
Isogenic Cell Lines Derived from Specific Organ Metastases Exhibit Divergent Cytogenomic Aberrations

Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Lines
2.2. Proteomics, RNA-Seq, and Exome-Associated SNPs
2.3. Karyotyping
2.4. Analysis of Genes/Proteins with Validated Functions in Cell Division
2.5. Mutation and Copy Number Variants (Amplifications/Gains and Losses) by DNA-Based Next-Generation Sequencing
3. Results
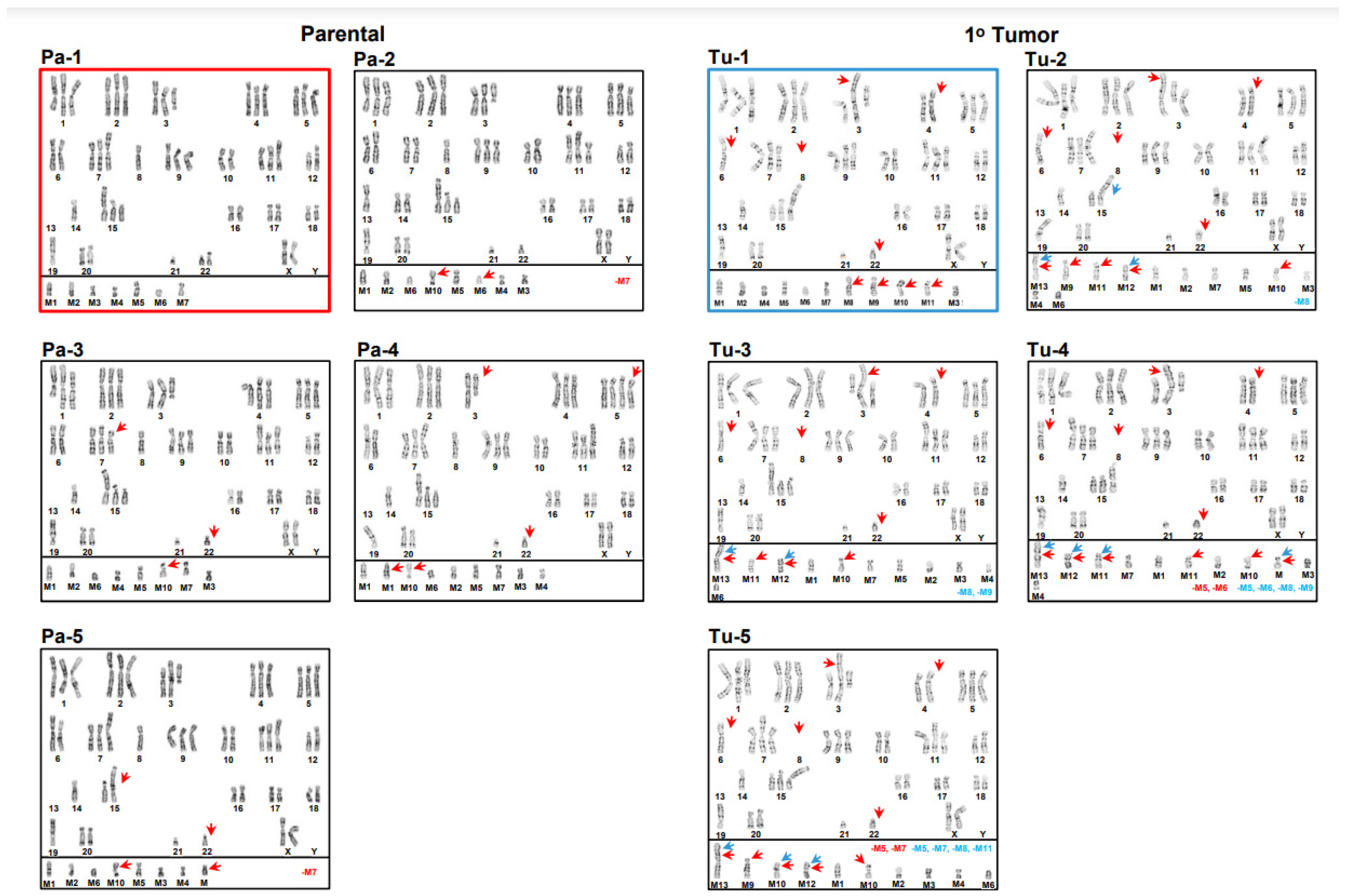
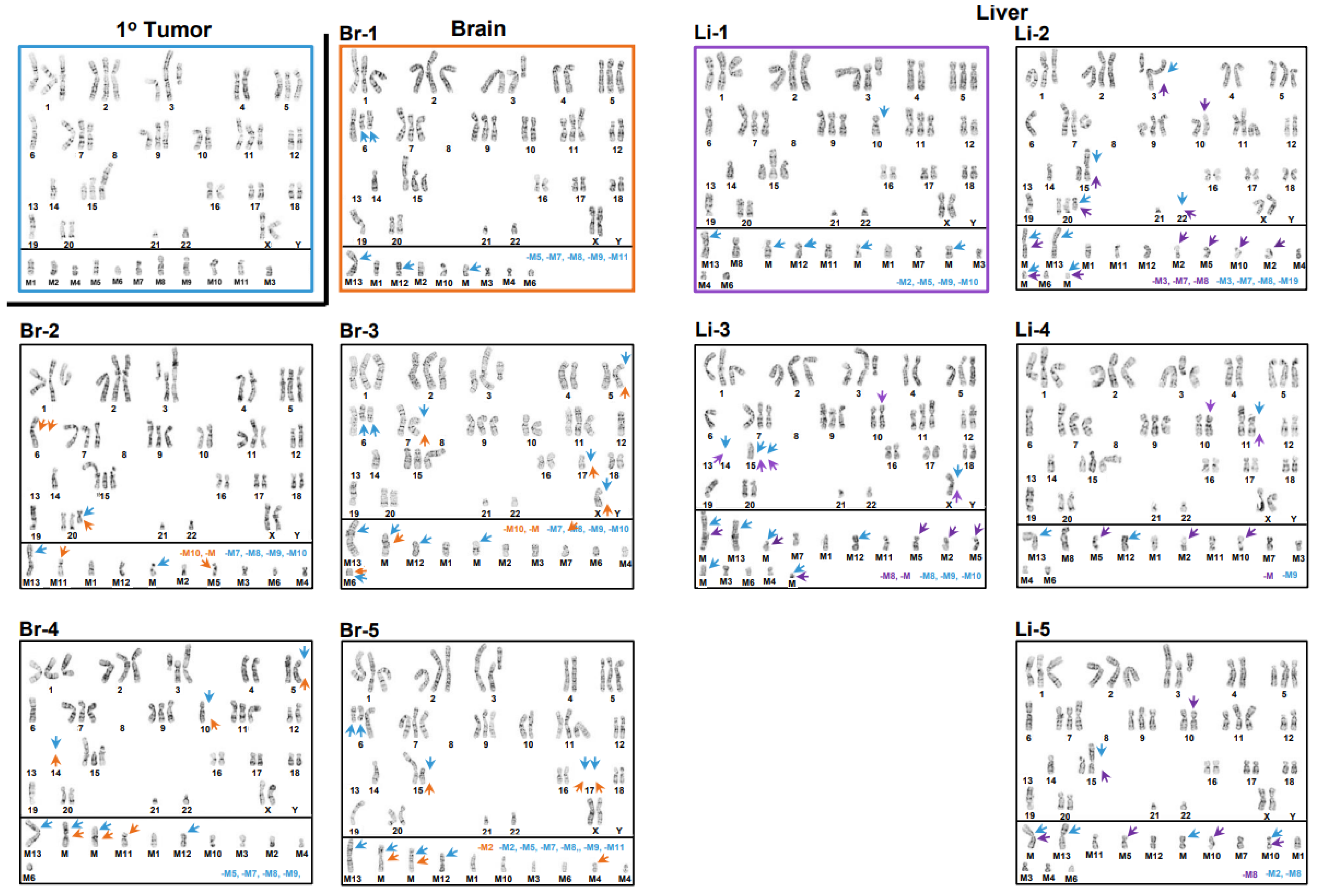
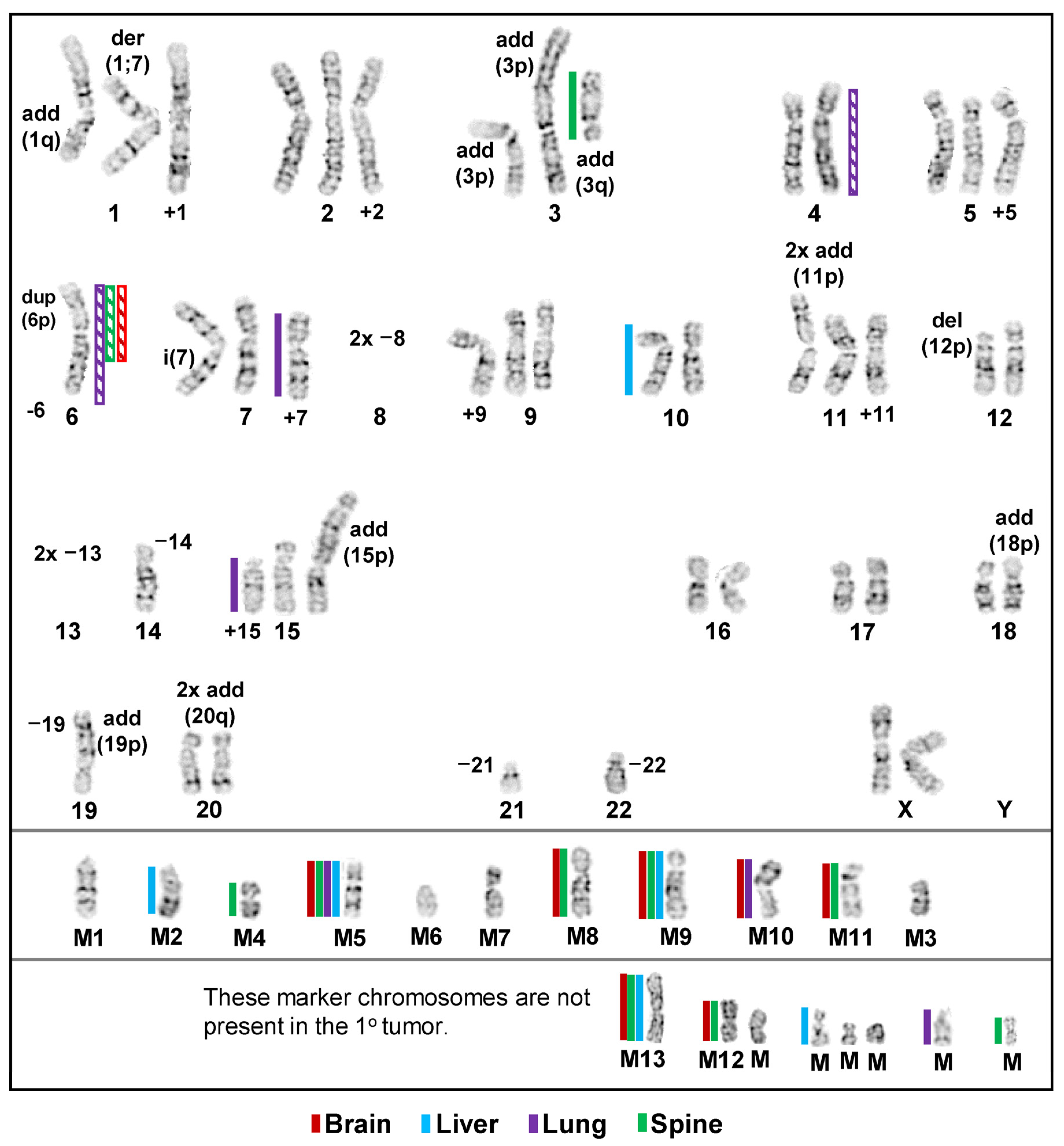
3.1. Inter- and Intrakaryotype Heterogeneities across All Cell Lines
3.2. Relative Differential Expression Levels of Proteins/Transcripts Associated with S/G2-Mitosis of the Cell Cycle
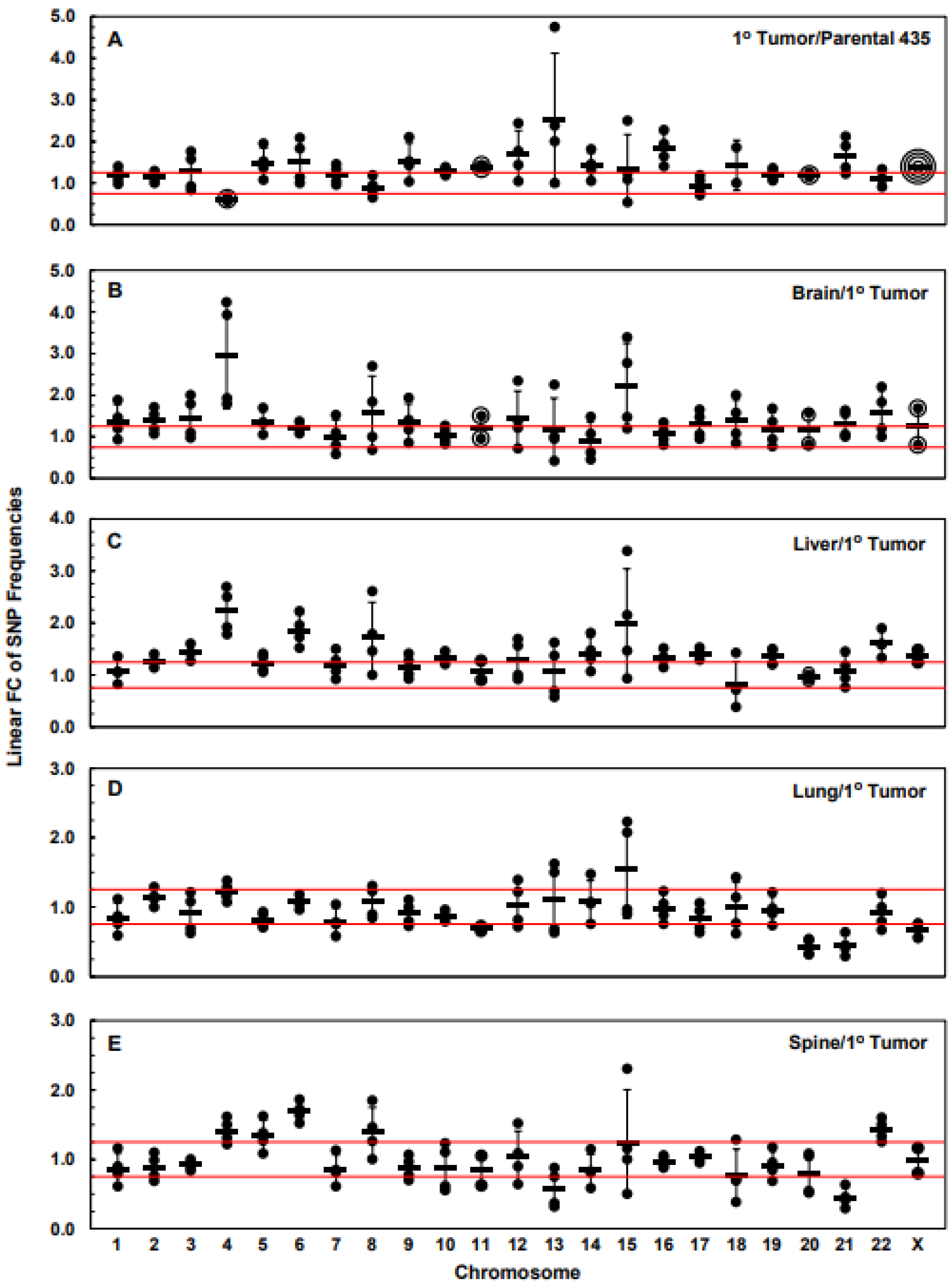
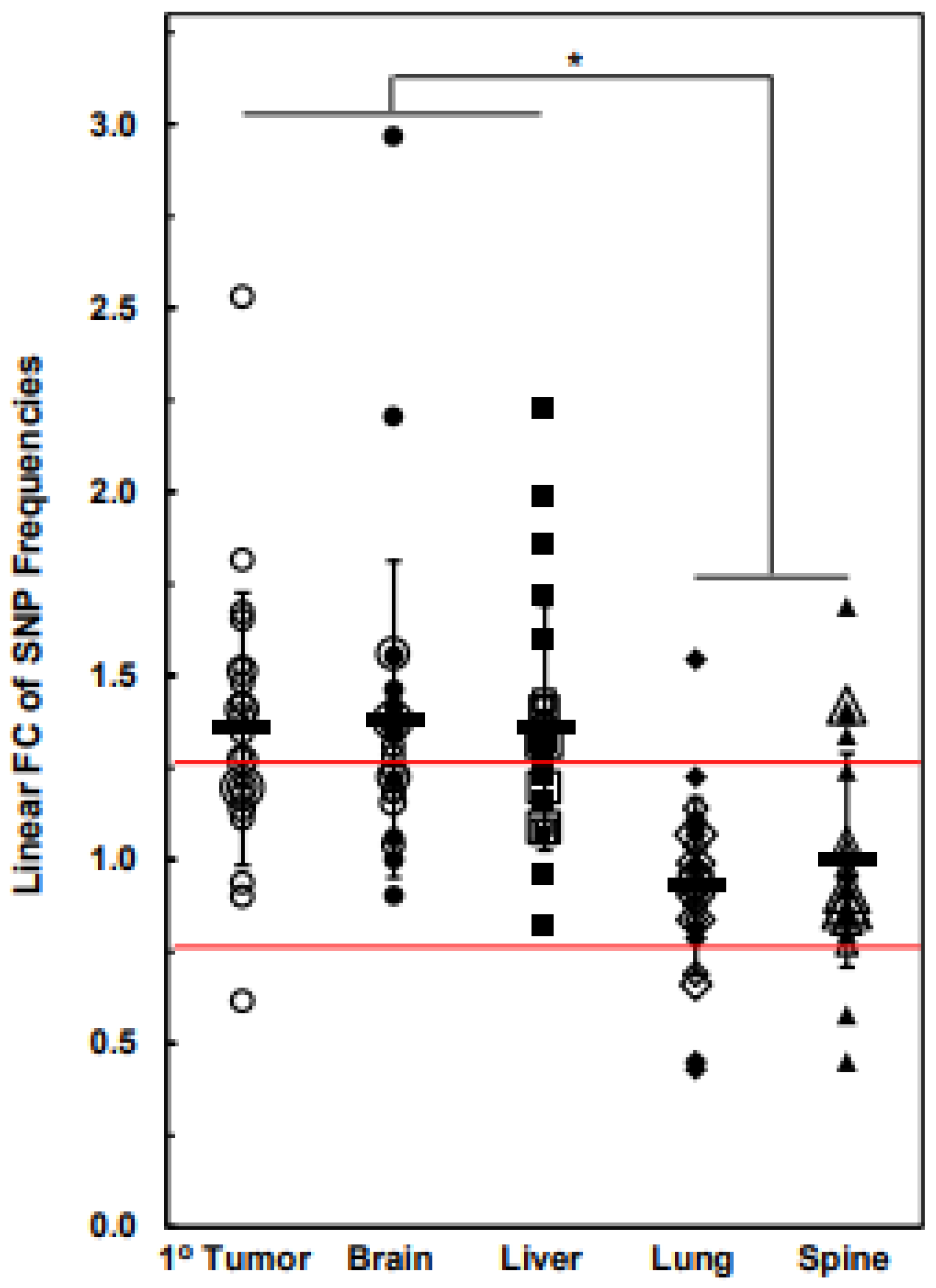
3.3. Numbers of Exome-Specific SNPs Differ in Each Chromosome across Isogenic Cell Lines
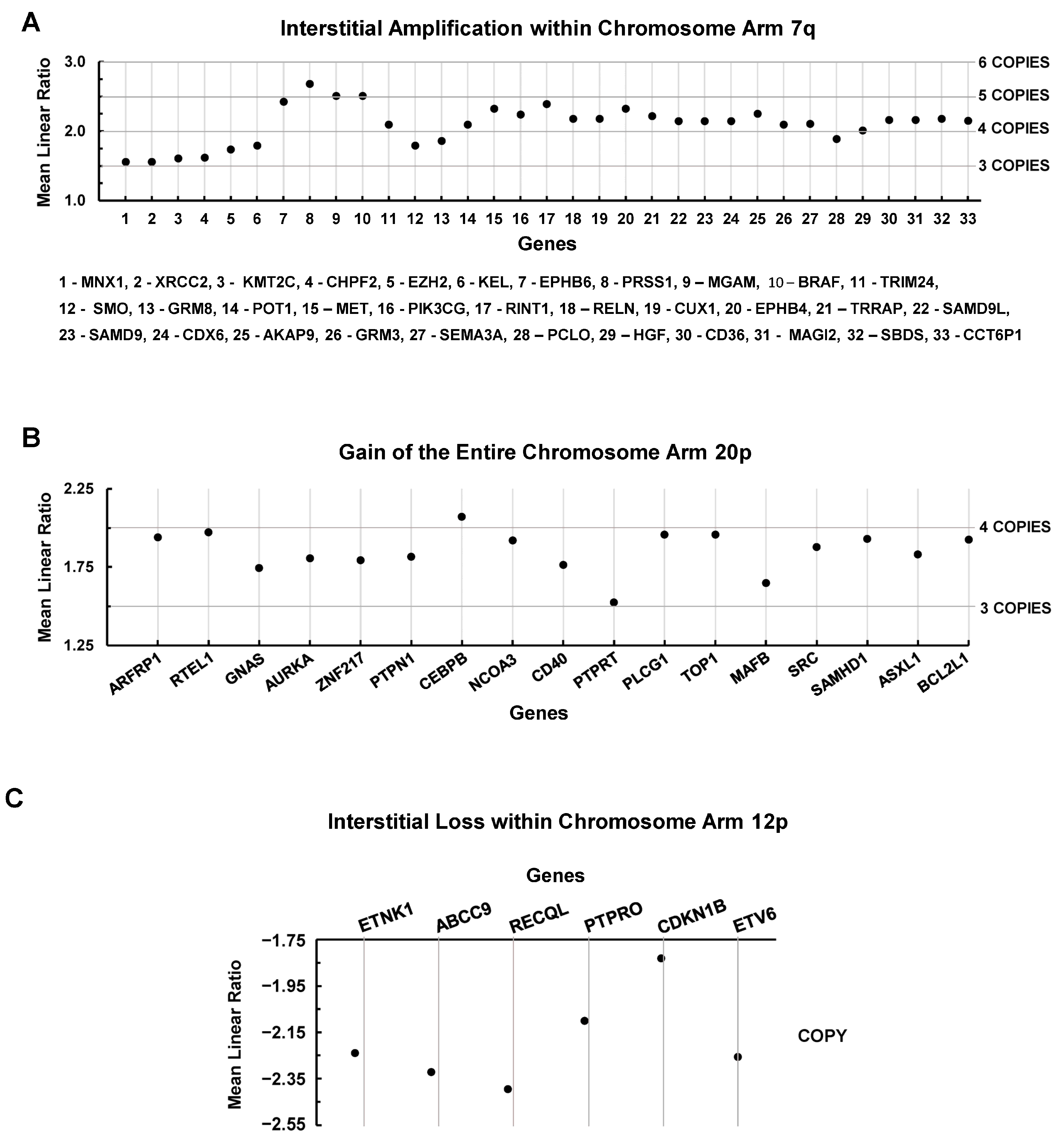
3.4. Chromosomal Level Amplifications, Gains, and Losses
3.5. Gene Variants by DNA-Based NGS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bignold, L.P.; Coghlan, B.L.; Jersmann, H.P. Hansemann, Boveri, chromosomes and the gametogenesis-related theories of tumours. Cell Biol. Int. 2006, 30, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Hardy, P.A.; Zacharias, H. Reappraisal of the Hansemann-Boveri hypothesis on the origin of tumors. Cell Biol. Int. 2005, 29, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Losing balance: The origin and impact of aneuploidy in cancer. EMBO Rep. 2012, 13, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinarsdottir, M.; Gudmundsson, I.H.; Jonasson, J.G.; Olafsdottir, E.J.; Eyfjord, J.E.; Ogmundsdottir, H.M. Cytogenetic polyclonality of breast carcinomas: Association with clinico-pathological characteristics and outcome. Genes Chromosom. Cancer 2011, 50, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e673. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef]
- Aze, A.; Zhou, J.C.; Costa, A.; Costanzo, V. DNA replication and homologous recombination factors: Acting together to maintain genome stability. Chromosoma 2013, 122, 401–413. [Google Scholar] [CrossRef]
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016, 16, 47. [Google Scholar] [CrossRef] [Green Version]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Toth, E.; et al. Corrigendum to: Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2019, 30, 1406. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Morris, B.B.; Smith, J.P.; Zhang, Q.; Jiang, Z.; Hampton, O.A.; Churchman, M.L.; Arnold, S.M.; Owen, D.H.; Gray, J.E.; Dillon, P.M.; et al. Replicative Instability Drives Cancer Progression. Biomolecules 2022, 12, 1570. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Bakker, B.; de Haan, L.; Tijhuis, A.E.; Wang, Y.; Kim, D.H.; Maldonado, M.; Demarest, M.A.; Artates, J.; Zhengyu, O.; et al. Transient genomic instability drives tumorigenesis through accelerated clonal evolution. Genes Dev. 2021, 35, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 2011, 108, 17974–17978. [Google Scholar] [CrossRef] [Green Version]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Bolkestein, M.; Wong, J.K.L.; Thewes, V.; Korber, V.; Hlevnjak, M.; Elgaafary, S.; Schulze, M.; Kommoss, F.K.F.; Sinn, H.P.; Anzeneder, T.; et al. Chromothripsis in Human Breast Cancer. Cancer Res. 2020, 80, 4918–4931. [Google Scholar] [CrossRef]
- Lin, C.L.; Tan, X.; Chen, M.; Kusi, M.; Hung, C.N.; Chou, C.W.; Hsu, Y.T.; Wang, C.M.; Kirma, N.; Chen, C.L.; et al. ERalpha-related chromothripsis enhances concordant gene transcription on chromosome 17q11.1-q24.1 in luminal breast cancer. BMC Med. Genom. 2020, 13, 69. [Google Scholar] [CrossRef]
- O’Neill, F.J.; Miles, C.P. Origin of nuclei in spontaneous HeLa cell chromosome pulverization. J. Natl. Cancer Inst. 1971, 46, 1085–1092. [Google Scholar]
- Sandberg, A.A.; Aya, T.; Ikeuchi, T.; Weinfeld, H. Definition and morphologic features of chromosome pulverization: A hypothesis to explain the phenomenon. J. Natl. Cancer Inst. 1970, 45, 615–623. [Google Scholar]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Brand, H.; Redin, C.E.; Hanscom, C.; Antolik, C.; Stone, M.R.; Glessner, J.T.; Mason, T.; Pregno, G.; Dorrani, N.; et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017, 18, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybytkowski, E.; Lenkiewicz, E.; Barrett, M.T.; Klein, K.; Nabavi, S.; Greenwood, C.M.; Basik, M. Chromosome-breakage genomic instability and chromothripsis in breast cancer. BMC Genom. 2014, 15, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320. [Google Scholar] [CrossRef] [PubMed]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 2018, 7, e37294. [Google Scholar] [CrossRef]
- Pinto, A.E.; Pereira, T.; Santos, M.; Branco, M.; Dias, A.; Silva, G.L.; Ferreira, M.C.; Andre, S. DNA ploidy is an independent predictor of survival in breast invasive ductal carcinoma: A long-term multivariate analysis of 393 patients. Ann. Surg. Oncol. 2013, 20, 1530–1537. [Google Scholar] [CrossRef]
- van Dijk, E.; van den Bosch, T.; Lenos, K.J.; El Makrini, K.; Nijman, L.E.; van Essen, H.F.B.; Lansu, N.; Boekhout, M.; Hageman, J.H.; Fitzgerald, R.C.; et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 2021, 12, 3188. [Google Scholar] [CrossRef]
- Sheltzer, J.M. A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 2013, 73, 6401–6412. [Google Scholar] [CrossRef] [Green Version]
- Lukow, D.A.; Sheltzer, J.M. Chromosomal instability and aneuploidy as causes of cancer drug resistance. Trends Cancer 2022, 8, 43–53. [Google Scholar] [CrossRef]
- Replogle, J.M.; Zhou, W.; Amaro, A.E.; McFarland, J.M.; Villalobos-Ortiz, M.; Ryan, J.; Letai, A.; Yilmaz, O.; Sheltzer, J.; Lippard, S.J.; et al. Aneuploidy increases resistance to chemotherapeutics by antagonizing cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30566–30576. [Google Scholar] [CrossRef]
- Yang, F.; Teoh, F.; Tan, A.S.M.; Cao, Y.; Pavelka, N.; Berman, J. Aneuploidy Enables Cross-Adaptation to Unrelated Drugs. Mol. Biol. Evol. 2019, 36, 1768–1782. [Google Scholar] [CrossRef]
- Cohen-Sharir, Y.; McFarland, J.M.; Abdusamad, M.; Marquis, C.; Bernhard, S.V.; Kazachkova, M.; Tang, H.; Ippolito, M.R.; Laue, K.; Zerbib, J.; et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 2021, 590, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Mustachio, L.M.; Liu, X.; Dmitrovsky, E. Engaging Anaphase Catastrophe Mechanisms to Eradicate Aneuploid Cancers. Mol. Cancer 2018, 17, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. OncoTargets Ther. 2017, 10, 5633–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scribano, C.M.; Wan, J.; Esbona, K.; Tucker, J.B.; Lasek, A.; Zhou, A.S.; Zasadil, L.M.; Molini, R.; Fitzgerald, J.; Lager, A.M.; et al. Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci. Transl. Med. 2021, 13, eabd4811. [Google Scholar] [CrossRef]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm. Sin. B 2021, 11, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- van Jaarsveld, R.H.; Kops, G. Difference Makers: Chromosomal Instability versus Aneuploidy in Cancer. Trends Cancer 2016, 2, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Guille, A.; Adelaide, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11, 4306. [Google Scholar] [CrossRef] [PubMed]
- Tyran, M.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaide, J.; Finetti, P.; Toulzian, J.; Viens, P.; Tallet, A.; Goncalves, A.; et al. A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers 2019, 11, 665. [Google Scholar] [CrossRef] [Green Version]
- Winnard, P.T., Jr.; Vesuna, F.; Muthukumar, S.; Raman, V. Divergent organ-specific isogenic metastatic cell lines identified using multi-omics exhibit differential drug sensitivity. PLoS ONE 2020, 15, e0242384. [Google Scholar] [CrossRef]
- Winnard, P.T., Jr.; Zhang, C.; Vesuna, F.; Kang, J.W.; Garry, J.; Dasari, R.R.; Barman, I.; Raman, V. Organ-specific isogenic metastatic breast cancer cell lines exhibit distinct Raman spectral signatures and metabolomes. Oncotarget 2017, 8, 20266–20287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Winnard, P.T., Jr.; Dasari, S.; Kominsky, S.L.; Doucet, M.; Jayaraman, S.; Raman, V.; Barman, I. Label-free Raman spectroscopy provides early determination and precise localization of breast cancer-colonized bone alterations. Chem. Sci. 2018, 9, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Winnard, P.T., Jr.; Kluth, J.B.; Raman, V. Noninvasive optical tracking of red fluorescent protein-expressing cancer cells in a model of metastatic breast cancer. Neoplasia 2006, 8, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schukken, K.M.; Foijer, F. CIN and Aneuploidy: Different Concepts, Different Consequences. Bioessays 2018, 40, 1700147. [Google Scholar] [CrossRef] [PubMed]
- Giotti, B.; Chen, S.H.; Barnett, M.W.; Regan, T.; Ly, T.; Wiemann, S.; Hume, D.A.; Freeman, T.C. Assembly of a parts list of the human mitotic cell cycle machinery. J. Mol. Cell Biol. 2019, 11, 703–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Pallavajjala, A.; Huang, J.; Haley, L.; Morsberger, L.; Stinnett, V.; Hardy, M.; Park, R.; Ament, C.; Finch, A.; et al. Clinical Utility of Targeted Next-Generation Sequencing Assay to Detect Copy Number Variants Associated with Myelodysplastic Syndrome in Myeloid Malignancies. J. Mol. Diagn. 2021, 23, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Pallavajjala, A.; Haley, L.; Stinnett, V.; Adams, E.; Pallavajjala, R.; Huang, J.; Morsberger, L.; Hardy, M.; Long, P.; Gocke, C.D.; et al. Utility of targeted next-generation sequencing assay to detect 1p/19q co-deletion in formalin-fixed paraffin-embedded glioma specimens. Hum. Pathol. 2022, 126, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Du, L.; Cai, S.; Lin, W.; Chen, C.; Still, M.; Yao, Z.; Coppes, R.P.; Pan, Y.; Zhang, D.; et al. Tyrosine Phosphatase PTPRO Deficiency in ERBB2-Positive Breast Cancer Contributes to Poor Prognosis and Lapatinib Resistance. Front. Pharm. 2022, 13, 838171. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Ma, L.; Gan, J.; Lin, W.; Chen, C.; Yao, Z.; Du, L.; Zheng, L.; Ke, C.; Huang, X.; et al. PTPRO represses ERBB2-driven breast oncogenesis by dephosphorylation and endosomal internalization of ERBB2. Oncogene 2017, 36, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, T.; Yuan, Y.; Sun, X. Identification of FOXE3 transcription factor as a potent oncogenic factor in triple-negative breast cancer. Biochem. Biophys. Res. Commun. 2020, 523, 78–85. [Google Scholar] [CrossRef]
- Huang, K.S.; Wang, Y.T.; Byadgi, O.; Huang, T.Y.; Tai, M.H.; Shaw, J.F.; Yang, C.H. Screening of Specific and Common Pathways in Breast Cancer Cell Lines MCF-7 and MDA-MB-231 Treated with Chlorophyllides Composites. Molecules 2022, 27, 3950. [Google Scholar] [CrossRef] [PubMed]
- Shivhare, S.; Das, A. Cell density modulates chemoresistance in breast cancer cells through differential expression of ABC transporters. Mol. Biol. Rep. 2022, 50, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Lu, X.; Lal, A.; Sharma, S. Genome-wide investigations on regulatory functions of RECQ1 helicase. Methods 2022, 204, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, Y.E.; Liu, H.; Zhou, X.; Yuan, M.; Hou, F.; Wang, L.; Jiang, W. Single-cell RNA-seq dissects the intratumoral heterogeneity of triple-negative breast cancer based on gene regulatory networks. Mol. Nucleic Acids 2021, 23, 682–690. [Google Scholar] [CrossRef]
- Gerling, M.; Nousiainen, K.; Hautaniemi, S.; Kruger, S.; Fritzsche, B.; Homann, N.; Bruch, H.P.; Auer, G.; Roblick, U.J.; Ried, T.; et al. Aneuploidy-associated gene expression signatures characterize malignant transformation in ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 691–703. [Google Scholar] [CrossRef]
- Shukla, A.; Nguyen, T.H.M.; Moka, S.B.; Ellis, J.J.; Grady, J.P.; Oey, H.; Cristino, A.S.; Khanna, K.K.; Kroese, D.P.; Krause, L.; et al. Chromosome arm aneuploidies shape tumour evolution and drug response. Nat. Commun. 2020, 11, 449. [Google Scholar] [CrossRef] [Green Version]
- Spurr, L.F.; Weichselbaum, R.R.; Pitroda, S.P. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers. Nat. Genet. 2022, 54, 1782–1785. [Google Scholar] [CrossRef]
- Adeyinka, A.; Pandis, N.; Nilsson, J.; Idvall, I.; Mertens, F.; Petersson, C.; Heim, S.; Mitelman, F. Different cytogenetic patterns in skeletal breast cancer metastases. Genes Chromosom. Cancer 1996, 16, 72–74. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Bloomfield, M.; Duesberg, P. Is cancer progression caused by gradual or simultaneous acquisitions of new chromosomes? Mol. Cytogenet. 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Ryu, N.M.; Kim, J.M. Centrobin plays a role in the cellular response to DNA damage. Cell Cycle 2019, 18, 2660–2671. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shim, J.; Ji, M.J.; Jung, Y.; Bong, S.M.; Jang, Y.J.; Yoon, E.K.; Lee, S.J.; Kim, K.G.; Kim, Y.H.; et al. The condensin component NCAPG2 regulates microtubule-kinetochore attachment through recruitment of Polo-like kinase 1 to kinetochores. Nat. Commun. 2014, 5, 4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.; Wie, M.; Baek, I.J.; Sohn, G.; Um, S.H.; Lee, S.G.; Seo, Y.; Ra, J.; Lee, E.A.; Kim, S.; et al. TRIP13 Participates in Immediate-Early Sensing of DNA Strand Breaks and ATM Signaling Amplification through MRE11. Cells 2022, 11, 4095. [Google Scholar] [CrossRef]
- Li, H.N.; Zheng, W.H.; Du, Y.Y.; Wang, G.; Dong, M.L.; Yang, Z.F.; Li, X.R. ZW10 interacting kinetochore protein may serve as a prognostic biomarker for human breast cancer: An integrated bioinformatics analysis. Oncol. Lett. 2020, 19, 2163–2174. [Google Scholar] [CrossRef] [Green Version]
- Job, A.; Schmitt, L.M.; von Wenserski, L.; Lankat-Buttgereit, B.; Gress, T.M.; Buchholz, M.; Gallmeier, E. Inactivation of PRIM1 Function Sensitizes Cancer Cells to ATR and CHK1 Inhibitors. Neoplasia 2018, 20, 1135–1143. [Google Scholar] [CrossRef]
- Yang, C.; Xie, S.; Wu, Y.; Ru, G.; He, X.; Pan, H.Y.; Wang, S.; Tong, X. Prognostic implications of cell division cycle protein 45 expression in hepatocellular carcinoma. PeerJ 2021, 9, e10824. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M. Molecular Link between DNA Damage Response and Microtubule Dynamics. Int. J. Mol. Sci. 2022, 23, 6986. [Google Scholar] [CrossRef]
- Nikolaev, A.; Yang, E.S. The Impact of DNA Repair Pathways in Cancer Biology and Therapy. Cancers 2017, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Rong, G.; Yi, Z.; Ma, F.; Guan, Y.; Xu, Y.; Li, L.; Xu, B. DNA damage response as a prognostic indicator in metastatic breast cancer via mutational analysis. Ann. Transl. Med. 2021, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Liegmann, A.S.; Heselmeyer-Haddad, K.; Lischka, A.; Hirsch, D.; Chen, W.D.; Torres, I.; Gemoll, T.; Rody, A.; Thorns, C.; Gertz, E.M.; et al. Single Cell Genetic Profiling of Tumors of Breast Cancer Patients Aged 50 Years and Older Reveals Enormous Intratumor Heterogeneity Independent of Individual Prognosis. Cancers 2021, 13, 3366. [Google Scholar] [CrossRef]
- Ramdzan, Z.M.; Vickridge, E.; Faraco, C.C.F.; Nepveu, A. CUT Domain Proteins in DNA Repair and Cancer. Cancers 2021, 13, 2953. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vadnais, C.; Livingstone, J.; Kwiatkowski, N.; Awan, A.; Cadieux, C.; Leduy, L.; Hallett, M.T.; Nepveu, A. Cut homeobox 1 causes chromosomal instability by promoting bipolar division after cytokinesis failure. Proc. Natl. Acad. Sci. USA 2011, 108, 1949–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process | Protein | Description | Chr Location | F.C. 1° Tumor |
|---|---|---|---|---|
| Cell Cycle Regulation | FZR1 | Fizzy/cell division cycle 20 related 1 | 19p13.3 | −1.36 |
| MELK | Maternal embryonic leucine zipper kinase | 9q13.2 | 1.30 | |
| PRR11 | Proline rich 11 | 17q22 | 1.25 | |
| UBE2C | Ubiquitin conjugating enzyme E2C | 20q13.12 | −1.38 | |
| UBE2S | Ubiquitin conjugating enzyme E2S | 19q13.42 | −1.25 | |
| Centrosome Regulation | CEP192 | Centrosomal protein 192kDa | 18p11.21 | 1.32 |
| CEP72 | Centrosomal protein 72kDa | 5p15.33 | 1.32 | |
| CEP76 | Centrosomal protein 76kDa | 18p11.21 | 1.26 | |
| CNTROB | Centrobin, centrosomal BRCA2 interacting protein | 17p13.1 | 3.32 | |
| Cytokinesis | ASPM | Abnormal spindle microtubule assembly | 1q31.3 | 1.36 |
| Chromosome Partition | NSL1 | NSL1, MIS12 kinetochore complex component | 1q32.3 | 1.73 |
| DNA Condensation | ESCO2 | Establishment of sister chromatid cohesion N-acetyltransferase 2 | 8q21.1 | 1.39 |
| NCAPD3 | Non-SMC condensin II complex subunit D3 | 11q25 | 1.25 | |
| NCAPG2 | Non-SMC condensin II complex subunit G2 | 7q36.3 | 1.49 | |
| Kinetochore Formation | CENPI | Centromere protein I | Xq22.1 | 1.27 |
| CENPQ | Centromere protein Q | 6p12.3 | −1.34 | |
| MIS18A | MIS18 kinetochore protein A | 21q22.11 | −1.52 | |
| KNTC1 | Kinetochore associated 1 | 12q24.31 | 1.85 | |
| Microtubule Regulation | TUBB3 | Tubulin, beta 3 class III | 16q24.3 | 1.32 |
| TUBGCP3 | Tubulin, gamma complex associated protein 3 | 13q24 | 1.44 | |
| PCNT | Pericentrin | 21q22.3 | 1.67 | |
| Spindle Assembly and Regulation | HAUS7 | HAUS augmin like complex subunit 7 | Xq28 | −1.43 |
| Spindle Checkpoint | ZW10 | ZW10 kinetochore protein | 11q23.2 | 1.36 |
| ZWILCH | ZWILCH kinetochore protein | 15q22.31 | 1.32 | |
| DNA Damage | MTBP | MDM2 binding protein | 8q24.12 | −1.32 |
| PARP2 | Poly(ADP-ribose) polymerase 2 | 14q11.2 | 1.24 | |
| TTI1 | TELO2 interacting protein 1 | 20q11.23 | 1.42 | |
| PCLAF | PCNA clamp associated factor | 15q22.31 | 1.41 | |
| DDX11 | DEAD/H (Asp-Glu-Ala-Asp/His) box helicase 11 | 12p11.21 | 1.32 | |
| POLE | Polymerase (DNA directed), epsilon, catalytic subunit | 12q24.33 | 1.49 | |
| CHEK2 | Checkpoint kinase 2 | 22q12.1 | 1.37 | |
| PCNA | Proliferating cell nuclear antigen | 20p12.3 | 1.30 | |
| INIP | INTS3 and NABP interacting protein | 9q32 | −1.35 | |
| NUCKS1 | Nuclear casein kinase and cyclin-dependent kinase substrate 1 | 1q32.1 | −1.83 | |
| RAD54B | RAD54 homolog B (S. cerevisiae) | 8q22.1 | −1.27 | |
| BRCA2 | Breast cancer 2 | 13q13.1 | 1.34 | |
| FANCD2 | Fanconi anemia complementation group D2 | 3p25.3 | 1.28 | |
| FANCI | Fanconi anemia complementation group I | 15q26.1 | 1.48 | |
| DNA Regulation | BLM | Bloom syndrome, RecQ helicase-like | 15q26.1 | 1.25 |
| POLE2 | Polymerase (DNA directed), epsilon 2, accessory subunit | 14q21.3 | 1.48 | |
| PRIM1 | Primase, DNA, polypeptide 1 (49 kDa) | 12q13.3 | 1.45 | |
| PRIM2 | Primase, DNA, polypeptide 2 (58 kDa) | 6p11.2 | 1.26 | |
| CDC45 | Cell division cycle 45 | 22q11.21 | 1.48 | |
| MCM5 | Minichromosome maintenance complex component 5 | 22q12.3 | 1.26 | |
| MCM7 | Minichromosome maintenance complex component 7 | 7q22.1 | 1.31 | |
| DNA Metabolism | DTYMK | Deoxythymidylate kinase | 2q37.3 | 1.28 |
| SLC29A1 | Solute carrier family 29 (equilibrative nucleoside transporter), member 1 | 6p21.1 | 1.27 | |
| TYMS | Thymidylate synthetase | 18p11.32 | −1.40 | |
| Chromatin Organization | HJURP | Holliday junction recognition protein | 2q37.1 | −1.25 |
| HIST1H2AC | Histone cluster 1, H2ac | 6p22.2 | −1.46 | |
| HIST1H3A | Histone cluster 1, H3a | 6p22.2 | 1.38 | |
| HIST1H3B | Histone cluster 1, H3b | 6p22.2 | 1.38 | |
| HIST1H3C | Histone cluster 1, H3c | 6p22.1 | 1.38 | |
| HIST1H3D | Histone cluster 1, H3d | 6p22.1 | 1.38 | |
| HIST1H3F | Histone cluster 1, H3f | 6p22.2 | 1.38 | |
| HIST1H3G | Histone cluster 1, H3g | 6p22.2 | 1.38 | |
| HIST1H3I | Histone cluster 1, H3i | 6p22.2 | 1.38 | |
| HIST1H3J | Histone cluster 1, H3j | 6p22.2 | 1.38 | |
| HIST2H2AA4 | Histone cluster 2 H2A family member a4 | 1q21.2 | −1.54 |
| Biological Process | Protein | Description | Chr Location | F.C. Brain | F.C. Liver | F.C. Lung | F.C. Spine |
|---|---|---|---|---|---|---|---|
| Cell Cycle Regulation | CDK4 | Cyclin-dependent kinase 4 | 12q14.1 | −1.51 | |||
| MELK | Maternal embryonic leucine zipper kinase | 9q13.2 | −1.51 | ||||
| PKMYT1 | Protein kinase, membrane associated tyrosine/threonine 1 | 16p13.3 | −1.68 | ||||
| RBL1 | Retinoblastoma-like 1 | 20q11.23 | −1.62 | ||||
| UBE2C | Ubiquitin conjugating enzyme E2C | 20q13.12 | −1.59 | ||||
| Centrosome Regulation | CCT3 | Chaperonin containing TCP1, subunit 3 gamma | 1q22 | −1.65 | −1.82 | −1.60 | |
| CEP192 | Centrosomal protein 192kDa | 18p11.21 | −1.56 | −1.97 | |||
| CEP72 | Centrosomal protein 72kDa | 5p15.33 | −1.67 | ||||
| CNTROB | Centrobin, centrosomal BRCA2 interacting protein | 17p13.1 | −2.70 | −3.45 | −3.03 | −3.33 | |
| KIF24 | Kinesin family member 24 | 9p13.3 | −1.96 | ||||
| Chromosome Partition | MIS18BP1 | MIS 18 binding protein 1 | 14q21.2 | −2.05 | |||
| DNA Condensation | ESCO2 | Establishment of sister chromatid cohesion N-acetyltransferase 2 | 8q21.1 | −1.50 | |||
| NCAPG2 | Non-SMC condensing II complex subunit G2 | 7q36.3 | −2.86 | −1.97 | −2.46 | −1.86 | |
| SMC2 | Structural maintenance of chromosomes 2 | 9q31.1 | −1.57 | ||||
| Kinetochore Formation | CENPE | Centromere protein E | 4q24 | −1.55 | |||
| CENPF | Centromere protein F | 1q41 | −1.53 | ||||
| KNTC1 | Kinetochore associated 1 | 12q24.31 | −1.96 | ||||
| Microtubule Regulation | DIAPH3 | Diaphanous related formin 3 | 13q21.2 | −1.54 | |||
| KIF4A | Kinesin family member 4A | Xq13.1 | −1.52 | ||||
| SKA3 | Spindle and kinetochore associated complex subunit 3 | 13q12.11 | −2.00 | ||||
| PCNT | Pericentrin | 21q22.3 | −1.52 | ||||
| Spindle Assembly and Regulation | KPNB1 | Karyopherin (importin) beta 1 | 17q21.32 | −1.58 | |||
| HAUS7 | HAUS augmin like complex subunit 7 | Xq28 | −1.52 | −1.74 | |||
| Spindle Checkpoint | TRIP13 | Thyroid hormone receptor interacter 13 | 5p15.33 | −1.68 | −1.82 | −1.50 | |
| ZW10 | ZW10 kinetochore protein | 11q23.2 | −1.87 | −1.92 | −1.78 | ||
| ZWILCH | ZWILCH kinetochore protein | 15q22.31 | −1.67 | ||||
| DNA Damage | RAD18 | RAD18, E3 ubiquitin protein ligase | 3p25.3 | −1.56 | |||
| TTI1 | TELO2 interacting protein 1 | 20q11.23 | −1.67 | −2.02 | |||
| PCLAF | PCNA clamp associated factor | 15q22.31 | −3.57 | −1.54 | |||
| DDX11 | DEAD/H (Asp-Glu-Ala-Asp/His) box helicase 11 | 12p11.21 | −1.54 | ||||
| CLSPN | Claspin | 1q34.3 | −1.76 | ||||
| TIMELESS | Timeless circadian clock | 12q13.3 | −1.57 | ||||
| TIPIN | TIMELESS interacting protein | 15q22.31 | −1.66 | ||||
| NUCKS 1 | Nuclear casein kinase & cyclin-dependent kinase substrate 1 | 1q32.1 | −1.81 | ||||
| RAD51AP1 | RAD51 associated protein 1 | 12p13.32 | −1.70 | −1.48 | |||
| TSN | Translin | 2q14.3 | −1.56 | ||||
| CHEK1 | Checkpoint kinase 1 | 11q24.2 | −1.72 | ||||
| ATRIP | ATR interacting protein | 3p21.31 | −1.60 | ||||
| UBE2T | Ubiquitin conjugating enzyme E2T | 1q32.1 | −1.60 | ||||
| DNA Replication | BAZ1B | Bromodomain adjacent to zinc finger domain 1B | 7q11.23 | −1.67 | |||
| HAT1 | Histone acetyltransferase 1 | 2q31.1 | −1.78 | −1.51 | |||
| RMI1 | RecQ mediated genome instability 1 | 9q21.32 | −1.61 | ||||
| POLA2 | Polymerase (DNA directed), α2, catalytic subunit | 11q13.1 | −1.51 | ||||
| POLE2 | Polymerase (DNA directed), ε2, catalytic subunit | 14q21.3 | −1.71 | ||||
| PRIM1 | Primase, DNA, polypeptide 1 (49 kDa) | 12q13.3 | −2.02 | −1.90 | −1.52 | ||
| CDC45 | Cell division cycle 45 | 22q11.21 | −2.11 | −1.54 | −1.64 | ||
| MCM10 | Minichromosome maintenance 10 replication initiation factor | 10p13 | −1.62 | ||||
| CDC6 | Cell division cycle 6 | 17q21.2 | −1.70 | ||||
| FAM111A | Family with sequence similarity 111 member A | 11q12.1 | −1.64 | ||||
| RFC3 | Replication factor C sununit 3 | 13q13.2 | −1.69 | −1.77 | −1.68 | ||
| DNA Metabolism | DCK | Deoxycytidine kinase | 4q13.3 | −1.64 | |||
| DHFR | Dihydrofolate reductase | 5q14.1 | −2.20 | ||||
| DUT | Deoxyuridine triphosphatase | 15q21.1 | −1.50 | ||||
| RRM1 | Ribnucleotide reductase M1 | 11p15.4 | −1.50 | ||||
| RRM2 | Ribonucleotide reductase M2 | 2q25.1 | −2.63 | −1.68 | |||
| SLC29A1 | Solute carrier family 29 (equilibrative nucleoside transporter), member 1 | 6p21.1 | −1.78 | ||||
| TK1 | Thymidine kinase 1, soluble | 17q25.3 | −1.54 | −1.65 | |||
| TYMS | Thymidylate synthetase | 18p11.32 | −2.39 | −1.58 | |||
| Chromatin Organization | ANP32E | Acidic nuclear phosphoprotein 32 family member E | 1q21.2 | −1.52 | |||
| UHRF1 | Ubiquitin-like with PHD & ring finger domains 1 | 19p13.3 | −2.13 | ||||
| HIST2H3A | Histone cluster 2, H3a | 1q21.2 | −1.62 |
| Proteins | Brain | Liver | Lung | Spine | |||||
|---|---|---|---|---|---|---|---|---|---|
| Biological Process | In Each Process | # Proteins | % | # Proteins | % | # Proteins | % | # Proteins | % |
| Cell Cycle Regulation | 68 | 15 | 22.1 | 4 | 5.9 | 5 | 7.4 | 1 | 1.5 |
| Centrosome Regulation | 31 | 6 | 19.4 | 3 | 9.7 | 3 | 9.7 | 4 | 12.9 |
| Cytokinesis | 20 | 6 | 30.0 | 0 | 0.0 | 2 | 10.0 | 0 | 0.0 |
| Chromosome Partition | 25 | 7 | 28.0 | 1 | 4.0 | 0 | 0.0 | 0 | 0.0 |
| DNA Condensation | 11 | 5 | 45.5 | 3 | 27.3 | 3 | 27.3 | 2 | 18.2 |
| Kinetochore Formation | 25 | 6 | 24.0 | 1 | 4.0 | 2 | 8.0 | 1 | 4.0 |
| Microtubule Regulation | 23 | 7 | 30.4 | 0 | 0.0 | 3 | 13.0 | 0 | 0.0 |
| Nuclear Envelope Regulation | 18 | 2 | 11.1 | 2 | 11.1 | 3 | 16.7 | 1 | 5.6 |
| Spindle Assembly and Regulation | 20 | 3 | 15.0 | 2 | 10.0 | 2 | 10.0 | 1 | 5.0 |
| Spindle Checkpoint | 14 | 7 | 50.0 | 3 | 21.4 | 3 | 21.4 | 2 | 14.3 |
| DNA Damage | 92 | 21 | 22.8 | 10 | 10.9 | 11 | 12.0 | 2 | 2.2 |
| DNA Replication | 50 | 14 | 28.0 | 8 | 16.0 | 4 | 8.0 | 1 | 2.0 |
| DNA Metabolism | 17 | 10 | 58.8 | 8 | 47.1 | 7 | 41.2 | 1 | 5.9 |
| Chromatin Organization | 55 | 1 | 1.8 | 14 | 25.5 | 1 | 1.8 | 0 | 0.0 |
| Proteins | Brain | Liver | Lung | Spine | |||||
|---|---|---|---|---|---|---|---|---|---|
| Biological Process | In Each Process | # Proteins | % | # Proteins | % | # Proteins | % | # Proteins | % |
| Cell Cycle Regulation | 68 | 9 | 13.2 | 25 | 36.8 | 19 | 27.9 | 14 | 20.6 |
| Centrosome Regulation | 31 | 3 | 9.7 | 21 | 67.7 | 11 | 35.5 | 7 | 22.6 |
| Cytokinesis | 20 | 2 | 10.0 | 9 | 45.0 | 5 | 25.0 | 0 | 0.0 |
| Chromosome Partition | 25 | 3 | 12.0 | 15 | 60.0 | 8 | 32.0 | 5 | 20.0 |
| DNA Condensation | 11 | 2 | 18.2 | 10 | 90.9 | 5 | 45.5 | 5 | 45.5 |
| Kinetochore Formation | 25 | 5 | 20.0 | 14 | 56.0 | 6 | 24.0 | 8 | 32.0 |
| Microtubule Regulation | 23 | 1 | 4.3 | 12 | 52.2 | 7 | 30.4 | 7 | 30.4 |
| Nuclear Envelope Regulation | 18 | 2 | 11.1 | 8 | 44.4 | 4 | 22.2 | 2 | 11.1 |
| Spindle Assembly and Regulation | 20 | 1 | 5.0 | 9 | 45.0 | 5 | 25.0 | 4 | 20.0 |
| Spindle Checkpoint | 14 | 5 | 35.7 | 6 | 42.9 | 3 | 21.4 | 2 | 14.3 |
| DNA Damage | 92 | 16 | 17.4 | 48 | 52.2 | 30 | 32.6 | 17 | 18.5 |
| DNA Replication | 50 | 16 | 32.0 | 36 | 72.0 | 19 | 38.0 | 13 | 26.0 |
| DNA Metabolism | 17 | 5 | 29.4 | 0 | 0.0 | 1 | 5.9 | 4 | 23.5 |
| Chromatin Organization | 55 | 1 | 1.8 | 6 | 10.9 | 6 | 10.9 | 3 | 5.5 |
| Chr | 1° Tumor | Brain | Liver | Lung | Spine |
|---|---|---|---|---|---|
| 1 | 1.18 | 1.37 | 1.07 | −1.20 | −1.16 |
| 2 | 1.14 | 1.38 | 1.27 | 1.14 | −1.13 |
| 3 | 1.27 | 1.46 | 1.43 | −1.11 | −1.09 |
| 4 | −1.64 | 2.97 | 2.23 | 1.22 | 1.41 |
| 5 | 1.48 | 1.35 | 1.23 | −1.23 | 1.34 |
| 6 | 1.51 | 1.22 | 1.86 | 1.07 | 1.69 |
| 7 | 1.20 | 1.00 | 1.20 | −1.27 | −1.18 |
| 8 | 0.90 | 1.55 | 1.72 | 1.07 | 1.39 |
| 9 | 1.52 | 1.34 | 1.16 | −1.11 | −1.14 |
| 10 | 1.28 | 1.04 | 1.33 | −1.14 | −1.14 |
| 11 | 1.38 | 1.23 | 1.09 | −1.45 | −1.19 |
| 12 | 1.67 | 1.42 | 1.30 | 1.04 | 1.04 |
| 13 | 2.53 | 1.15 | 1.07 | 1.11 | −1.73 |
| 14 | 1.41 | 0.90 | 1.42 | 1.09 | −1.19 |
| 15 | 1.34 | 2.21 | 1.98 | 1.54 | 1.24 |
| 16 | 1.82 | 1.06 | 1.32 | −1.02 | −1.03 |
| 17 | 0.94 | 1.28 | 1.41 | −1.20 | 1.03 |
| 18 | 1.43 | 1.37 | 0.82 | −1.01 | −1.30 |
| 19 | 1.21 | 1.19 | 1.35 | −1.04 | −1.09 |
| 20 | 1.20 | 1.18 | 0.96 | −2.33 | −1.25 |
| 21 | 1.65 | 1.31 | 1.09 | −2.23 | −2.22 |
| 22 | 1.11 | 1.56 | 1.60 | −1.09 | 1.42 |
| X | 1.39 | 1.24 | 1.36 | −1.52 | −1.02 |
| Chr | Br vs. 1° T | Li vs. 1° T | Lu vs. 1° T | Sp vs. 1° T | Br vs. Li | Br vs. Lu | Br vs. Sp | Li vs. Lu | Li vs. Sp | Lu vs. Sp |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ----- | ----- | ----- | ----- | ----- | 0.06 | ----- | ----- | ----- | ----- |
| 2 | ----- | ----- | ----- | ----- | ----- | ----- | 0.03 | ----- | 0.005 | 0.06 |
| 3 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | 0.015 | 0.0005 | ----- |
| 4 | 0.04 | 0.003 | 0.001 | 0.003 | ----- | ----- | ----- | 0.01 | 0.01 | ----- |
| 5 | ----- | ----- | 0.01 | ----- | ----- | 0.009 | ----- | 0.008 | ----- | 0.006 |
| 6 | ----- | ----- | ----- | ----- | 0.008 | ----- | 0.002 | 0.008 | ----- | 0.0004 |
| 7 | ----- | ----- | 0.04 | ----- | ----- | ----- | ----- | 0.04 | ----- | ----- |
| 8 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| 9 | ----- | ----- | 0.04 | 0.03 | ----- | ----- | ----- | ----- | ----- | ----- |
| 10 | ----- | ----- | 0.0004 | ----- | 0.04 | ----- | ----- | 0.0004 | 0.04 | ----- |
| 11 | ----- | ----- | 0.004 | 0.005 | ----- | 0.04 | ----- | 0.04 | ----- | ----- |
| 12 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| 13 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| 14 | ----- | ----- | ----- | 0.03 | ----- | ----- | ----- | ----- | 0.02 | ----- |
| 15 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| 16 | 0.02 | 0.05 | 0.008 | 0.02 | ----- | ----- | ----- | 0.035 | 0.007 | ----- |
| 17 | ----- | 0.009 | ----- | ----- | ----- | 0.06 | ----- | 0.002 | 0.001 | ----- |
| 18 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| 19 | ----- | ----- | ----- | 0.06 | ----- | ----- | ----- | 0.015 | 0.02 | ----- |
| 20 | ----- | ----- | 0.00002 | ----- | ----- | 0.04 | ----- | 0.0003 | ----- | ----- |
| 21 | ----- | 0.002 | 0.006 | 0.006 | ----- | 0.01 | 0.003 | 0.04 | 0.009 | ----- |
| 22 | ----- | 0.01 | ----- | 0.04 | ----- | ----- | ----- | 0.006 | ----- | 0.01 |
| X | ----- | ----- | 0.001 | 0.03 | ----- | ----- | ----- | 0.0003 | 0.02 | 0.04 |
| Transcript | 1° T/ PCL | Brain/ PCL | Liver/ PCL | Lung/ PCL | Spine/ PCL | Brain/ 1° T | Liver/ 1° T | Lung/ 1° T | Spine/ 1° T |
|---|---|---|---|---|---|---|---|---|---|
| KMT2C | −1.41 | 1.56 | 1.36 | 1.30 | 1.33 | ----- † | ----- | ----- | ----- |
| CHPF2 | ----- | ----- | 1.29 | -----. | ----- | ----- | 1.35 | ----- | ----- |
| EZH2 | ----- | ----- | −1.28 | ----- | ----- | ----- | −1.30 | ----- | ----- |
| KEL | ----- | 2.12 | 1.30 | ----- | ----- | 2.11 | 1.37 | ----- | 1.26 |
| EPHB6 | −1.39 | 2.02 | ----- | ----- | ----- | ----- | −1.34 | ----- | ----- |
| MGAM | ----- | −1.41 | ----- | −1.56 | 2.08 | ----- | 1.35 | −1.43 | ----- |
| BRAF | ----- | ----- | ----- | 1.51 | ----- | ----- | ----- | 1.40 | ----- |
| SMO | 3.76 | −6.25 | ----- | −3.85 | −3.70 | −1.43 | 2.15 | ----- | ----- |
| GRM8 | -----. | -----. | -----. | -----. | -----. | N.I.L. ‡ | N.I.L. | -----. | -----. |
| POT1 | ----- | ----- | ----- | ----- | 1.27 | ----- | ----- | ----- | ----- |
| MET | ----- | ----- | ----- | 1.49 | ----- | ----- | ----- | 1.46 | ----- |
| PIK3CG | ----- | N.I.L. | ----- | N.I.L. | N.I.L. | ----- | ----- | ----- | ----- |
| RINT1 | ----- | ----- | ----- | ----- | ----- | ----- | −1.37 | ----- | ----- |
| RELN | ----- | 1.78 | ----- | ----- | 1.27 | 1.33 | ----- | ----- | ----- |
| CUX1 | −1.30 | 1.33 | 1.26 | 1.30 | 1.40 | ----- | ----- | ----- | ----- |
| EPHB4 | ----- | 1.43 | 1.46 | ----- | ----- | ----- | ----- | −1.46 | ----- |
| TRRAP | ----- | ----- | ----- | 1.36 | ----- | ----- | ----- | 1.26 | ----- |
| SAMD9L | −2.00 | 1.26 | 2.60 | 1.32 | 1.90 | −1.54 | 1.28 | ----- | 1.26 |
| SAMD9 | −1.35 | ----- | 1.52 | 1.30 | 1.70 | −1.30 | ----- | ----- | ----- |
| CDK6 | −1.33 | 1.37 | 1.26 | 1.68 | 1.37 | ----- | ----- | 1.27 | ----- |
| AKAP9 | ----- | ----- | ----- | 1.38 | ----- | ----- | ----- | ----- | ----- |
| PCLO | −4.17 | 6.35 | 2.13 | 1.76 | 3.78 | 1.51 | −1.26 | −2.27 | ----- |
| HGF | N.I.L. | N.I.L. | ----- | N.I.L. | N.I.L. | N.I.L. | ----- | N.I.L. | N.I.L. |
| CD36 | 1.38 | ----- | ----- | ----- | ----- | ----- | ----- | 1.32 | ----- |
| MAGI2 | ----- | ----- | ----- | ----- | 1.31 | ----- | ----- | ----- | 1.27 |
| SBDS | ----- | −1.39 | ----- | 1.34 | ----- | ----- | ----- | 1.62 | ----- |
| SEMA3A | ----- | ----- | ----- | 1.89 | ----- | −1.26 | ----- | 1.65 | −1.27 |
| Transcript | 1° T/ PCL | Brain/ PCL | Liver/ PCL | Lung/ PCL | Spine/ PCL | Brain/ 1° T | Liver/ 1° T | Lung/ 1° T | Spine/ 1° T |
|---|---|---|---|---|---|---|---|---|---|
| ARFRP1 | ----- † | ----- | ----- | ----- | ----- | ----- | ----- | 1.29 | ----- |
| RTEL1 | ----- | ----- | ----- | -----. | −1.39 | ----- | ----- | ----- | −1.38 |
| GNAS | 1.33 | −1.35 | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| AURKA | ----- | −1.39 | −1.43 | −1.30 | ----- | ----- | ----- | ----- | ----- |
| ZNF217 | ----- | ----- | ----- | 1.27 | ----- | ----- | ----- | ----- | ----- |
| PTPN1 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | 1.25 | ----- |
| CEBPB | ----- | ----- | ----- | 1.78 | ----- | ----- | ----- | 2.06 | ----- |
| NCOA3 | ----- | ----- | ----- | 1.59 | ----- | ----- | ----- | 1.74 | ----- |
| CD40 | ----- | ----- | 1.27 | ----- | 1.25 | ----- | ----- | −1.42 | ----- |
| PTPRT | ----- | 1.40 | ----- | N.I.L. ‡ | ----- | ----- | ----- | N.I.L. | ----- |
| PLCG1 | ----- | ----- | 1.37 | ----- | ----- | ----- | ----- | ----- | ----- |
| TOP1 | ----- | ----- | ----- | 1.54 | ----- | ----- | ----- | 1.56 | ----- |
| MAFB | ----- | 1.61 | 1.25 | ----- | 1.70 | 1.34 | −1.48 | −1.40 | 1.42 |
| SRC | ----- | ----- | −1.30 | ----- | −1.35 | ----- | N.I.L. | −1.46 | ----- |
| SAMHD1 | ----- | ----- | 1.40 | ----- | ----- | 1.27 | ----- | ----- | ----- |
| BCL2L1 | ----- | ----- | 1.75 | 1.27 | 1.70 | ----- | 1.68 | ----- | ----- |
| Transcript | 1° T/ PCL | Brain/ PCL | Liver/ PCL | Lung/ PCL | Spine/ 435 | Brain/ 1° T | Liver/ 1° T | Lung/ 1° T | Spine/ 1° T |
|---|---|---|---|---|---|---|---|---|---|
| ABCC9 | ----- † | 1.25 | ----- | 1.26 | ----- | ----- | ----- | ----- | ----- |
| RECQL | ----- | ----- | 1.37 | 1.31 | ----- | −1.25 | −1.33 | 1.29 | ----- |
| PTPRO | ----- | ----- | ----- | 1.39 | ----- | ----- | ----- | 1.53 | ----- |
| CDKN1B | ----- | ----- | 1.26 | ----- | ----- | ----- | 1.31 | ----- | ----- |
| Transcript | 1° T/ PCL | Brain/ PCL | Liver/ PCL | Lung/ PCL | Spine/ PCL | Brain/ 1° T | Liver/ 1° T | Lung/ 1° T | Spine/ 1° T |
|---|---|---|---|---|---|---|---|---|---|
| KMT2C | ----- † | −1.48 | ----- | −1.32 | −1.27 | −1.29 | ----- | ----- | ----- |
| EZH2 | ----- | ----- | ----- | ----- | ----- | −1.33 | ----- | ----- | ----- |
| EPHB6 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | 1.42 | 1.32 |
| PRSS1 | 1.30 | ----- | ----- | ----- | ----- | −1.25 | ----- | ----- | ----- |
| BRAF | ----- | ----- | ----- | 1.51 | ----- | ----- | ----- | 1.40 | ----- |
| TRIM24 | ----- | ----- | ----- | ----- | 1.33 | ----- | ----- | ----- | ----- |
| MET | ----- | −1.31 | ----- | ----- | ----- | −1.25 | ----- | ----- | ----- |
| RINT1 | 1.40 | ----- | ----- | ----- | ----- | −1.47 | −1.45 | −1.32 | ----- |
| CUX1 | ----- | 1.35 | ----- | ----- | ----- | 1.40 | ----- | ----- | ----- |
| SAMD9L | 2.14 | 1.37 | 1.64 | 2.13 | 1.42 | −1.56 | −1.31 | ----- | −1.52 |
| SAMD9 | 1.63 | ----- | 1.76 | 2.02 | 1.72 | −1.56 | ----- | ----- | ----- |
| CDK6 | 1.30 | ----- | ----- | ----- | ----- | −1.43 | −1.37 | ----- | ----- |
| PCLO | 5.20 | 4.45 | 6.87 | 2.90 | 4.25 | ----- | 1.32 | −1.79 | ----- |
| HGF | 2.73 | 1.90 | 3.48 | 1.43 | 1.42 | −1.47 | 1.25 | −1.96 | −1.33 |
| SBDS | ----- | −1.37 | −1.61 | ----- | ----- | −1.39 | −1.64 | ----- | ----- |
| Transcript | 1° T/PCL | Brain/PCL | Liver/ PCL | Lung/ PCL | Spine/ PCL | Brain/ 1° T | Liver/ 1° T | Lung/ 1° T | Spine/ 1° T |
|---|---|---|---|---|---|---|---|---|---|
| GNAS | ----- † | −1.26 | −1.37 | −1.43 | −1.37 | ----- | ----- | ----- | ----- |
| AURKA | ----- | −1.37 | ----- | -----. | ----- | −1.29 | ----- | ----- | ----- |
| ZNF217 | ----- | ----- | −1.28 | ----- | ----- | ----- | −1.32 | ----- | ----- |
| NCOA3 | ----- | ----- | ----- | ----- | ----- | ----- | ----- | 1.25 | ----- |
| PLCG1 | ----- | 1.38 | ----- | ----- | ----- | ----- | ----- | ----- | ----- |
| SRC | ----- | ----- | ----- | ----- | ----- | ----- | ----- | 1.29 | ----- |
| SAMHD1 | ----- | −1.28 | ----- | 1.60 | ----- | −1.56 | ----- | 1.30 | ----- |
| BCL2L1 | ----- | 1.27 | ----- | −1.25 | ----- | ----- | ----- | ----- | −1.38 |
| Chr | Base | CDS | AA | 1° | Metastatic | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | IGV ‡ Link | Location | Change | Change | Change | 435 | Tumor | Brain | Liver | Lung | Spine |
| DDX41 | Chr 5: 176940410 | 5q35.3 | T → C | 1174 A → G | K392E | 16.83 | ----- | ----- | ----- | ----- | ----- |
| GRIN2A | Chr 16: 9858034 † | 16p13.2 | C → A | 3367 G → T | E1123 * | 26.97 | ----- | ----- | ----- | ----- | ----- |
| LILRB1 | Chr 19: 55143437 | 19q13.42 | C → A | 410 C → A | S137 * | 6.23 | ----- | ----- | ----- | ----- | ----- |
| PLCG1 | Chr 20: 39794170 | 20q12 | G → C | 1590 G → C | E530D | 12.85 | ----- | ----- | ----- | ----- | ----- |
| PCLO • | Chr 7: 82583661 | 7q21.11 | AG → A | 6607 del C | L2203 * | 9.18 | ----- | ----- | ----- | ----- | ----- |
| NHS | Chr X: 17745083 | Xq22.13 | G → A | 2794 G → A | D932N | ----- | ----- | ----- | ----- | 6.62 | ----- |
| PIK3R1 | Chr 5: 67593265 | 5q13.1 | G → C | 2011 G → C | V671L | ----- | ----- | ----- | ----- | 5.70 | ----- |
| EIF4A1 | Chr 17: 7478539 | 17p13.1 | C → T | 308 C → T | A103V | ----- | ----- | ----- | 11.51 | ----- | ----- |
| EPHA2 | Chr 1: 16455960 | 1q36.13 | C → T | 2794 G → A | A932T | ----- | ----- | ----- | 32.05 | ----- | 12.60 |
| ERCC3 | Chr 2: 128046383 | 2q14.3 | C → T | 688 G → A | E230K | ----- | ----- | ----- | 31.8 | ----- | 11.76 |
| MKI67 | Chr 10: 129905527 | 10q26.2 | G → T | 4577 C → A | A1526E | ----- | 31.21 | 33.06 | ----- | ----- | 17.95 |
| PRKN | Chr 6: 162683593 | 6q26 | C → T | 376 G → A | D126N | ----- | 48.11 | 46.83 | ----- | ----- | 29.26 |
| PCLO • | Chr 7: 82580341 | 7q21.11 | G → A | 9563 C → T | S3188F | ----- | 21.52 | 22.54 | ----- | ----- | 13.20 |
| PCLO • | Chr 7: 82595727 | 7q21.11 | G → A | 3377 C → T | P1126L | ----- | 18.17 | 20.62 | 18.41 | ----- | 19.05 |
| POLE | Chr 12: 133225951 | 12q24.33 | C → T | 3946 G → A | G1316R | ----- | 47.56 | 46.92 | 48.44 | ----- | 47.90 |
| CDKN1C | Chr 11: 2905327 | 11q15.4 | C → T | 293 G → A | S98N | ----- | 31.48 | 34.38 | 30.84 | ----- | 33.15 |
| IGSF3 | Chr 1: 117122200 | 1q13.1 | C → A | 3208 G → T | E1070 * | ----- | 54.31 | 53.63 | 53.28 | 45.95 | 52.67 |
| MED12 | Chr X: 70350053 | Xq13.1 | C → G | 4036 C → G | R1346G | ----- | 44.67 | 45.56 | 50.27 | 43.53 | 49.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winnard, P.T., Jr.; Morsberger, L.; Yonescu, R.; Jiang, L.; Zou, Y.S.; Raman, V. Isogenic Cell Lines Derived from Specific Organ Metastases Exhibit Divergent Cytogenomic Aberrations. Cancers 2023, 15, 1420. https://doi.org/10.3390/cancers15051420
Winnard PT Jr., Morsberger L, Yonescu R, Jiang L, Zou YS, Raman V. Isogenic Cell Lines Derived from Specific Organ Metastases Exhibit Divergent Cytogenomic Aberrations. Cancers. 2023; 15(5):1420. https://doi.org/10.3390/cancers15051420
Chicago/Turabian StyleWinnard, Paul T., Jr., Laura Morsberger, Raluca Yonescu, Liqun Jiang, Ying S. Zou, and Venu Raman. 2023. "Isogenic Cell Lines Derived from Specific Organ Metastases Exhibit Divergent Cytogenomic Aberrations" Cancers 15, no. 5: 1420. https://doi.org/10.3390/cancers15051420
APA StyleWinnard, P. T., Jr., Morsberger, L., Yonescu, R., Jiang, L., Zou, Y. S., & Raman, V. (2023). Isogenic Cell Lines Derived from Specific Organ Metastases Exhibit Divergent Cytogenomic Aberrations. Cancers, 15(5), 1420. https://doi.org/10.3390/cancers15051420