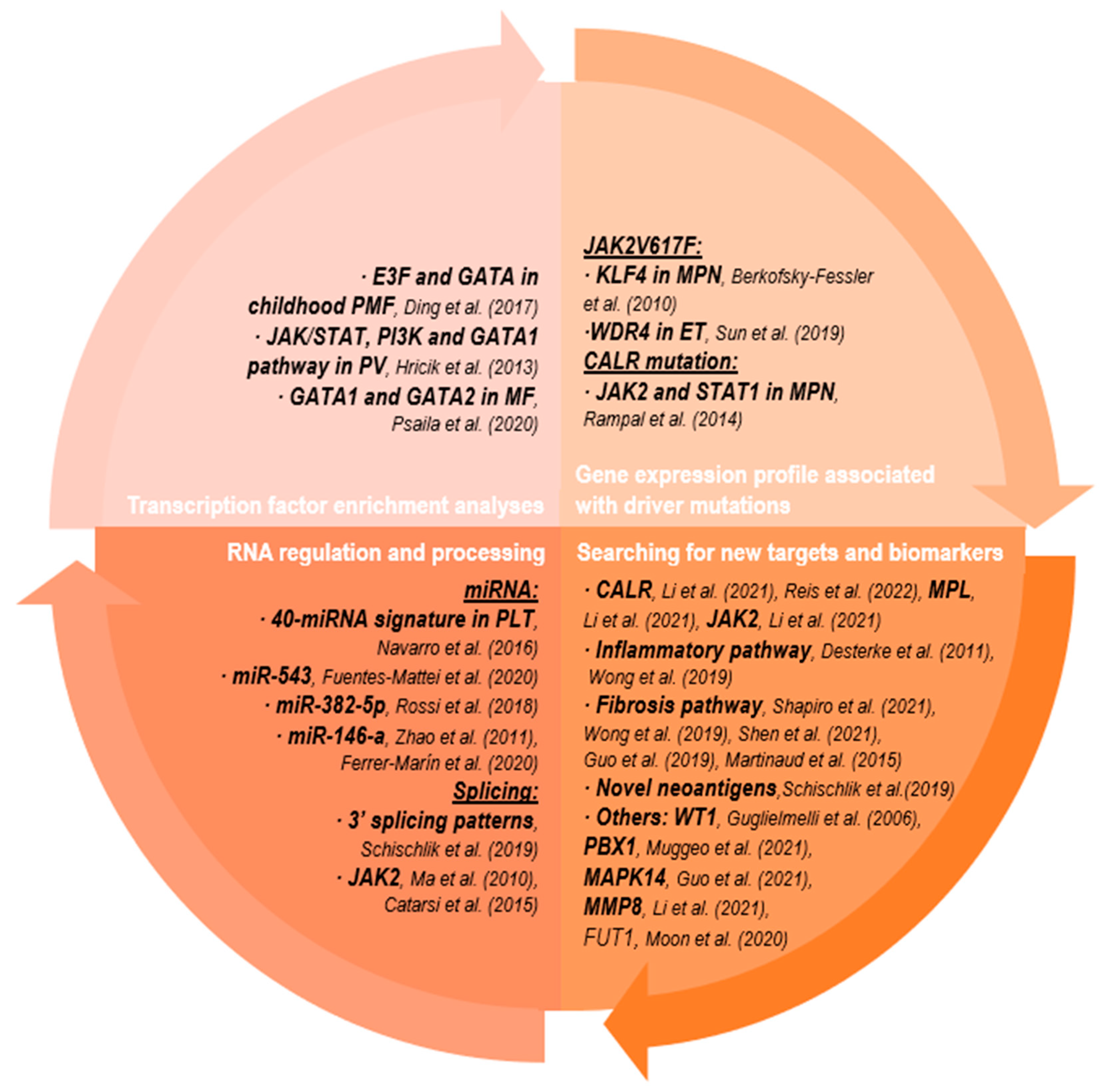{kind=link}
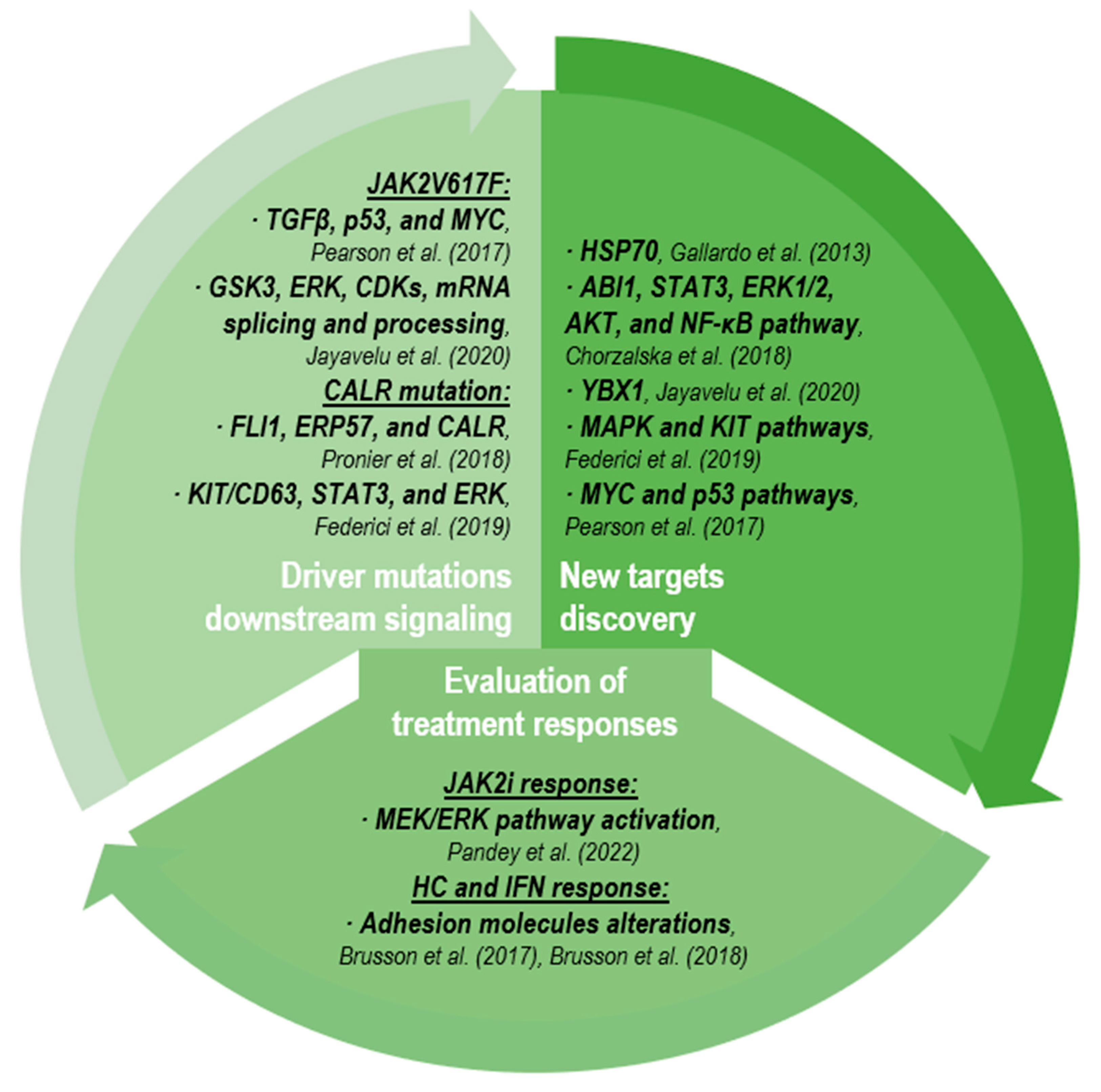{kind=link}
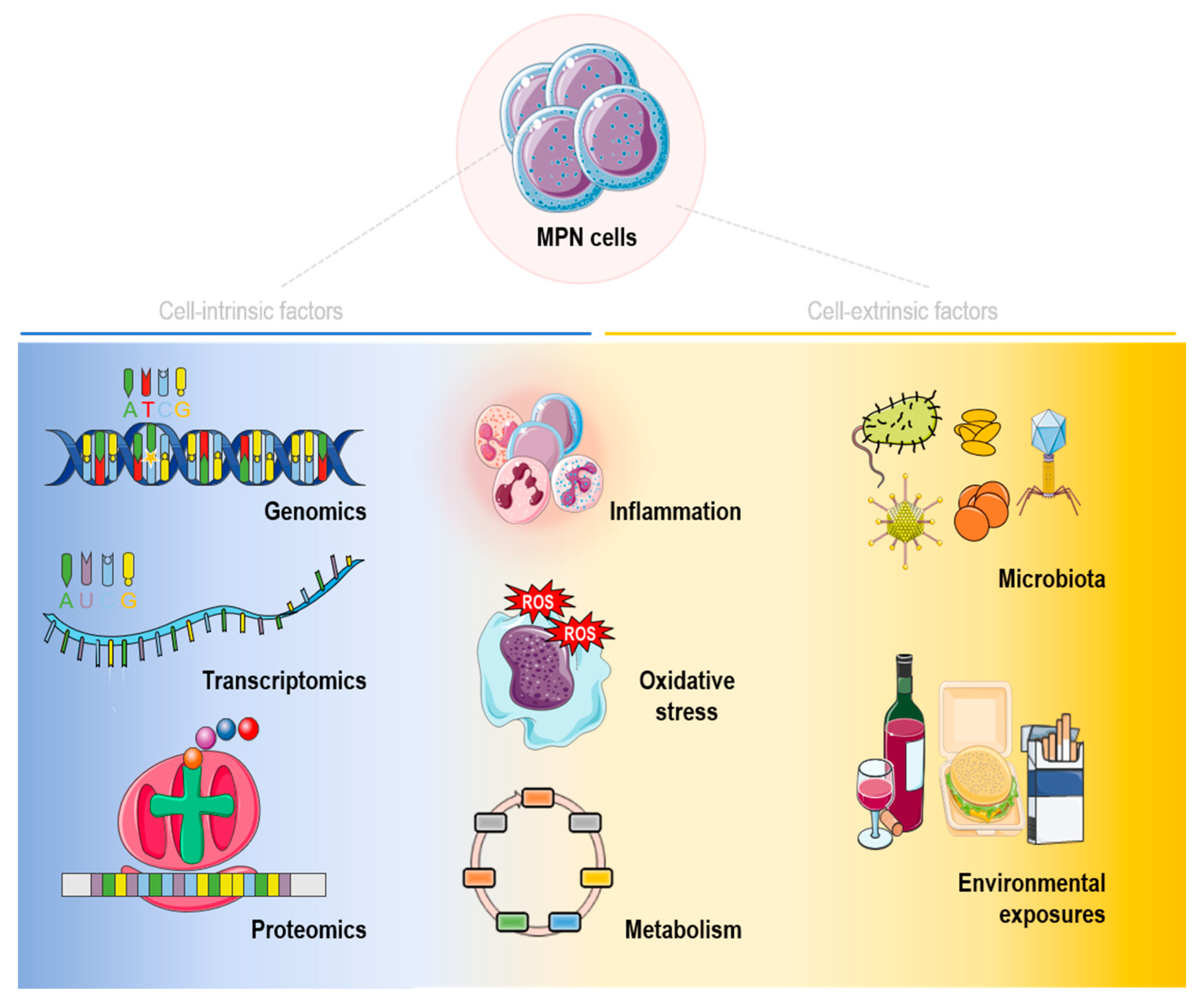{kind=link}
1. Introduction
2. Intrinsic Factors
2.1. Genomic Studies
2.1.1. Classical Driver Mutations
2.1.2. Triple Negative Patients
2.1.3. Mutations in Other Myeloid Genes
Mutations in Genes Involved in DNA Methylation
Mutations in Splicing Components
Mutations in Genes Involved in DNA Repair and Other Signaling Pathways
2.2. Transcriptomic Analyses

2.2.1. Transcription Factor Enrichment Analyses
2.2.2. Gene Expression Profile Associated with Driver Mutation
2.2.3. Searching for New Targets and Biomarkers
2.2.4. RNA Regulation and Processing
2.3. Proteomic and Post-Translational Modifications

2.3.1. Driver Mutation Downstream Signaling
2.3.2. New Target Discovery
2.3.3. Evaluation of Treatment Responses
3. Extrinsic Factors
3.1. Microbiome
3.2. Other Extrinsic Factors
4. Interaction between Intrinsic and Extrinsic Factors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Rolles, B.; Mullally, A. Molecular Pathogenesis of Myeloproliferative Neoplasms. Curr. Hematol. Malign Rep. 2022, 17, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Kristinsson, S.Y.; Andersson, T.M.-L.; Landgren, O.; Eloranta, S.; Derolf, R.; Dickman, P.W.; Björkholm, M. Patterns of Survival Among Patients With Myeloproliferative Neoplasms Diagnosed in Sweden From 1973 to 2008: A Population-Based Study. J. Clin. Oncol. 2012, 30, 2995–3001. [Google Scholar] [CrossRef] [Green Version]
- Hultcrantz, M.; Wilkes, S.R.; Kristinsson, S.Y.; Andersson, T.M.-L.; Derolf, R.; Eloranta, S.; Samuelsson, J.; Landgren, O.; Dickman, P.W.; Lambert, P.C.; et al. Risk and Cause of Death in Patients Diagnosed With Myeloproliferative Neoplasms in Sweden Between 1973 and 2005: A Population-Based Study. J. Clin. Oncol. 2015, 33, 2288–2295. [Google Scholar] [CrossRef]
- McLornan, D.P.; Yakoub-Agha, I.; Robin, M.; Chalandon, Y.; Harrison, C.N.; Kroger, N. State-of-the-art review: Allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica 2019, 104, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Wright, K.L.; Epling-Burnette, P.K.; Reuther, G.W. Metabolic Vulnerabilities and Epigenetic Dysregulation in Myeloproliferative Neoplasms. Front. Immunol. 2020, 11, 604142. [Google Scholar] [CrossRef]
- Zoi, K.; Cross, N. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLOS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. SomaticCALRMutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Jacquelin, S.; Kramer, F.; Mullally, A.; Lane, S.W. Murine Models of Myelofibrosis. Cancers 2020, 12, 2381. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Loscocco, G.G.; Mannarelli, C.; Rossi, E.; Mannelli, F.; Ramundo, F.; Coltro, G.; Betti, S.; Maccari, C.; Ceglie, S.; et al. JAK2V617F variant allele frequency >50% identifies patients with polycythemia vera at high risk for venous thrombosis. Blood Cancer J. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Guglielmelli, P.; Gangat, N.; Rossi, E.; Mannarelli, C.; Betti, S.; Maccari, C.; Ramundo, F.; Jadoon, Y.; Gesullo, F.; et al. Clinical and molecular predictors of fibrotic progression in essential thrombocythemia: A multicenter study involving 1607 patients. Am. J. Hematol. 2021, 96, 1472–1480. [Google Scholar] [CrossRef]
- Defour, J.-P.; Chachoua, I.; Pecquet, C.; Constantinescu, S. Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: Implications for myeloproliferative neoplasms. Leukemia 2015, 30, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.M.; Bench, A.J.; Goday-Fernández, A.; Anand, S.; Vaghela, K.J.; Beer, P.; Scott, M.A.; Bareford, D.; Green, A.R.; Huntly, B.; et al. Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br. J. Haematol. 2010, 149, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Komatsu, H.; Wakita, A.; Kato-Uranishi, M.; Ito, M.; Satoh, A.; Tsuboi, K.; Nitta, M.; Miyazaki, H.; Iida, S.; et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood 2004, 103, 4198–4200. [Google Scholar] [CrossRef]
- Jia, R.; Kralovics, R. Progress in elucidation of molecular pathophysiology of myeloproliferative neoplasms and its application to therapeutic decisions. Int. J. Hematol. 2019, 111, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansier, O.; Paz, D.L.; Ianotto, J.-C.; Le Bris, Y.; Chauveau, A.; Boyer, F.; Conejero, C.; Fitoussi, O.; Riou, J.; Adiko, D.; et al. Clinical and biological characterization of MPN patients harboring two driver mutations, a French intergroup of myeloproliferative neoplasms (FIM) study. Am. J. Hematol. 2018, 93, E84–E86. [Google Scholar] [CrossRef] [PubMed]
- Hermange, G.; Rakotonirainy, A.; Bentriou, M.; Tisserand, A.; El-Khoury, M.; Girodon, F.; Marzac, C.; Vainchenker, W.; Plo, I.; Cournède, P.-H. Inferring the initiation and development of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2022, 119, e2120374119. [Google Scholar] [CrossRef] [PubMed]
- Sousos, N.; Leathlobhair, M.N.; Karali, C.S.; Louka, E.; Bienz, N.; Royston, D.; Clark, S.-A.; Hamblin, A.; Howard, K.; Mathews, V.; et al. In utero origin of myelofibrosis presenting in adult monozygotic twins. Nat. Med. 2022, 28, 1207–1211. [Google Scholar] [CrossRef]
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell 2021, 28, 514–523.e9. [Google Scholar] [CrossRef]
- Williams, N.; Lee, J.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; Campbell, P.J.; et al. Phylogenetic Reconstruction of Myeloproliferative Neoplasm Reveals Very Early Origins and Lifelong Evolution. Biol. Med. BioRxiv 2020. [Google Scholar] [CrossRef]
- Langabeer, S.E. Chasing down the triple-negative myeloproliferative neoplasms: Implications for molecular diagnostics. Jak-Stat 2016, 5, e1248011. [Google Scholar] [CrossRef] [Green Version]
- Alimam, S.; Villiers, W.; Dillon, R.; Simpson, M.; Runglall, M.; Smith, A.; Chatzikyriakou, P.; Lavender, P.; Kanda, A.; Mills, K.; et al. Patients with triple-negative, JAK2V617F- and CALR-mutated essential thrombocythemia share a unique gene expression signature. Blood Adv. 2021, 5, 1059–1068. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Godfrey, A.L.; Nangalia, J. Genomic heterogeneity in myeloproliferative neoplasms and applications to clinical practice. Blood Rev. 2020, 42, 100708. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Perdomo, M.N.S.S.; Perera, M.D.M.; De la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Gangat, N.; Coltro, G.; Lasho, T.L.; Loscocco, G.G.; Finke, C.M.; Morsia, E.; Sordi, B.; Szuber, N.; Hanson, C.A.; et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021, 11, 1–4. [Google Scholar] [CrossRef]
- Bartels, S.; Vogtmann, J.; Schipper, E.; Büsche, G.; Schlue, J.; Lehmann, U.; Kreipe, H. Combination of myeloproliferative neoplasm driver gene activation with mutations of splice factor or epigenetic modifier genes increases risk of rapid blastic progression. Eur. J. Haematol. 2021, 106, 520–528. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Morishita, S.; Hashimoto, Y.; Furuya, C.; Edahiro, Y.; Ochiai, T.; Shirane, S.; Inano, T.; Yasuda, H.; Ando, M.; Araki, M.; et al. Non-driver gene mutation analysis in a large cohort of polycythemia vera and essential thrombocythemia. Eur. J. Haematol. 2022, 110, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.; Pancrazzi, A.; Wassie, E.; Ketterling, R.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hautin, M.; Mornet, C.; Chauveau, A.; Bernard, D.G.; Corcos, L.; Lippert, E. Splicing Anomalies in Myeloproliferative Neoplasms: Paving the Way for New Therapeutic Venues. Cancers 2020, 12, 2216. [Google Scholar] [CrossRef]
- Schischlik, F.; Jäger, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Guglielmelli, P.; Mannelli, F.; Mora, B.; Mannarelli, C.; Rotunno, G.; Pancani, F.; Maccari, C.; Bartalucci, N.; Romagnoli, S.; et al. SF3B1 mutations in primary and secondary myelofibrosis: Clinical, molecular and prognostic correlates. Am. J. Hematol. 2022, 97, E347–E349. [Google Scholar] [CrossRef]
- Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Jimma, T.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SF3B1 mutations in primary myelofibrosis: Clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012, 26, 1135–1137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Meira, A.; Rahman, H.; Norfo, R.; Wen, W.; Chédeville, A.; O’Sullivan, J.; Wang, G.; Paterson, A.; Louka, E.; Brierley, C.K.; et al. Single-Cell Multi-Omics Reveals the Genetic, Cellular and Molecular Landscape of TP53 Mutated Leukemic Transformation in MPN. Blood 2021, 138, 3. [Google Scholar] [CrossRef]
- Ding, N.; Zhang, Z.; Yang, W.; Ren, L.; Zhang, Y.; Zhang, J.; Li, Z.; Zhang, P.; Zhu, X.; Chen, X.; et al. Transcriptome Analysis of Monozygotic Twin Brothers with Childhood Primary Myelofibrosis. Genom. Proteom. Bioinform. 2017, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hricik, T.; Federici, G.; Zeuner, A.; Alimena, G.; Tafuri, A.; Tirelli, V.; Varricchio, L.; Masiello, F.; Ciaffoni, F.; Vaglio, S.; et al. Transcriptomic and phospho-proteomic analyzes of erythroblasts expanded in vitro from normal donors and from patients with polycythemia vera. Am. J. Hematol. 2013, 88, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Wang, G.; Rodriguez-Meira, A.; Li, R.; Heuston, E.F.; Murphy, L.; Yee, D.; Hitchcock, I.S.; Sousos, N.; O’Sullivan, J.; et al. Single-Cell Analyses Reveal Megakaryocyte-Biased Hematopoiesis in Myelofibrosis and Identify Mutant Clone-Specific Targets. Mol. Cell 2020, 78, 477–492.e8. [Google Scholar] [CrossRef]
- Berkofsky-Fessler, W.; Buzzai, M.; Kim, M.K.-H.; Fruchtman, S.; Najfeld, V.; Min, D.-J.; Costa, F.F.; Bischof, J.M.; Soares, M.B.; McConnell, M.J.; et al. Transcriptional Profiling of Polycythemia Vera Identifies Gene Expression Patterns Both Dependent and Independent from the Action of JAK2V617F. Clin. Cancer Res. 2010, 16, 4339–4352. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Ju, M.; Dai, X.; Dong, H.; Gu, W.; Gao, Y.; Fu, R.; Liu, X.; Huang, Y.; Liu, W.; et al. Multilevel defects in the hematopoietic niche in essential thrombocythemia. Haematologica 2019, 105, 661–673. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.; Murphy, L.; Psaila, B. Message in a platelet: Decoding platelet transcriptomes in myeloproliferative neoplasms. Cell Rep. Med. 2021, 2. [Google Scholar] [CrossRef] [PubMed]
- Desterke, C.; Bilhou-Nabéra, C.; Guerton, B.; Martinaud, C.; Tonetti, C.; Clay, D.; Guglielmelli, P.; Vannucchi, A.; Bordessoule, D.; Hasselbalch, H.; et al. FLT3-Mediated p38–MAPK Activation Participates in the Control of Megakaryopoiesis in Primary Myelofibrosis. Cancer Res 2011, 71, 2901–2915. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.J.; Baltay, M.; Getz, A.; Fuhrman, K.; Aster, J.C.; Hasserjian, R.P.; Pozdnyakova, O. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLoS ONE 2019, 14, e0216810. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Du, W.; Perkins, C.; Fechter, L.; Natu, V.; Maecker, H.; Rowley, J.; Gotlib, J.; Zehnder, J.; Krishnan, A. Platelet transcriptome identifies progressive markers and potential therapeutic targets in chronic myeloproliferative neoplasms. Cell Rep. Med. 2021, 2, 100425. [Google Scholar] [CrossRef]
- Guo, B.B.; Linden, M.D.; Fuller, K.A.; Phillips, M.; Mirzai, B.; Wilson, L.; Chuah, H.; Liang, J.; Howman, R.; Grove, C.S.; et al. Platelets in myeloproliferative neoplasms have a distinct transcript signature in the presence of marrow fibrosis. Br. J. Haematol. 2019, 188, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Martinaud, C.; Desterke, C.; Konopacki, J.; Pieri, L.; Torossian, F.; Golub, R.; Schmutz, S.; Anginot, A.; Guerton, B.; Rochet, N.; et al. Osteogenic Potential of Mesenchymal Stromal Cells Contributes to Primary Myelofibrosis. Cancer Res. 2015, 75, 4753–4765. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Zini, R.; Bogani, C.; Salati, S.; Pancrazzi, A.; Bianchi, E.; Mannelli, F.; Ferrari, S.; Le Bousse-Kerdilès, M.-C.; Bosi, A.; et al. Molecular Profiling of CD34+ Cells in Idiopathic Myelofibrosis Identifies a Set of Disease-Associated Genes and Reveals the Clinical Significance of Wilms’ Tumor Gene 1 (WT1). Stem Cells 2006, 25, 165–173. [Google Scholar] [CrossRef]
- Muggeo, S.; Crisafulli, L.; Uva, P.; Fontana, E.; Ubezio, M.; Morenghi, E.; Colombo, F.S.; Rigoni, R.; Peano, C.; Vezzoni, P.; et al. PBX1-directed stem cell transcriptional program drives tumor progression in myeloproliferative neoplasm. Stem Cell Rep. 2021, 16, 2607–2616. [Google Scholar] [CrossRef]
- Guo, C.; Gao, Y.-Y.; Ju, Q.-Q.; Wang, M.; Zhang, C.-X.; Gong, M.; Li, Z.-L. MAPK14 over-expression is a transcriptomic feature of polycythemia vera and correlates with adverse clinical outcomes. J. Transl. Med. 2021, 19, 1–13. [Google Scholar] [CrossRef]
- Li, W.; Zhao, Y.; Wang, D.; Ding, Z.; Li, C.; Wang, B.; Xue, X.; Ma, J.; Deng, Y.; Liu, Q.; et al. Transcriptome research identifies four hub genes related to primary myelofibrosis: A holistic research by weighted gene co-expression network analysis. Aging 2021, 13, 23284–23307. [Google Scholar] [CrossRef]
- Li, W.; Yuan, B.; Zhao, Y.; Lu, T.; Zhang, S.; Ding, Z.; Wang, D.; Zhong, S.; Gao, G.; Yan, M. Transcriptome profiling reveals target in primary myelofibrosis together with structural biology study on novel natural inhibitors regarding JAK2. Aging 2021, 13, 8248–8275. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.C.; Gim, J.-A.; Kim, D.S.; Choi, C.W.; Yoon, J.; Yoon, S.-Y. Total Platelet Transcriptomics and Its Network Analysis by RNA-Seq and miRNA-Seq and PCA Application in Essential Thrombocythaemia. Acta Haematol. 2020, 144, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.; Buonpane, R.; Celik, H.; Marty, C.; Lei, A.; Jobe, F.; Rupar, M.; Zhang, Y.; DiMatteo, D.; Awdew, R.; et al. Discovery of INCA033989, a Monoclonal Antibody That Selectively Antagonizes Mutant Calreticulin Oncogenic Function in Myeloproliferative Neoplasms (MPNs). Blood 2022, 140, 14–15. [Google Scholar] [CrossRef]
- Navarro, A.; Pairet, S.; Álvarez-Larrán, A.; Pons, A.; Ferrer, G.; Longarón, R.; Fernández-Rodríguez, C.; Camacho, L.; Monzó, M.; Besses, C.; et al. miR-203 and miR-221 regulate SOCS1 and SOCS3 in essential thrombocythemia. Blood Cancer J. 2016, 6, e406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Mattei, E.; Bayraktar, R.; Manshouri, T.; Silva, A.M.; Ivan, C.; Gulei, D.; Fabris, L.; Amaral, N.S.D.; Mur, P.; Perez, C.; et al. miR-543 regulates the epigenetic landscape of myelofibrosis by targeting TET1 and TET2. J. Clin. Investig. 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, C.; Zini, R.; Rontauroli, S.; Ruberti, S.; Prudente, Z.; Barbieri, G.; Bianchi, E.; Salati, S.; Genovese, E.; Bartalucci, N.; et al. Role of TGF -β1/miR-382-5p/ SOD 2 axis in the induction of oxidative stress in CD 34+ cells from primary myelofibrosis. Mol. Oncol. 2018, 12, 2102–2123. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-κB dysregulation in microRNA-146a–deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Marín, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernández-Rivas, J.M.; Hernández-Boluda, J.C.; Fernandez-Rodriguez, C.; Luño, E.; Hernandez, C.G.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 34, 2648–2659. [Google Scholar] [CrossRef]
- Ma, W.; Kantarjian, H.; Zhang, X.; Wang, X.; Zhang, Z.; Yeh, C.-H.; O’Brien, S.; Giles, F.; Bruey, J.M.; Albitar, M. JAK2 Exon 14 Deletion in Patients with Chronic Myeloproliferative Neoplasms. PLoS ONE 2010, 5, e12165. [Google Scholar] [CrossRef] [Green Version]
- Catarsi, P.; Rosti, V.; Morreale, G.; Poletto, V.; Villani, L.; Bertorelli, R.; Pedrazzini, M.; Zorzetto, M.; Barosi, G. AGIMM Investigators JAK2 Exon 14 Skipping in Patients with Primary Myelofibrosis: A Minor Splice Variant Modulated by the JAK2-V617F Allele Burden. PLoS ONE 2015, 10, e0116636. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Guglielmelli, P.; Pietra, D.; Rumi, E.; Rossi, C.; Rontauroli, S.; Genovese, E.; Fanelli, T.; Calabresi, L.; Bianchi, E.; et al. CALR mutational status identifies different disease subtypes of essential thrombocythemia showing distinct expression profiles. Blood Cancer J. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, H.; Cardozo, C.; Raza, A. MicroRNAs in myeloproliferative neoplasms. Br. J. Haematol. 2013, 161, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Gao, W.; Li, Q.; Liu, Y.; Chen, H.; Cui, Y.; Sun, Z.; Liu, Z. Comprehensive characterization of posttranscriptional impairment-related 3′-UTR mutations in 2413 whole genomes of cancer patients. NPJ Genom. Med. 2022, 7, 1–12. [Google Scholar] [CrossRef]
- Quattrocchi, A.; Quattrocchi, A.; Maiorca, C.; Maiorca, C.; Billi, M.; Billi, M.; Tomassini, S.; Tomassini, S.; De Marinis, E.; De Marinis, E.; et al. Genetic lesions disrupting calreticulin 3′-untranslated region in JAK2 mutation-negative polycythemia vera. Am. J. Hematol. 2020, 95. [Google Scholar] [CrossRef]
- Pearson, S.; Williamson, A.J.K.; Blance, R.; Somervaille, T.C.P.; Taylor, S.; Azadbakht, N.; Whetton, A.D.; Pierce, A. Proteomic analysis of JAK2V617F-induced changes identifies potential new combinatorial therapeutic approaches. Leukemia 2017, 31, 2717–2725. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; Lacava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef]
- Federici, G.; Varricchio, L.; Martelli, F.; Falchi, M.; Picconi, O.; Francescangeli, F.; Contavalli, P.; Girelli, G.; Tafuri, A.; Petricoin, E.F.I.; et al. Phosphoproteomic Landscaping Identifies Non-canonical cKIT Signaling in Polycythemia Vera Erythroid Progenitors. Front. Oncol. 2019, 9, 1245. [Google Scholar] [CrossRef]
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia Vera. Mol. Cancer 2013, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Chorzalska, A.; Morgan, J.; Ahsan, N.; Treaba, D.O.; Olszewski, A.J.; Petersen, M.; Kingston, N.; Cheng, Y.; Lombardo, K.; Schorl, C.; et al. Bone marrow–specific loss of ABI1 induces myeloproliferative neoplasm with features resembling human myelofibrosis. Blood 2018, 132, 2053–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, G.; Kuykendall, A.T.; Reuther, G.W. JAK2 inhibitor persistence in MPN: Uncovering a central role of ERK activation. Blood Cancer J. 2022, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brusson, M.; Cochet, S.; Leduc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; Le Van Kim, C.; Cassinat, B.; Kiladjian, J.-J.; et al. Enhanced calreticulin expression in red cells of polycythemia vera patients harboring the JAK2V617F mutation. Haematologica 2017, 102, e241–e244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusson, M.; De Grandis, M.; Cochet, S.; Bigot, S.; Marin, M.; LeDuc, M.; Guillonneau, F.; Mayeux, P.; Peyrard, T.; Chomienne, C.; et al. Impact of hydroxycarbamide and interferon-α on red cell adhesion and membrane protein expression in polycythemia vera. Haematologica 2018, 103, 972–981. [Google Scholar] [CrossRef]
- Mossuz, P.; Arlotto, M.; Hermouet, S.; Bouamrani, A.; Lippert, E.; Girodon, F.; Dobo, I.; Vincent, P.; Cahn, J.Y.; Berger, F. Proteomic study of the impact of the JAK2–V617F mutation on the phenotype of essential thrombocythemia. Exp. Hematol. 2008, 36, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020, 42, 100711. [Google Scholar] [CrossRef]
- Ramanathan, G.; Fleischman, A.G. The Microenvironment in Myeloproliferative Neoplasms. Hematol. Clin. North Am. 2020, 35, 205–216. [Google Scholar] [CrossRef]
- Catani, L.; Cavo, M.; Palandri, F. The Power of Extracellular Vesicles in Myeloproliferative Neoplasms: “Crafting” a Microenvironment That Matters. Cells 2021, 10, 2316. [Google Scholar] [CrossRef]
- Zhan, H.; Kaushansky, K. The Hematopoietic Microenvironment in Myeloproliferative Neoplasms: The Interplay Between Nature (Stem Cells) and Nurture (the Niche). Tumor Microenviron. Hematop. Cells Part B 2020, 1273, 135–145. [Google Scholar] [CrossRef]
- Epstein, M.; Achong, B.; Barr, Y. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Azevedo, M.M.; Pina-Vaz, C.; Baltazar, F. Microbes and Cancer: Friends or Faux? Int. J. Mol. Sci. 2020, 21, 3115. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Herranz, M.; Klein-González, N.; Rodríguez-Lobato, L.G.; Juan, M.; de Larrea, C.F. Gut Microbiota Influence in Hematological Malignancies: From Genesis to Cure. Int. J. Mol. Sci. 2021, 22, 1026. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Garrett, W.S.; Trinchieri, G.; Wargo, J. The cancer microbiome. Nat. Rev. Cancer 2019, 19, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.E.; Markey, K.A. The contribution of the intestinal microbiome to immune recovery after HCT. Front. Immunol. 2022, 13, 4562. [Google Scholar] [CrossRef] [PubMed]
- Goodman, B.; Gardner, H. The microbiome and cancer. J. Pathol. 2018, 244, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Woerner, J.; Huang, Y.; Hutter, S.; Gurnari, C.; Sánchez, J.M.H.; Wang, J.; Huang, Y.; Schnabel, D.; Aaby, M.; Xu, W.; et al. Circulating microbial content in myeloid malignancy patients is associated with disease subtypes and patient outcomes. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Santisteban, M.; Kim, S.; Pepine, C.; Raizada, M.K. Brain–Gut–Bone Marrow Axis. Circ. Res. 2016, 118, 1327–1336. [Google Scholar] [CrossRef]
- Barone, M.; Barone, M.; Ricci, F.; Auteri, G.; Corradi, G.; Fabbri, F.; Papa, V.; Bandini, E.; Cenacchi, G.; Tazzari, P.L.; et al. An Abnormal Host/Microbiomes Signature of Plasma-Derived Extracellular Vesicles Is Associated to Polycythemia Vera. Front. Oncol. 2021, 11, 4993. [Google Scholar] [CrossRef]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Elalaoui, K.; Weihe, C.; Oliver, M.A.; Craver, M.B.; Lai, H.Y.; Brooks, S.; Kim, D.; Martiny, J.; Whiteson, K.; Fleischman, A. Investigating the Role of the Gut Microbiome in the Inflammatory State of Myeloproliferative Neoplasms. Blood 2018, 132, 3051. [Google Scholar] [CrossRef]
- Oliver, A.; El Alaoui, K.; Haunschild, C.; Avelar-Barragan, J.; Luque, L.F.M.; Whiteson, K.; Fleischman, A.G. Fecal Microbial Community Composition in Myeloproliferative Neoplasm Patients Is Associated with an Inflammatory State. Microbiol. Spectr. 2022, 10, e00032-22. [Google Scholar] [CrossRef]
- Pedersen, K.M.; Bak, M.; Sørensen, A.L.; Zwisler, A.-D.; Ellervik, C.; Larsen, M.K.; Hasselbalch, H.C.; Tolstrup, J.S. Smoking is associated with increased risk of myeloproliferative neoplasms: A general population-based cohort study. Cancer Med. 2018, 7, 5796–5802. [Google Scholar] [CrossRef] [Green Version]
- Leal, A.; Thompson, C.A.; Wang, A.H.; Vierkant, R.; Habermann, T.M.; Ross, J.A.; Mesa, R.A.; Virnig, B.A.; Cerhan, J.R. Anthropometric, medical history and lifestyle risk factors for myeloproliferative neoplasms in The Iowa Women’s Health Study cohort. Int. J. Cancer 2013, 134, 1741–1750. [Google Scholar] [CrossRef] [Green Version]
- Iho, S.; Tanaka, Y.; Takauji, R.; Kobayashi, C.; Muramatsu, I.; Iwasaki, H.; Nakamura, K.; Sasaki, Y.; Nakao, K.; Takahashi, T. Nicotine induces human neutrophils to produce IL-8 through the generation of peroxynitrite and subsequent activation of NF-κB. J. Leukoc. Biol. 2003, 74, 942–951. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, K.M.; Çolak, Y.; Bojesen, S.E.; Nordestgaard, B.G. Low high-density lipoprotein and increased risk of several cancers: 2 population-based cohort studies including 116,728 individuals. J. Hematol. Oncol. 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Gleitz, H.F.; Benabid, A.; Schneider, R.K. Still a burning question: The interplay between inflammation and fibrosis in myeloproliferative neoplasms. Curr. Opin. Hematol. 2021, 28, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Luque, L.F.M.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr. Hematol. Malign Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, A.; García-Vicente, R.; Morales, M.; Ortiz-Ruiz, A.; Martínez-López, J.; Linares, M. Protein Carbonylation and Lipid Peroxidation in Hematological Malignancies. Antioxidants 2020, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef]
- Durmus, A.; Mentese, A.; Yilmaz, M.; Sumer, A.; Akalin, I.; Topal, C.; Alver, A. Increased oxidative stress in patients with essential thrombocythemia. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2860–2866. [Google Scholar] [PubMed]
- Vener, C.; Novembrino, C.; Catena, F.B.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S.; et al. Oxidative stress is increased in primary and post−polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef]
- Balaian, E.; Wobus, M.; Bornhäuser, M.; Chavakis, T.; Sockel, K. Myelodysplastic Syndromes and Metabolism. Int. J. Mol. Sci. 2021, 22, 11250. [Google Scholar] [CrossRef] [PubMed]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.M.; Fernandes, M.S.; Deshpande, A.; Weisberg, E.; Inguilizian, H.V.; Abdel-Wahab, O.; Kung, A.L.; Levine, R.L.; Griffin, J.D.; Sattler, M. The JAK2V617F oncogene requires expression of inducible phosphofructokinase/fructose-bisphosphatase 3 for cell growth and increased metabolic activity. Leukemia 2011, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Rao, T.N.; Hansen, N.; Hilfiker, J.; Rai, S.; Majewska, J.-M.; Leković, D.; Gezer, D.; Andina, N.; Galli, S.; Cassel, T.; et al. JAK2-mutant hematopoietic cells display metabolic alterations that can be targeted to treat myeloproliferative neoplasms. Blood 2019, 134, 1832–1846. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Ciano, K.; Dong, K.; Zucker, S. Targeting glutamine metabolism in myeloproliferative neoplasms. Blood Cells Mol. Dis. 2015, 55, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Cebrián, N.; Rojas-Benedicto, A.; Albors-Vaquer, A.; Bellosillo, B.; Besses, C.; Martínez-López, J.; Pineda-Lucena, A.; Puchades-Carrasco, L. Polycythemia Vera and Essential Thrombocythemia Patients Exhibit Unique Serum Metabolic Profiles Compared to Healthy Individuals and Secondary Thrombocytosis Patients. Cancers 2021, 13, 482. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales, M.L.; Ferrer-Marín, F. Deepening Our Understanding of the Factors Affecting Landscape of Myeloproliferative Neoplasms: What Do We Know about Them? Cancers 2023, 15, 1348. https://doi.org/10.3390/cancers15041348
Morales ML, Ferrer-Marín F. Deepening Our Understanding of the Factors Affecting Landscape of Myeloproliferative Neoplasms: What Do We Know about Them? Cancers. 2023; 15(4):1348. https://doi.org/10.3390/cancers15041348
Chicago/Turabian StyleMorales, María Luz, and Francisca Ferrer-Marín. 2023. "Deepening Our Understanding of the Factors Affecting Landscape of Myeloproliferative Neoplasms: What Do We Know about Them?" Cancers 15, no. 4: 1348. https://doi.org/10.3390/cancers15041348
APA StyleMorales, M. L., & Ferrer-Marín, F. (2023). Deepening Our Understanding of the Factors Affecting Landscape of Myeloproliferative Neoplasms: What Do We Know about Them? Cancers, 15(4), 1348. https://doi.org/10.3390/cancers15041348