
Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells

 , ,
, ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
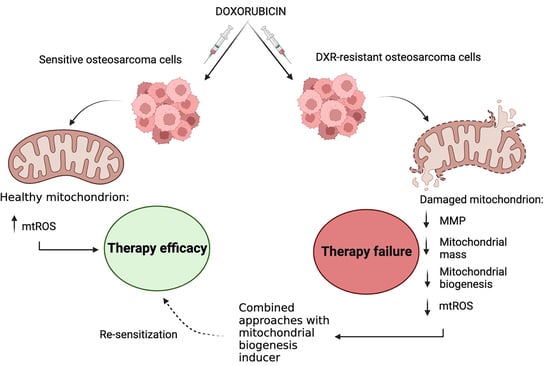
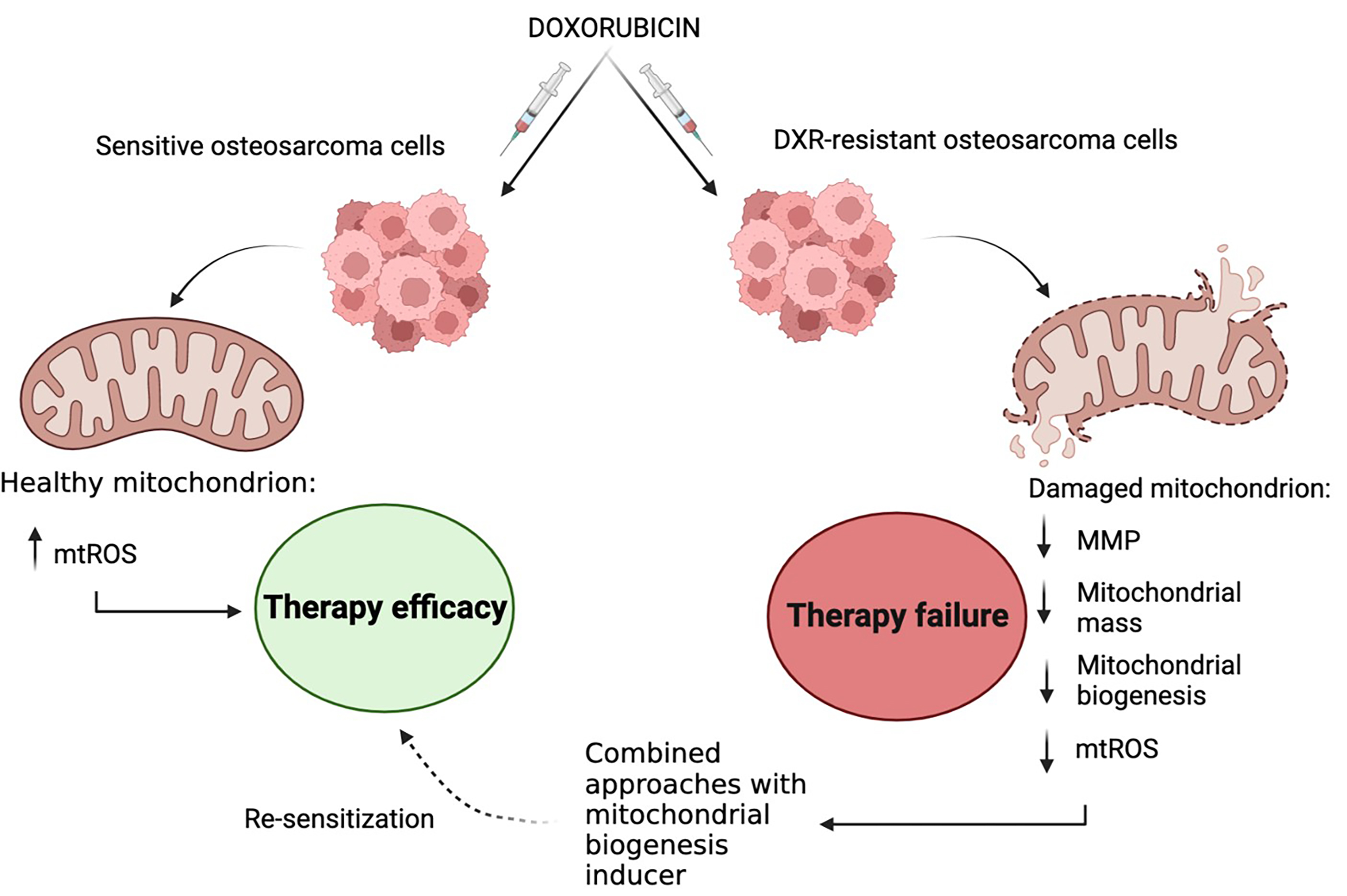
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mitochondrial Network by Confocal Microscopy
2.3. Mitochondrial Membrane Potential by Flow Cytometry
2.4. Mitochondrial Mass by Flow Cytometry
2.5. Quantitative Real-Time PCR
2.6. Western Blotting
2.7. Mitochondrial ROS Levels by Flow Cytometry
2.8. Apoptosis Detection by Flow Cytometry
2.9. Cell Viability Assays
2.9.1. Crystal Violet Assay
2.9.2. Trypan Blue Assay
2.9.3. Hoechst Cell Count in Hypoxic Conditions
2.10. Statistical Analysis
3. Results
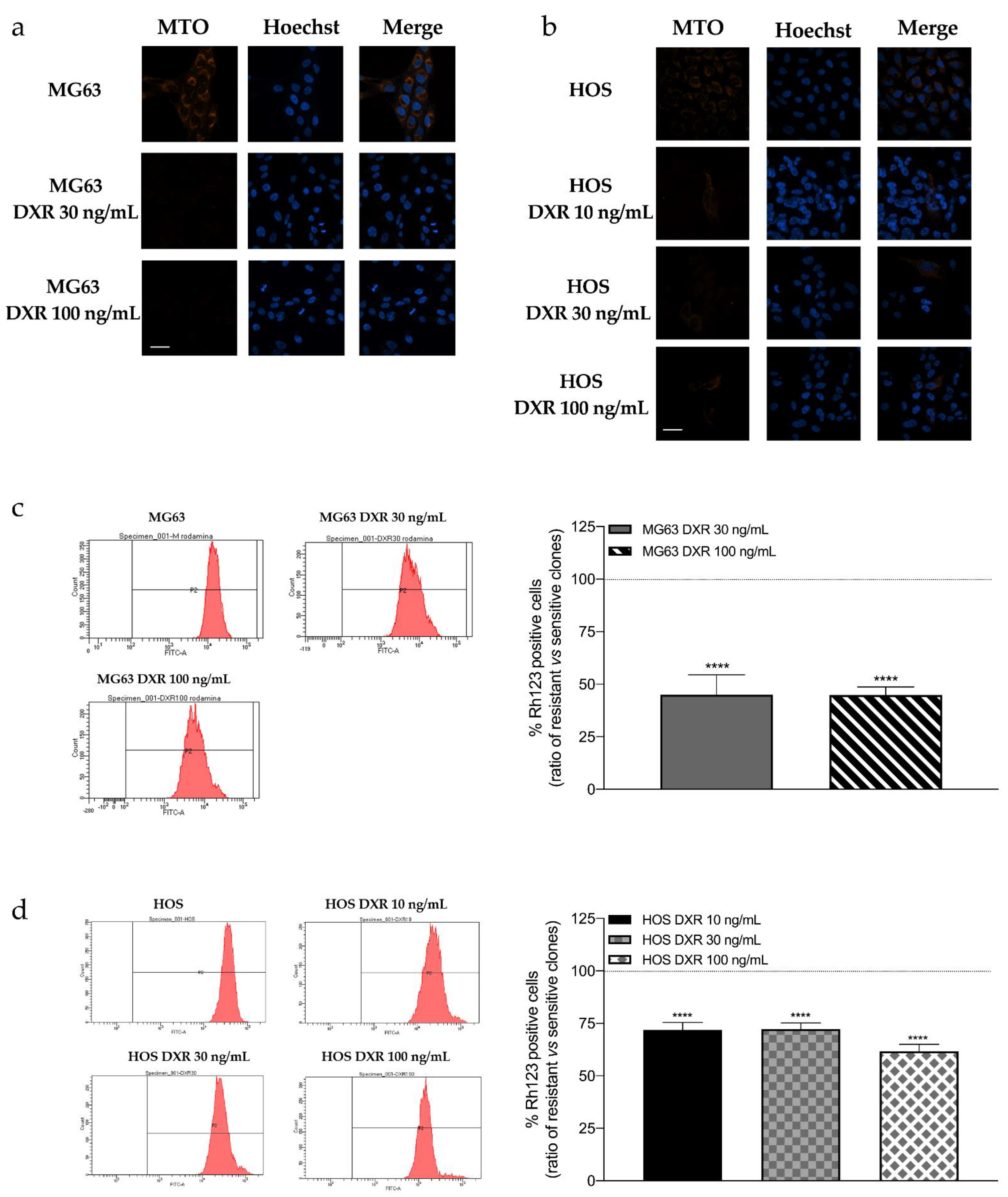
3.1. DXR-Resistant Clones Demonstrate Changes in Mitochondrial Morphology and Loss in Mitochondrial Membrane Potential
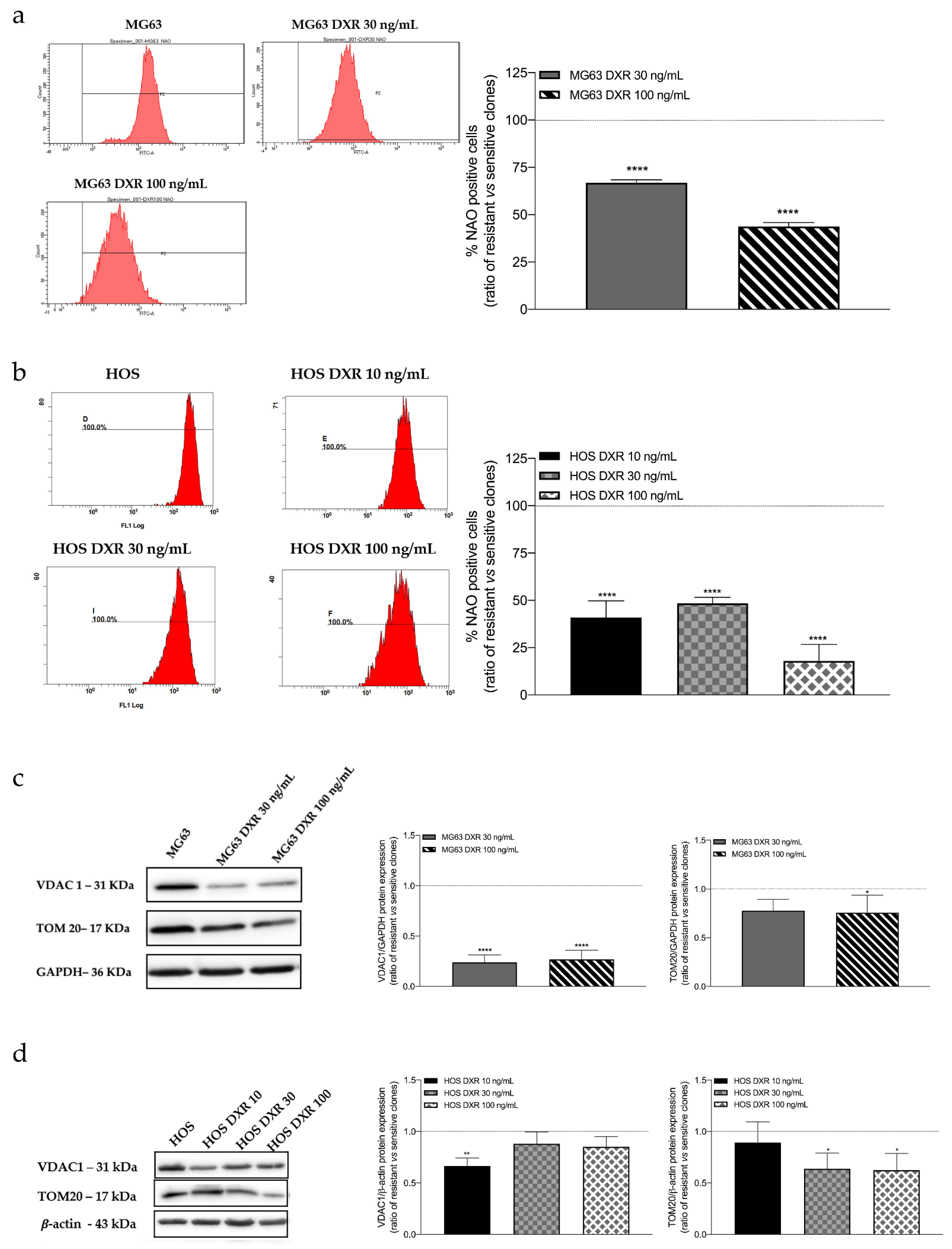
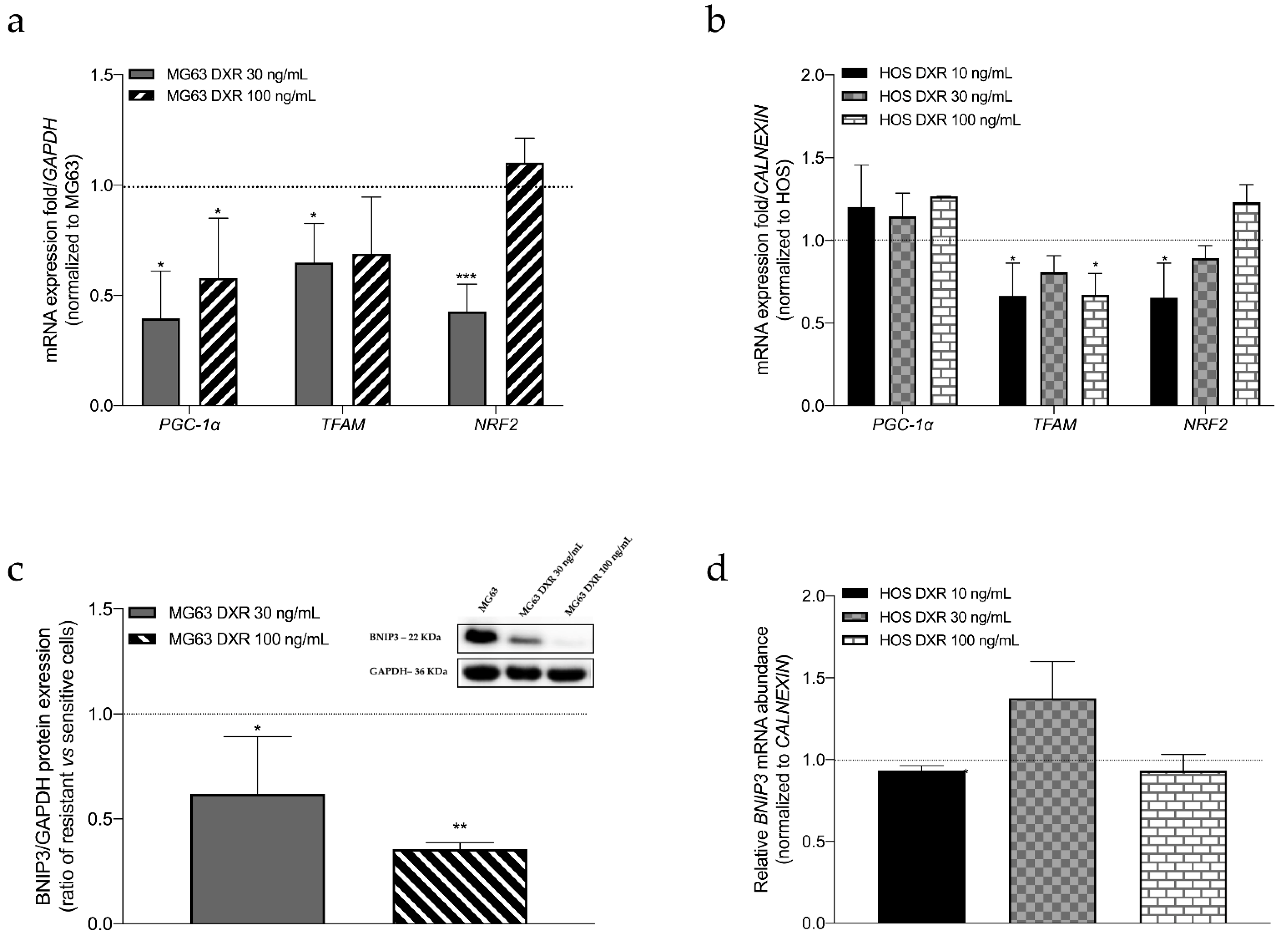
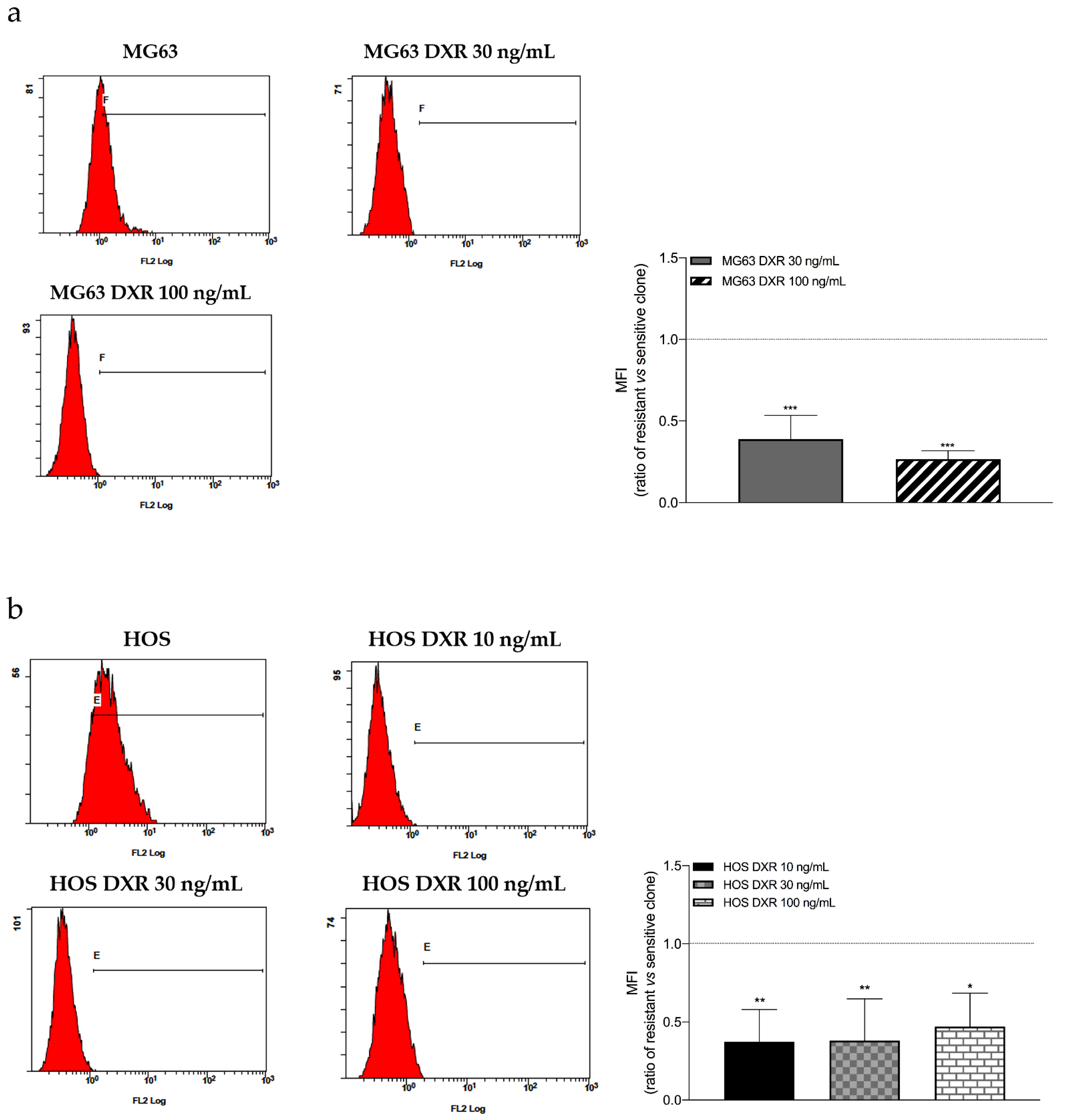
3.2. The Mitochondrial Mass Was Lower in DXR-Resistant Clones than in Sensitive Cells: Is Mitochondrial Biogenesis or Mitophagy the Involved Pathway?
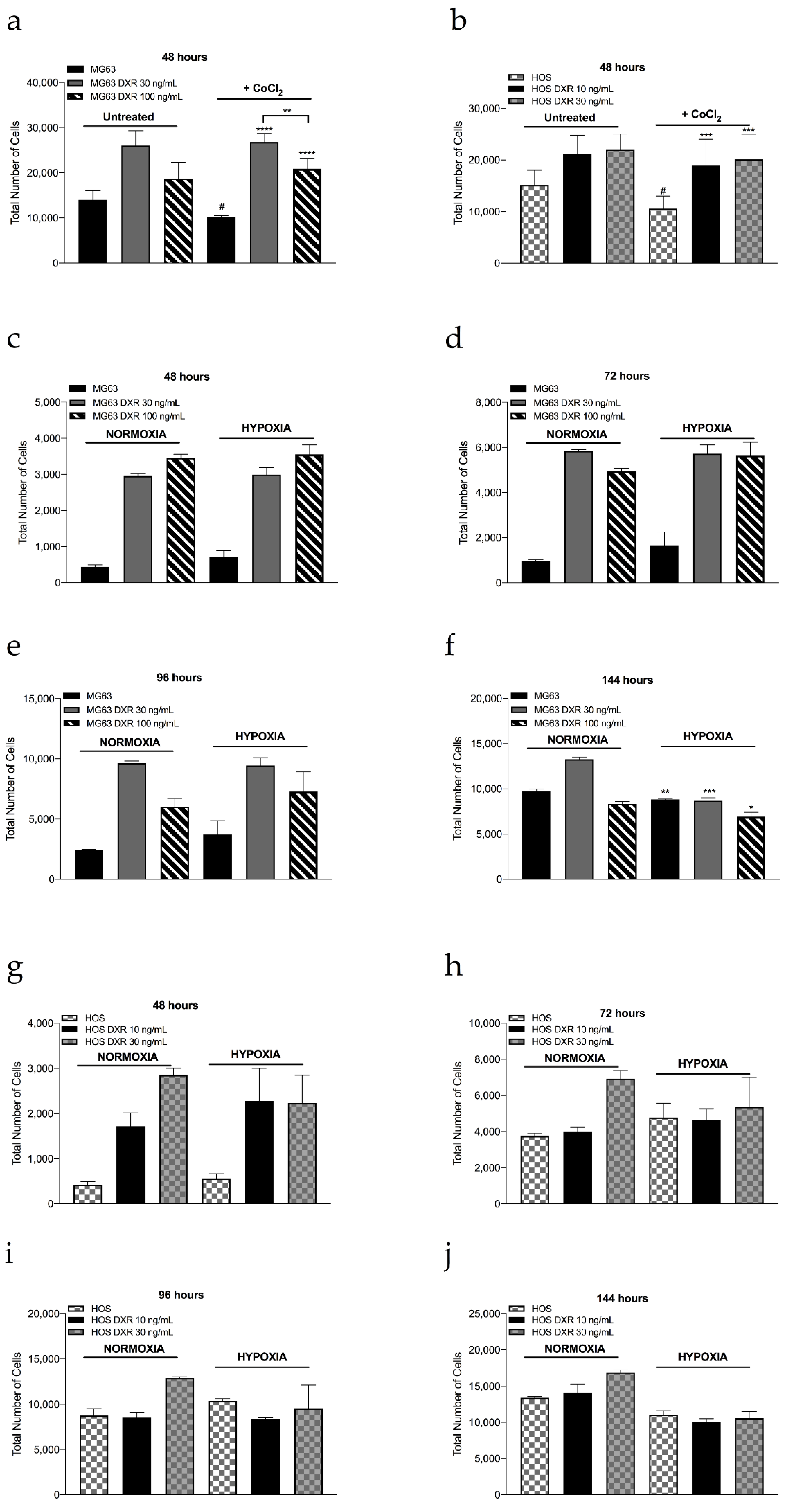
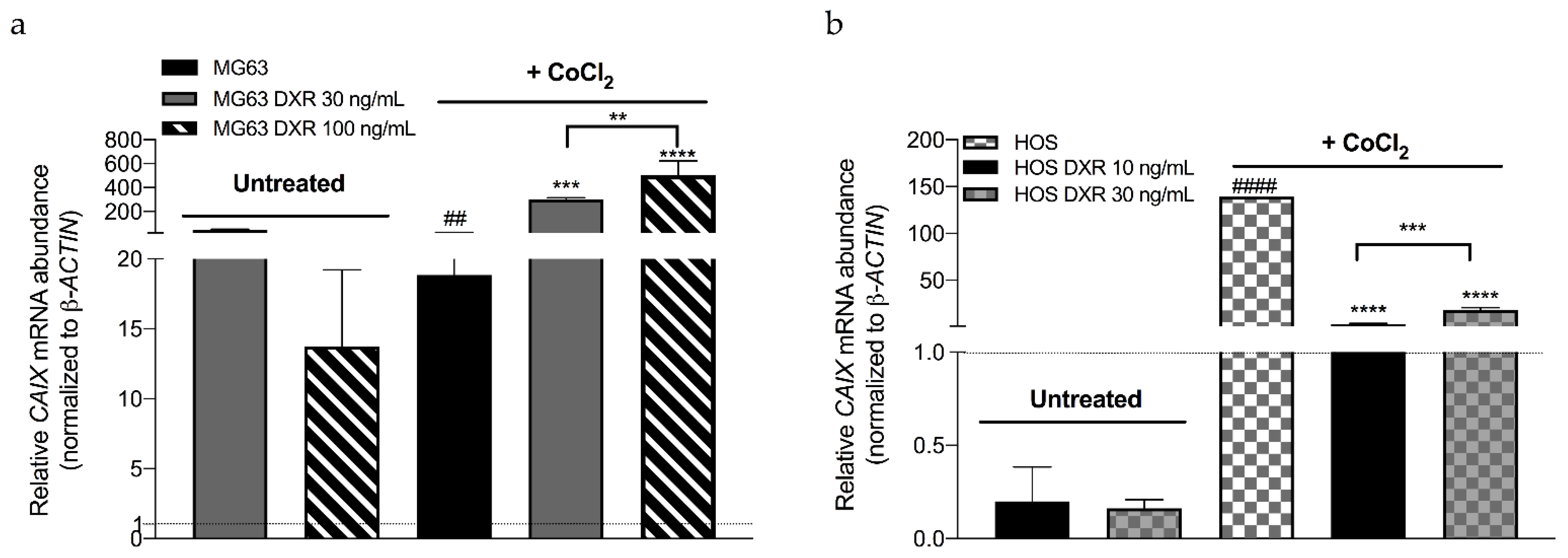
3.3. DXR Osteosarcoma Cells Show Decreased mtROS Levels and Different Response to Hypoxia
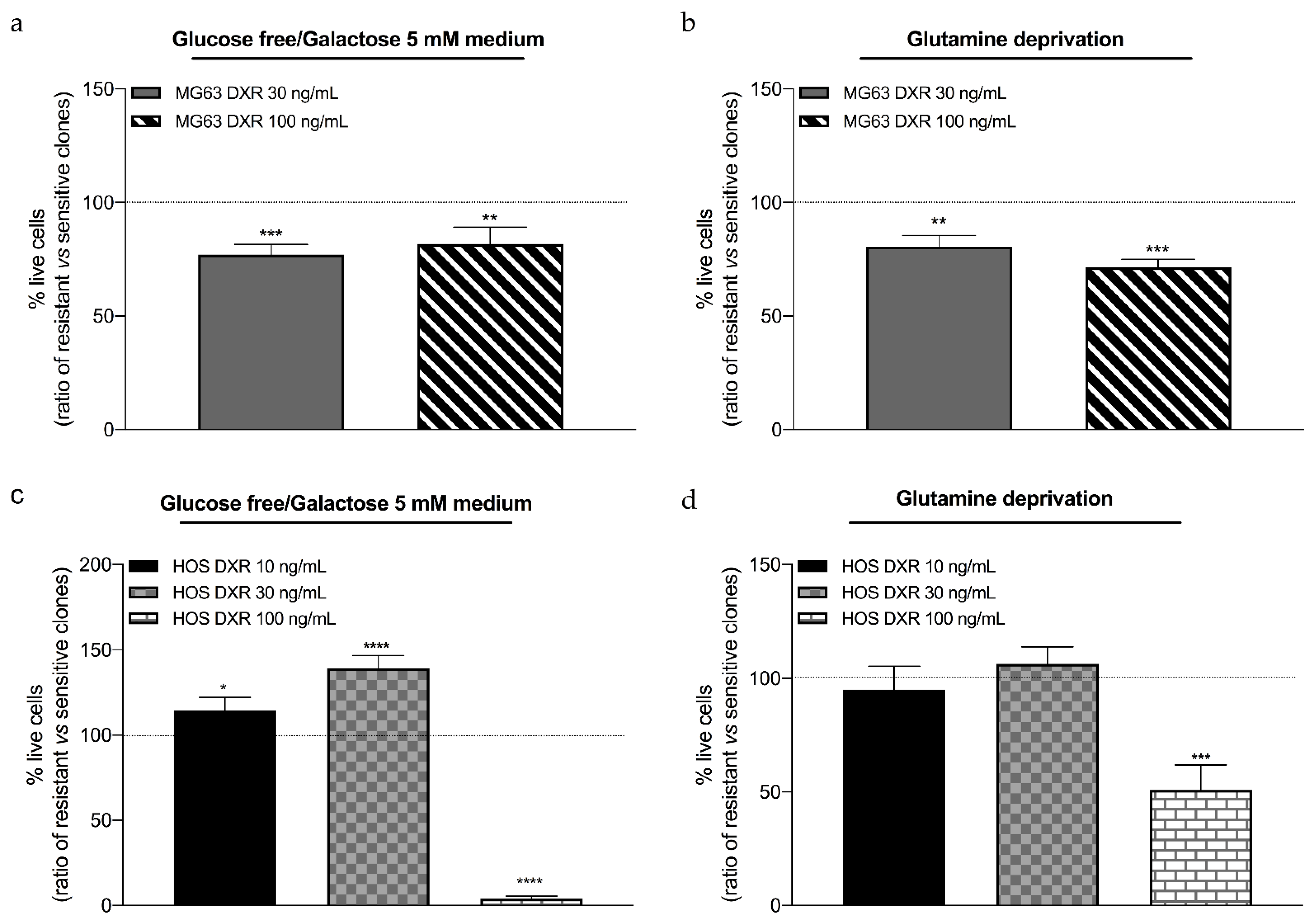
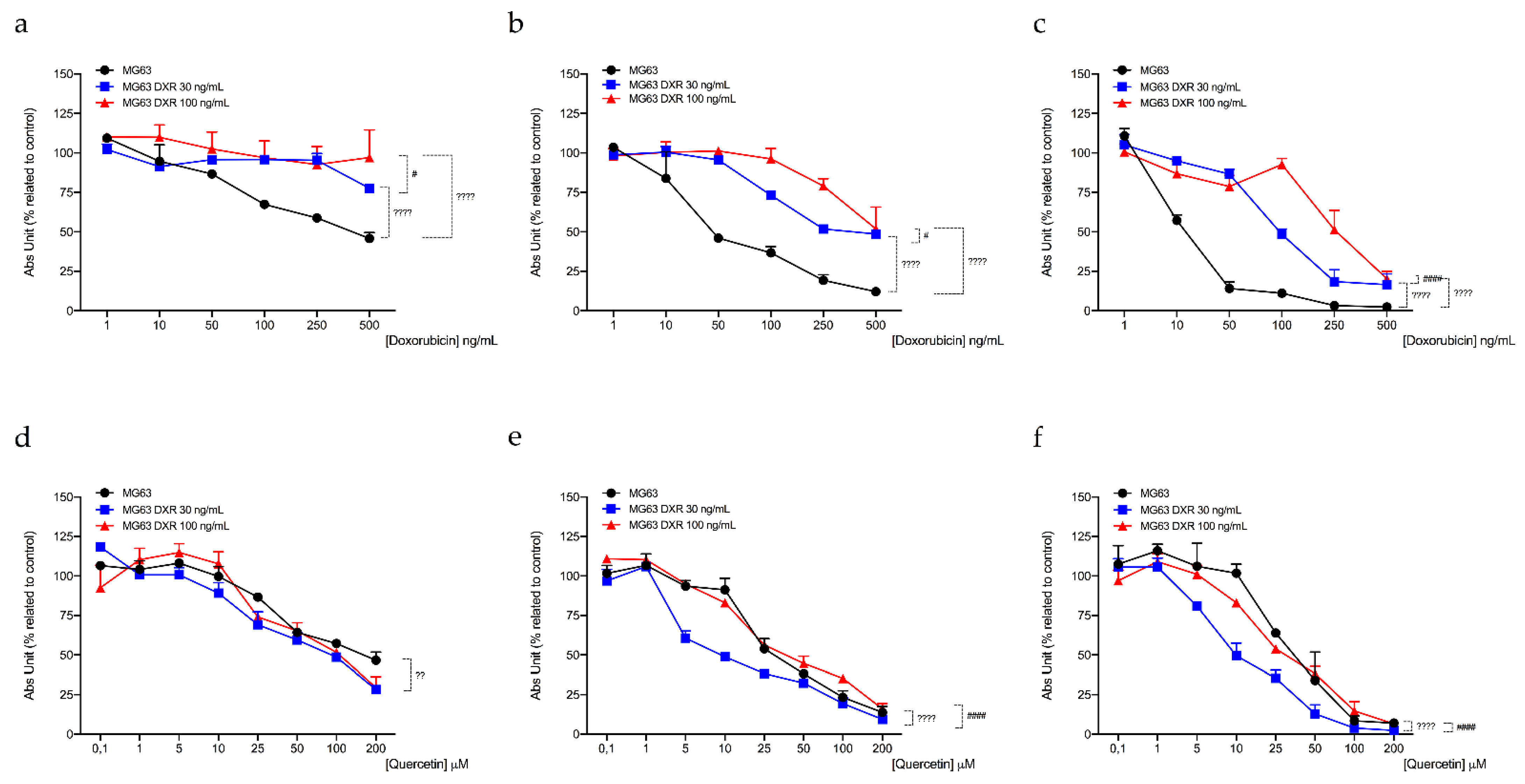
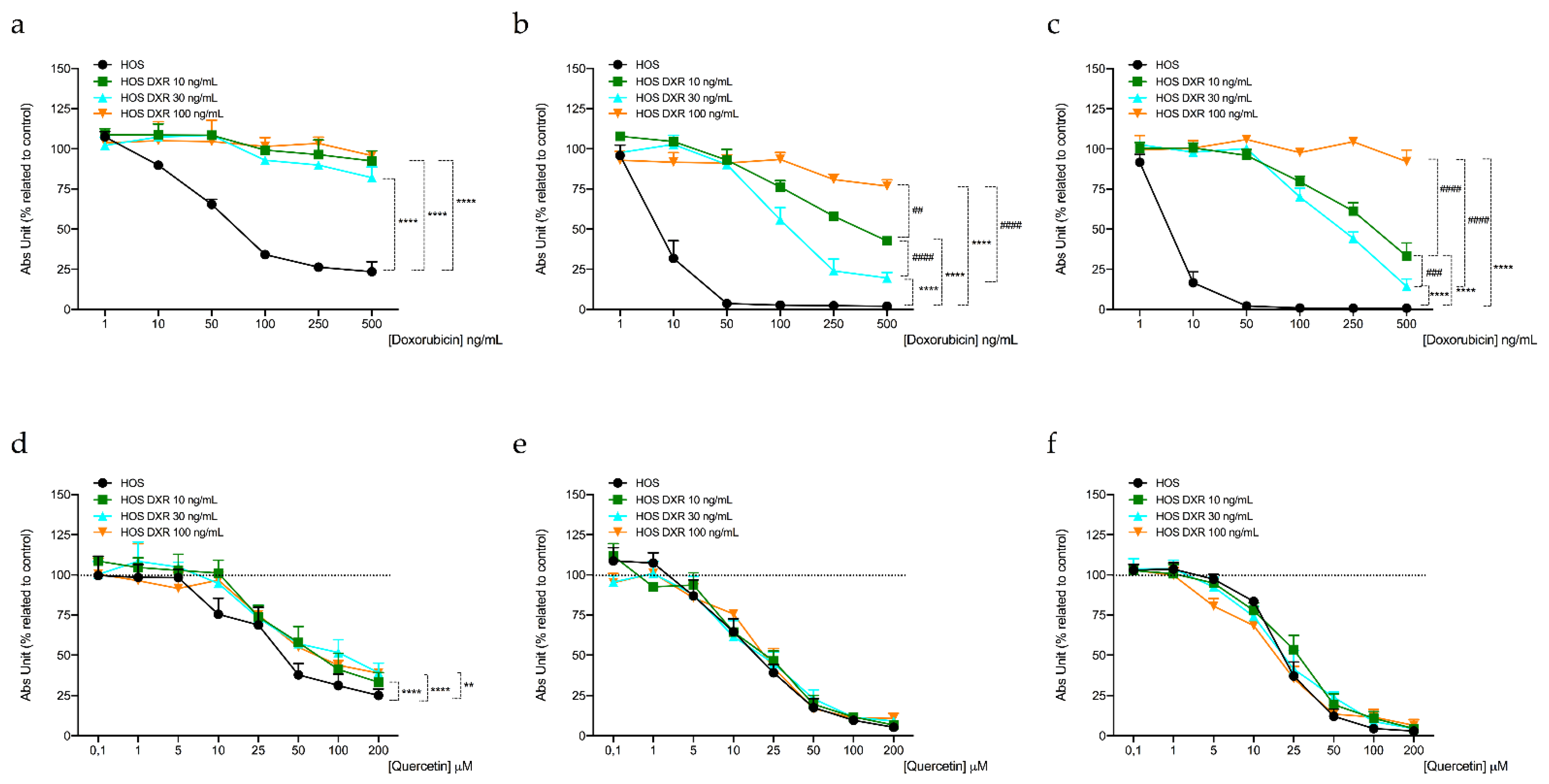
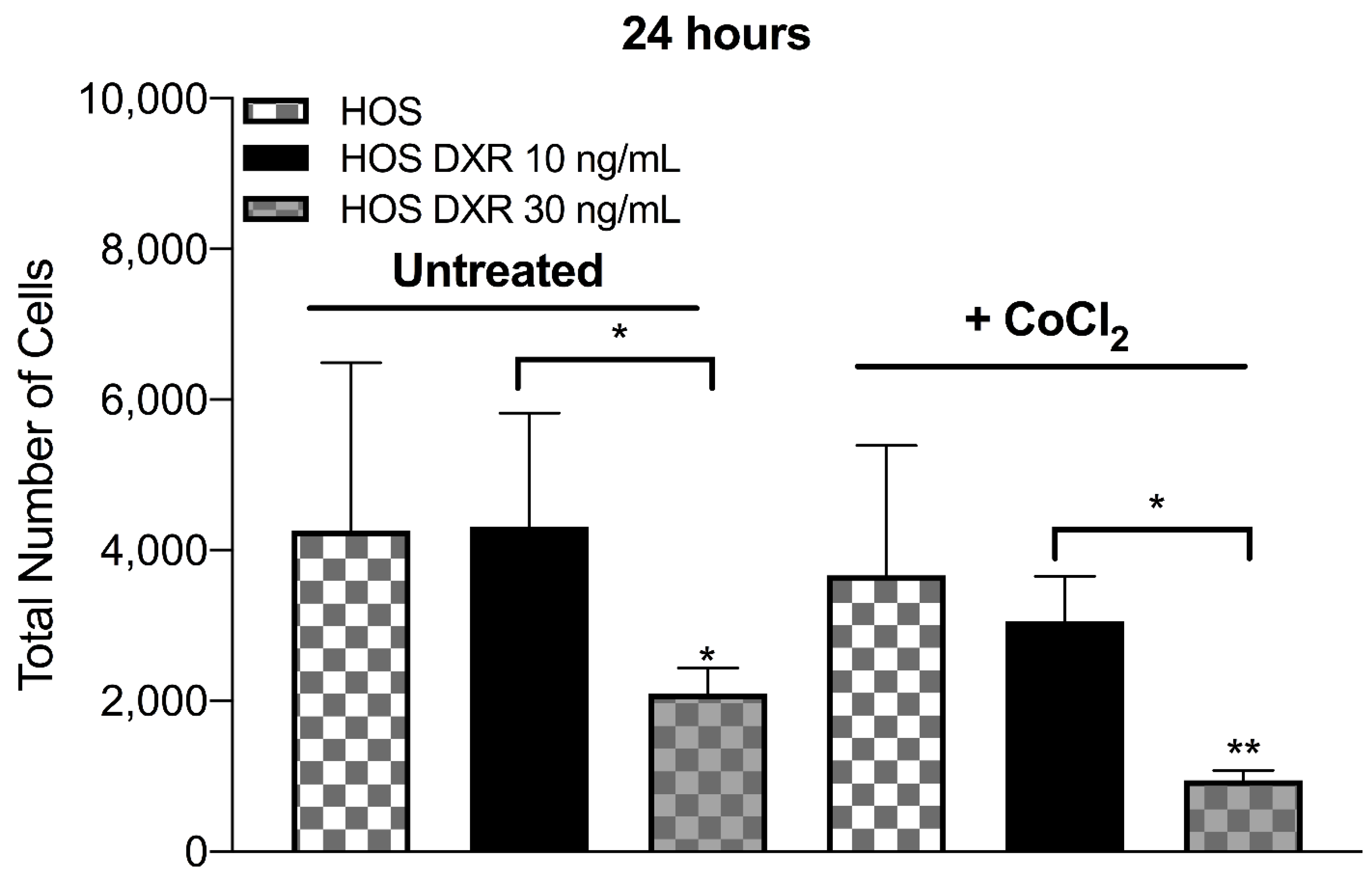
3.4. DXR-Resistant Osteosarcoma Cells Show Different Behavior under Metabolic Stresses
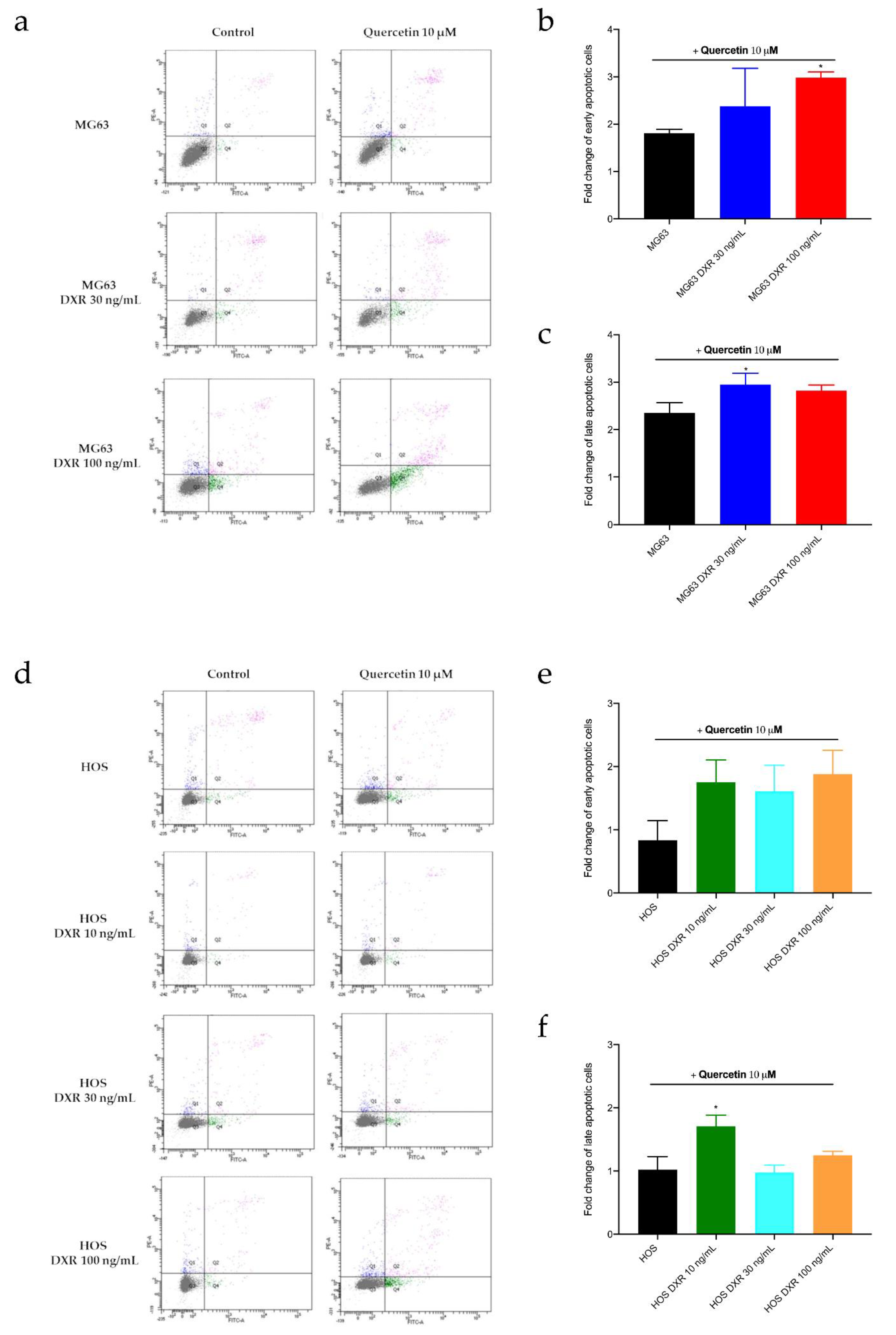
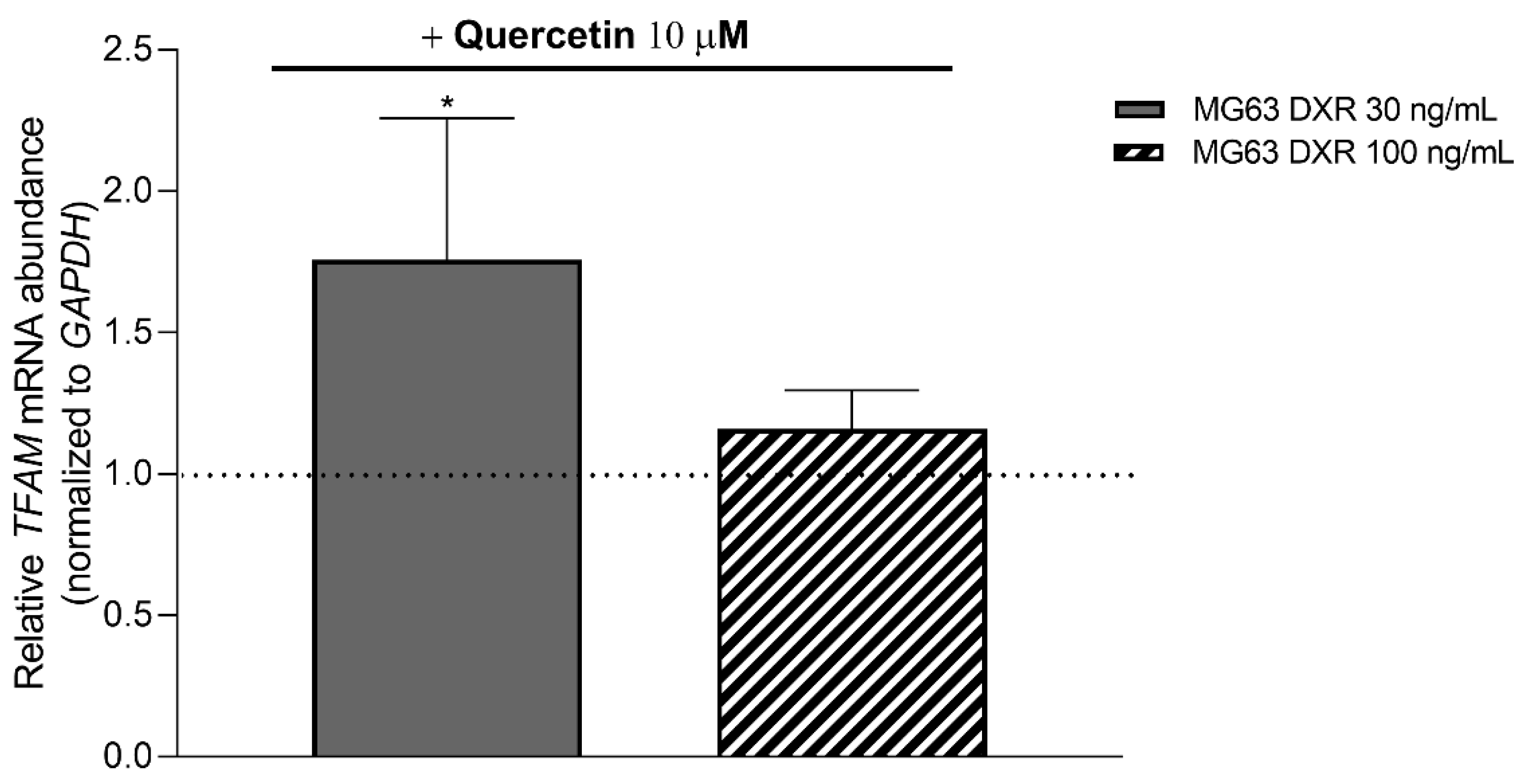
3.5. Targeting Mitochondrial Biogenesis to Restore DXR Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Appendix A




References
- Tobeiha, M.; Rajabi, A.; Raisi, A.; Mohajeri, M.; Yazdi, S.M.; Davoodvandi, A.; Aslanbeigi, F.; Vaziri, M.; Hamblin, M.R.; Mirzaei, H. Potential of natural products in osteosarcoma treatment: Focus on Molecular Mechanisms. Biomed. Pharmacother. 2021, 144, 112257. [Google Scholar] [CrossRef] [PubMed]
- Marchandet, L.; Lallier, M.; Charrier, C.; Baud’huin, M.; Ory, B.; Lamoureux, F. Mechanisms of resistance to conventional therapies for osteosarcoma. Cancers 2021, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- Barani, M.; Mukhtar, M.; Rahdar, A.; Sargazi, S.; Pandey, S.; Kang, M. Recent advances in nanotechnology—based diagnosis and treatments of human osteosarcoma. Biosensors 2021, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Argenziano, M.; Tortora, C.; Pota, E.; Di Paola, A.; Di Martino, M.; Di Leva, C.; Di Pinto, D.; Rossi, F. Osteosarcoma in children: Not only chemotherapy. Pharmaceuticals 2021, 14, 923. [Google Scholar] [CrossRef] [PubMed]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Baldini, N. Multidrug resistance—A multiplex phenomenon. Nat. Med. 1997, 3, 378–380. [Google Scholar] [CrossRef]
- Baldini, N.; Scotlandi, K.; Barbanti-Bròdano, G.; Manara, M.C.; Maurici, D.; Bacci, G.; Bertoni, F.; Picci, P.; Sottili, S.; Campanacci, M.; et al. Expression of p-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N. Engl. J. Med. 1995, 333, 1380–1385. [Google Scholar] [CrossRef]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef] [Green Version]
- Ha, J.S.; Byun, J.; Ahn, D.-R. Overcoming Doxorubicin Resistance of Cancer Cells by Cas9-Mediated Gene Disruption. Sci. Rep. 2016, 6, 22847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget 2015, 6, 30102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vianello, C.; Cocetta, V.; Catanzaro, D.; Dorn, G.W.; De Milito, A.; Rizzolio, F.; Canzonieri, V.; Cecchin, E.; Roncato, R.; Toffoli, G.; et al. Cisplatin resistance can be curtailed by blunting bnip3-mediated mitochondrial autophagy. Cell Death Dis. 2022, 13, 398. [Google Scholar] [CrossRef] [PubMed]
- Fotia, C.; Avnet, S.; Kusuzaki, K.; Roncuzzi, L.; Baldini, N. Acridine orange is an effective anti-cancer drug that affects mitochondrial function in osteosarcoma cells. Curr. Pharm. Des. 2015, 21, 4088–4094. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1 α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- McDonald, P.C.; Dedhar, S. Carbonic Anhydrase IX (CAIX) as a Mediator of Hypoxia-Induced Stress Response in Cancer Cells. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S.C., McKenna, R., Eds.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; Volume 75, pp. 255–269. ISBN 978-94-007-7358-5. [Google Scholar]
- Rayamajhi, N.; Kim, S.-K.; Go, H.; Joe, Y.; Callaway, Z.; Kang, J.-G.; Ryter, S.W.; Chung, H.T. Quercetin induces mitochondrial biogenesis through activation of HO-1 in HepG2 cells. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Chatterjee, A.; Dasgupta, S.; Sidransky, D. Mitochondrial subversion in cancer. Cancer Prev. Res. 2011, 4, 638–654. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.-M.; Hu, X.; Ming, Q.-L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Aung, L.H.H.; Li, R.; Prabhakar, B.S.; Maker, A.V.; Li, P. Mitochondrial protein 18 (MTP18) plays a pro-apoptotic role in chemotherapy—Induced gastric cancer cell apoptosis. Oncotarget 2017, 8, 56582–56597. [Google Scholar] [CrossRef]
- Locatelli, L.; Cazzaniga, A.; Fedele, G.; Zocchi, M.; Scrimieri, R.; Moscheni, C.; Castiglioni, S.; Maier, J.A. A Comparison of doxorubicin-resistant colon cancer LoVo and leukemia HL60 cells: Common features, different underlying mechanisms. Curr. Issues Mol. Biol. 2021, 43, 163–175. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.G.; Schadt, E.E. The role of macromolecular damage in aging and age-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S28–S32. [Google Scholar] [CrossRef] [PubMed]
- Detmer, F.J.; Alpert, N.M.; Moon, S.-H.; Dhaynaut, M.; Guerrero, J.L.; Guehl, N.J.; Xing, F.; Brugarolas, P.; Shoup, T.M.; Normandin, M.D.; et al. PET imaging of mitochondrial function in acute doxorubicin-induced cardiotoxicity: A proof-of-principle study. Sci. Rep. 2022, 12, 6122. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell. Mol. Med. 2020, 24, 6534–6557. [Google Scholar] [CrossRef]
- Xie, D.; Wu, X.; Lan, L.; Shangguan, F.; Lin, X.; Chen, F.; Xu, S.; Zhang, Y.; Chen, Z.; Huang, K.; et al. Downregulation of TFAM inhibits the tumorigenesis of non-small cell lung cancer by activating ROS-mediated JNK/P38MAPK signaling and reducing cellular bioenergetics. Oncotarget 2016, 7, 11609–11624. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Xu, J.; Hu, W.; Wang, F.; Zhou, Y.; Xu, W.; Gong, W.; Shao, L. TFAM depletion overcomes hepatocellular carcinoma resistance to doxorubicin and sorafenib through AMPK activation and mitochondrial dysfunction. Gene 2020, 753, 144807. [Google Scholar] [CrossRef]
- Shen, L.; Sun, B.; Sheng, J.; Yu, S.; Li, Y.; Xu, H.; Su, J.; Sun, L. PGC1α Promotes cisplatin resistance in human ovarian carcinoma cells through upregulation of mitochondrial biogenesis. Int. J. Oncol. 2018, 53, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Cortini, M.; Armirotti, A.; Columbaro, M.; Longo, D.L.; Di Pompo, G.; Cannas, E.; Maresca, A.; Errani, C.; Longhi, A.; Righi, A.; et al. Exploring Metabolic adaptations to the acidic microenvironment of osteosarcoma cells unveils sphingosine 1-phosphate as a valuable therapeutic target. Cancers 2021, 13, 311. [Google Scholar] [CrossRef]
- Koshinaka, K.; Honda, A.; Masuda, H.; Sato, A. Effect of quercetin treatment on mitochondrial biogenesis and exercise-induced AMP-activated protein kinase activation in rat skeletal muscle. Nutrients 2020, 12, 729. [Google Scholar] [CrossRef] [Green Version]
- McKay, T.B.; Lyon, D.; Sarker-Nag, A.; Priyadarsini, S.; Asara, J.M.; Karamichos, D. Quercetin Attenuates lactate production and extracellular matrix secretion in keratoconus. Sci. Rep. 2015, 5, 9003. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 Quercetin (μM)a | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| MG63 | 124.87 ± 9.03 | 68.42 ± 4.53 | 64.71 ± 3.87 |
| MG63 DXR30 ng/mL | 110.87 ± 2.46 | 47.03 ± 8.13 * | 35.70 ± 2.34 ** |
| MG63 DXR100 ng/mL | 100.62 ± 7.50 ° | 84.41 ± 7.54 | 56.84 ± 8.60 |
| Cell Line | IC50 Quercetin (μM) a | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| HOS | 339.23 ± 6.70 | 226.80 ± 10.07 | 224.24 ± 4.72 |
| HOS DXR10 ng/mL | 536.25 ± 11.46 **** | 265.30 ± 0.95 *** | 286.04 ± 1.00 **** |
| HOS DXR30 ng/mL | 458.51 ± 3.02 °°°° | 270.48 ± 3.57 °°°° | 262.12 ± 3.39 °°°° |
| HOS DXR100 ng/mL | 792.65 ± 7.36 #### | 236.77 ± 4.16 | 248.01 ± 2.88 #### |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomini, I.; Cortini, M.; Tinazzi, M.; Baldini, N.; Cocetta, V.; Ragazzi, E.; Avnet, S.; Montopoli, M. Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells. Cancers 2023, 15, 1370. https://doi.org/10.3390/cancers15051370
Giacomini I, Cortini M, Tinazzi M, Baldini N, Cocetta V, Ragazzi E, Avnet S, Montopoli M. Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells. Cancers. 2023; 15(5):1370. https://doi.org/10.3390/cancers15051370
Chicago/Turabian StyleGiacomini, Isabella, Margherita Cortini, Mattia Tinazzi, Nicola Baldini, Veronica Cocetta, Eugenio Ragazzi, Sofia Avnet, and Monica Montopoli. 2023. "Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells" Cancers 15, no. 5: 1370. https://doi.org/10.3390/cancers15051370
APA StyleGiacomini, I., Cortini, M., Tinazzi, M., Baldini, N., Cocetta, V., Ragazzi, E., Avnet, S., & Montopoli, M. (2023). Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells. Cancers, 15(5), 1370. https://doi.org/10.3390/cancers15051370