
Transgenic HA-1-Specific CD8+ T-Lymphocytes Selectively Target Leukemic Cells

 , ,
, ,  , and
, and 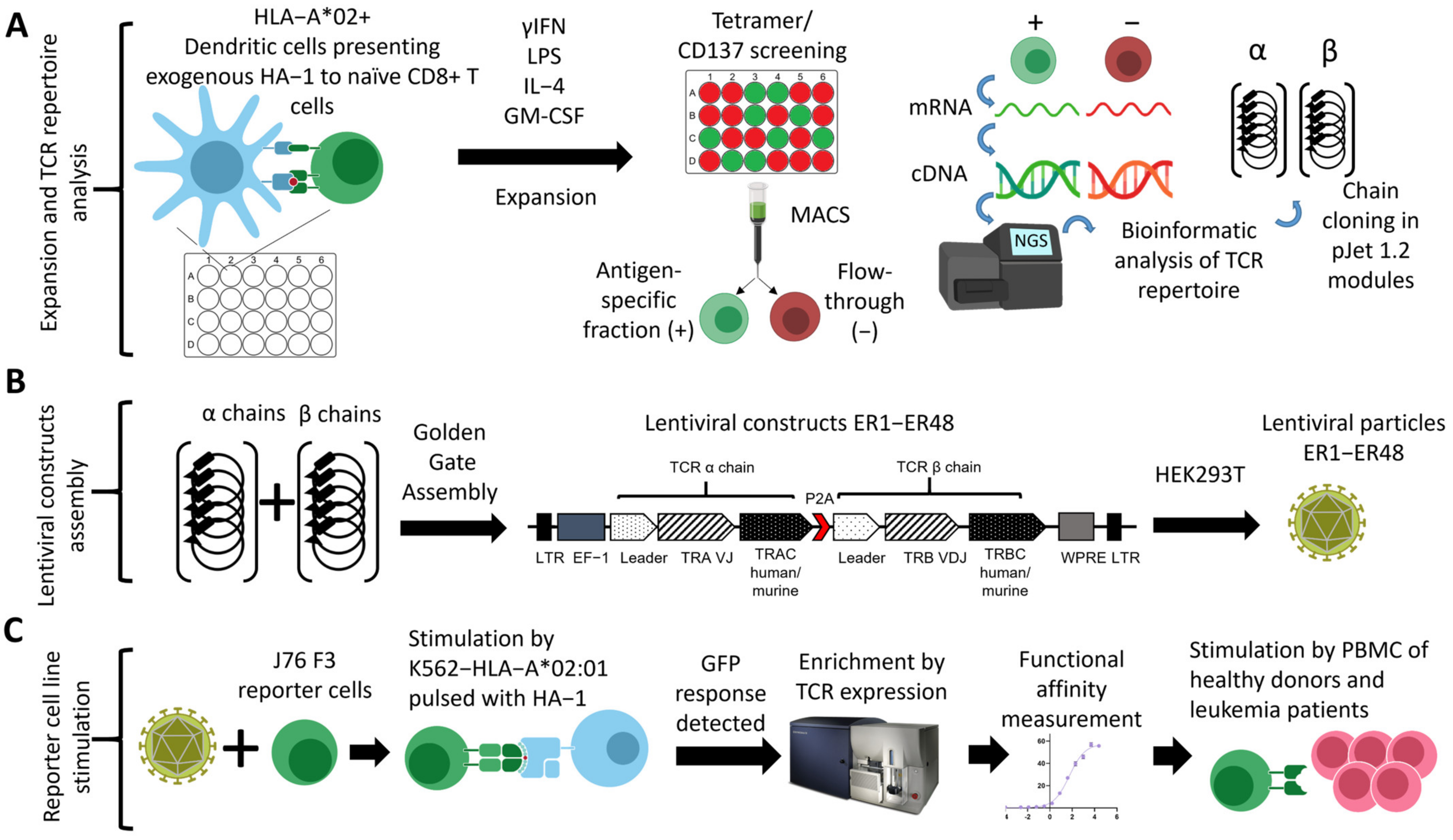{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Generation of HA-1-Specific T Cell Clones
2.3. HA-1-Specific TCR Repertoire Sequencing and Bioinformatic Analysis
2.4. Determining Functionality and Affinity of HA-1-Specific TCR
2.5. CD8+ T Cell Activity Assays
2.6. Transgenic TCR Assembly
2.7. Lentiviral Transduction, Purification, and Expansion of Primary T cells
2.8. CRISPR/Cas Knockout of Endogenous TCR
3. Results
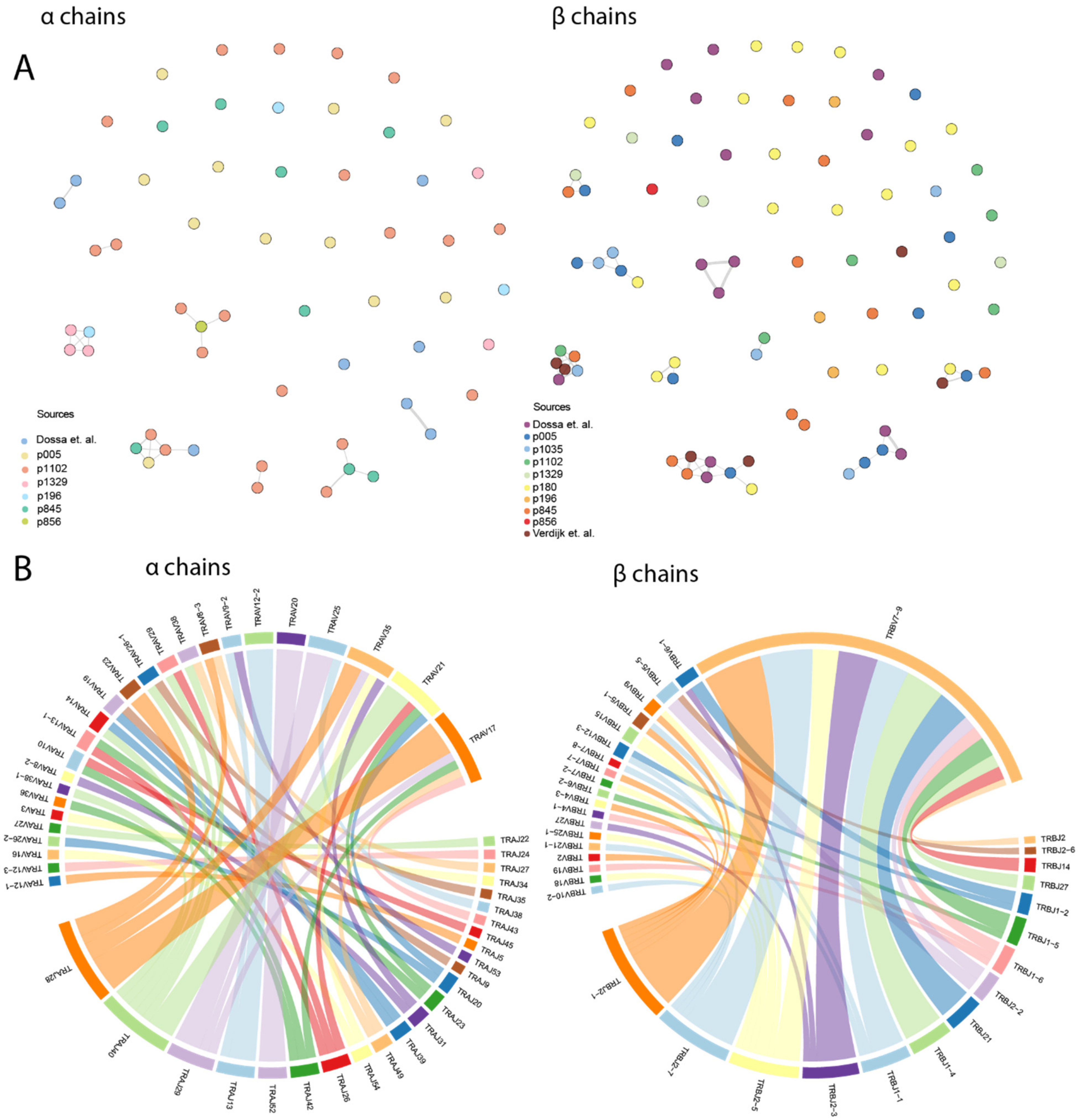
3.1. Bioinformatic Analysis Revealed the Low Degree of CDR3 Similarity in HA-1-Specific TCR Repertoire and Predominant Usage of TRBV7-9 Gene
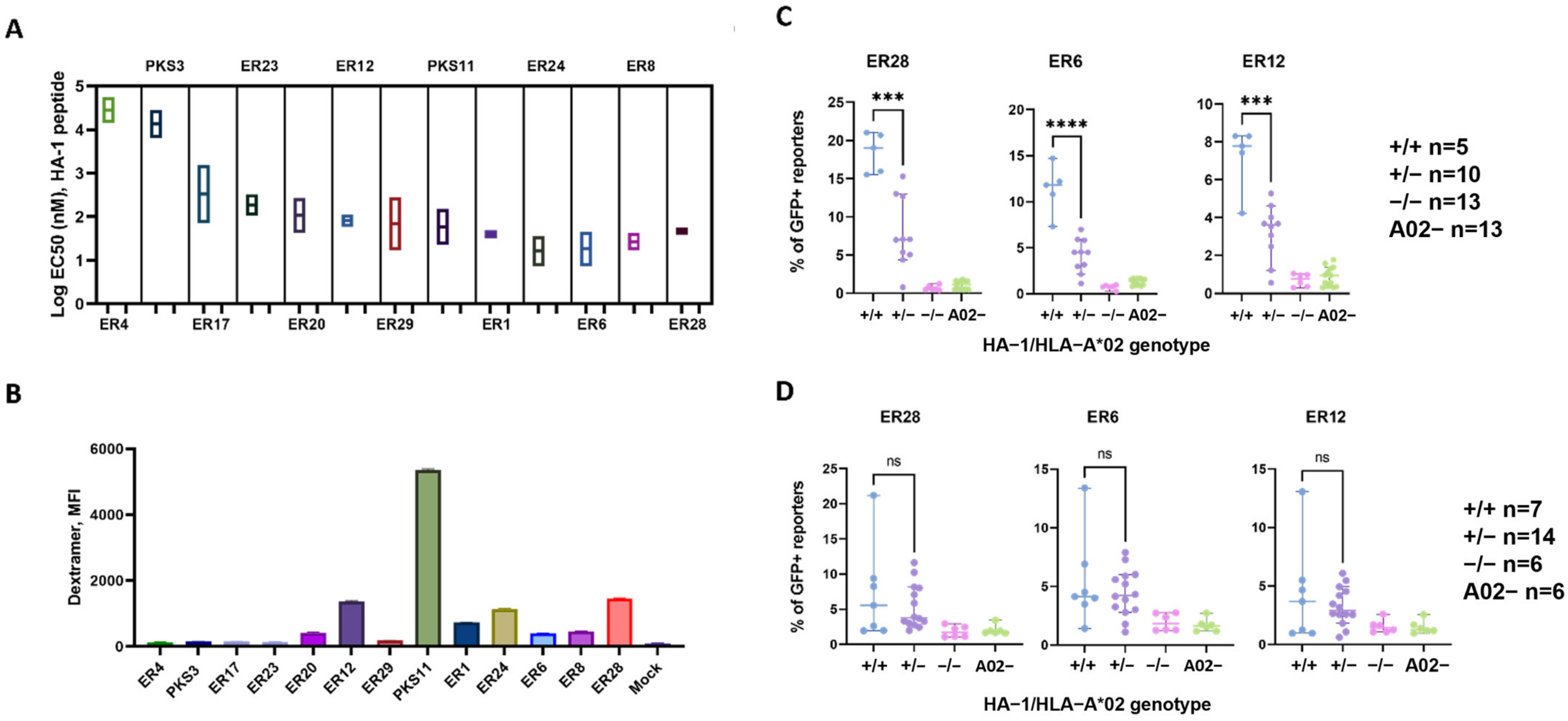
3.2. J76 Cell Reporter Assay Revealed Functional Chain Combinations and Estimated the Avidity of HA-1-Specific TCRs
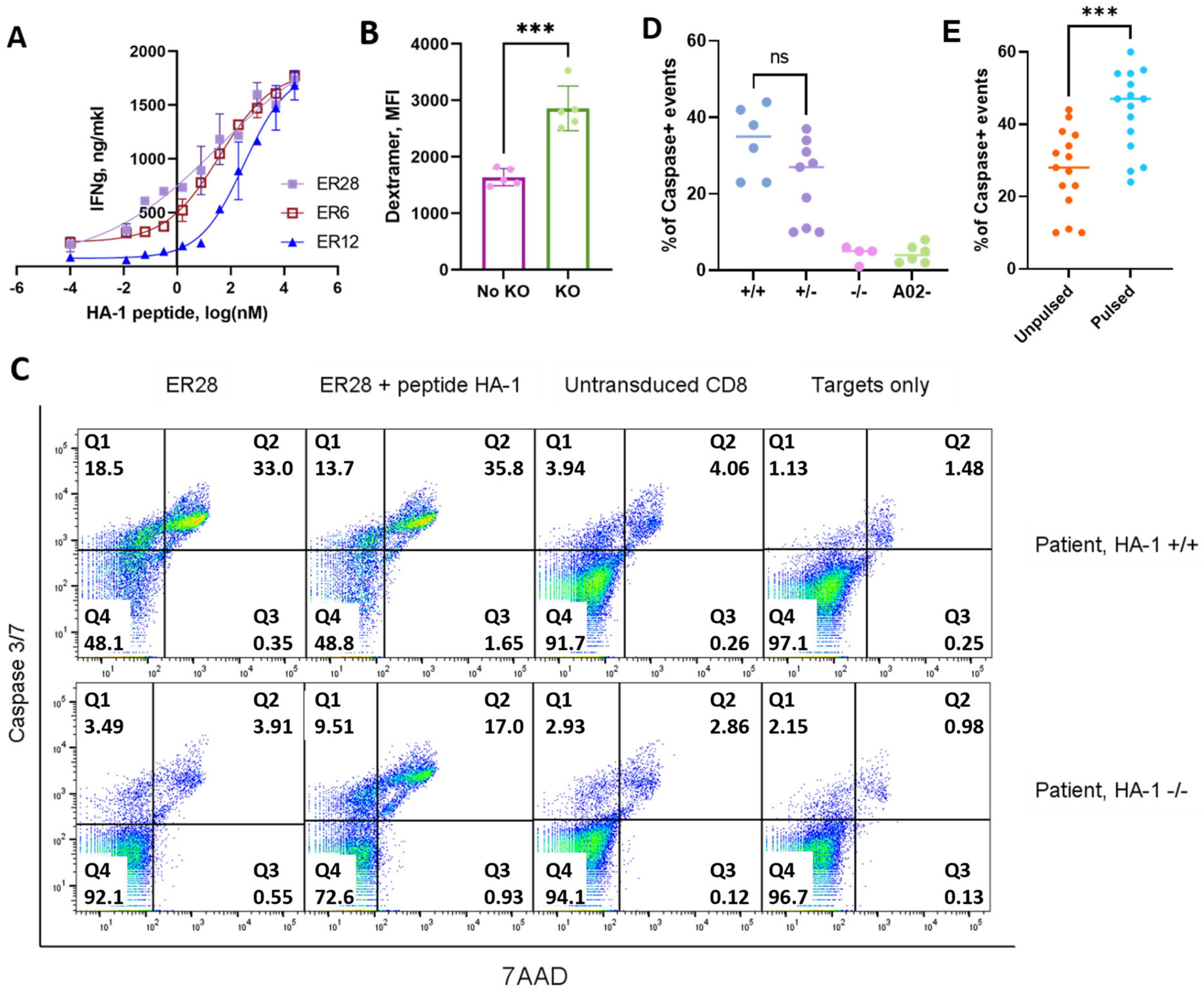
3.3. Selected Transgenic TCRs Specifically Recognized Endogenously Processed HA-1 Peptide
3.4. CD8+ T cells with Murinized Transgenic HA-1–Specific TCRs and CRISPR/Cas Knockout of Endogenous TCR Showed Specific Lysis of PBMC from HA-1+ Patients with Various Hematological Malignancies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loke, J.; Buka, R.; Craddock, C. Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia: Who, When, and How? Front. Immunol 2021, 12, 659595. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.-M. Allogeneic Stem Cell Transplantation for Adult Acute Lymphoblastic Leukemia: When and How. Haematologica 2011, 96, 1083–1086. [Google Scholar] [CrossRef]
- Butturini, A.; Bortin, M.M.; Gale, R.P. Graft-versus-Leukemia Following Bone Marrow Transplantation. Bone Marrow Transpl. 1987, 2, 233–242. [Google Scholar]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic Effect of Graft-versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. New Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Ambinder, R.; Piantadosi, S.; Santos, G. Evidence of a Graft-versus-Lymphoma Effect Associated with Allogeneic Bone Marrow Transplantation. Blood 1991, 77, 649–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giebel, S.; Labopin, M.; Potter, M.; Poiré, X.; Sengeloev, H.; Socié, G.; Huynh, A.; Afanasyev, B.V.; Schanz, U.; Ringden, O.; et al. Comparable Results of Autologous and Allogeneic Haematopoietic Stem Cell Transplantation for Adults with Philadelphia-Positive Acute Lymphoblastic Leukaemia in First Complete Molecular Remission: An Analysis by the Acute Leukemia Working Party of the EBMT. Eur. J. Cancer 2018, 96, 73–81. [Google Scholar] [CrossRef]
- Schmid, C.; de Wreede, L.C.; Biezen, A.v.; Finke, J.; Ehninger, G.; Ganser, A.; Volin, L.; Niederwieser, D.; Beelen, D.; Alessandrino, P.; et al. Outcome after Relapse of Myelodysplastic Syndrome and Secondary Acute Myeloid Leukemia Following Allogeneic Stem Cell Transplantation: A Retrospective Registry Analysis on 698 Patients by the Chronic Malignancies Working Party of the European Society of Blood and Marrow Transplantation. Haematologica 2018, 103, 237–245. [Google Scholar] [CrossRef]
- Rautenberg, C.; Germing, U.; Haas, R.; Kobbe, G.; Schroeder, T. Relapse of Acute Myeloid Leukemia after Allogeneic Stem Cell Transplantation: Prevention, Detection, and Treatment. Int. J. Mol. Sci. 2019, 20, 228. [Google Scholar] [CrossRef] [Green Version]
- Schmid, C.; Labopin, M.; Nagler, A.; Niederwieser, D.; Castagna, L.; Tabrizi, R.; Stadler, M.; Kuball, J.; Cornelissen, J.; Vorlicek, J.; et al. Treatment, Risk Factors, and Outcome of Adults with Relapsed AML after Reduced Intensity Conditioning for Allogeneic Stem Cell Transplantation. Blood 2012, 119, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- McDonald, G.B.; Sandmaier, B.M.; Mielcarek, M.; Sorror, M.; Pergam, S.A.; Cheng, G.-S.; Hingorani, S.; Boeckh, M.; Flowers, M.D.; Lee, S.J.; et al. Survival, Nonrelapse Mortality, and Relapse-Related Mortality After Allogeneic Hematopoietic Cell Transplantation: Comparing 2003–2007 Versus 2013–2017 Cohorts. Ann. Intern. Med. 2020, 172, 229. [Google Scholar] [CrossRef]
- Jabbour, E.; O’Brien, S.; Ravandi, F.; Kantarjian, H. Monoclonal Antibodies in Acute Lymphoblastic Leukemia. Blood 2015, 125, 4010–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.A.; Law, A.; Hunyadkurti, J.; Desilets, S.; Leyton, J.V.; Keating, A. Antibody Therapies for Acute Myeloid Leukemia: Unconjugated, Toxin-Conjugated, Radio-Conjugated and Multivalent Formats. J. Clin. Med. 2019, 8, 1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kröger, N. Prevention and Treatment of Relapse by Drugs. In The EBMT Handbook, Hematopoietic Stem Cell Transplantation and Cellular Therapies; Springer: Berlin/Heidelberg, Germany, 2018; pp. 437–442. [Google Scholar] [CrossRef]
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr. Treat. Option. Oncol. 2017, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr. Treat. Option. Oncol. 2020, 21, 66. [Google Scholar] [CrossRef]
- Brancati, S.; Gozzo, L.; Romano, G.L.; Vetro, C.; Dulcamare, I.; Maugeri, C.; Parisi, M.; Longo, L.; Vitale, D.C.; Raimondo, F.D.; et al. Venetoclax in Relapsed/Refractory Acute Myeloid Leukemia: Are Supporting Evidences Enough? Cancers 2021, 14, 22. [Google Scholar] [CrossRef]
- Aldoss, I.; Yang, D.; Aribi, A.; Ali, H.; Sandhu, K.; Malki, M.M.A.; Mei, M.; Salhotra, A.; Khaled, S.; Nakamura, R.; et al. Efficacy of the Combination of Venetoclax and Hypomethylating Agents in Relapsed/Refractory Acute Myeloid Leukemia. Haematologica 2018, 103, e404–e407. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet. Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.; Yacko, L.R.; Glode, A.E. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pract. Oncol. 2019, 10, 68–82. [Google Scholar]
- Webster, J.A.; Luznik, L.; Gojo, I. Treatment of AML Relapse After Allo-HCT. Front. Oncol. 2021, 11, 812207. [Google Scholar] [CrossRef]
- Goulmy, E. Human Minor Histocompatibility Antigens: New Concepts for Marrow Transplantation and Adoptive Immunotherapy. Immunol. Rev. 1997, 157, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Kernan, N.A.; Bartsch, G.; Ash, R.C.; Beatty, P.G.; Champlin, R.; Filipovich, A.; Gajewski, J.; Hansen, J.A.; Henslee-Downey, J.; McCullough, J.; et al. Analysis of 462 Transplantations from Unrelated Donors Facilitated by the National Marrow Donor Program. N. Engl. J. Med. 1993, 328, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Falkenburg, J.H.F.; Marijt, W.A.F.; Heemskerk, M.H.M.; Willemze, R. Minor Histocompatibility Antigens as Targets of Graft-versus-Leukemia Reactions. Curr. Opin. Hematol. 2002, 9, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Den Haan, J.M.M.; Meadows, L.M.; Wang, W.; Pool, J.; Blokland, E.; Bishop, T.L.; Reinhardus, C.; Shabanowitz, J.; Offringa, R.; Hunt, D.F.; et al. The Minor Histocompatibility Antigen HA-1: A Diallelic Gene with a Single Amino Acid Polymorphism. Science 1998, 279, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Wilke, M.; Dolstra, H.; Maas, F.; Pool, J.; Brouwer, R.; Falkenburg, F.J.; Rebello, A.; Lamers, F.; Schuuring, E.; Kluin, P.; et al. Quantification of the HA-1 Gene Product at the RNA Level; Relevance for Immunotherapy of Hematological Malignancies. Hematol. J. 2003, 4, 315–320. [Google Scholar] [CrossRef]
- Summers, C.; Sheth, V.S.; Bleakley, M. Minor Histocompatibility Antigen-Specific T Cells. Front. Pediatr. 2020, 8, 284. [Google Scholar] [CrossRef]
- Fujii, N.; Hiraki, A.; Ikeda, K.; Ohmura, Y.; Nozaki, I.; Shinagawa, K.; Ishimaru, F.; Kiura, K.; Shimizu, N.; Tanimoto, M.; et al. Expression of Minor Histocompatibility Antigen, HA-1, in Solid Tumor Cells. Transplantation 2002, 73, 1137–1141. [Google Scholar] [CrossRef]
- Spierings, E.; Gras, S.; Reiser, J.-B.; Mommaas, B.; Almekinders, M.; Kester, M.G.; Chouquet, A.; Gorrec, M.; Drijfhout, J.W.; Ossendorp, F.; et al. Steric Hindrance and Fast Dissociation Explain the Lack of Immunogenicity of the Minor Histocompatibility HA-1Arg Null Allele. J. Immunol. Baltim. Md. 2009, 182, 4809–4816. [Google Scholar] [CrossRef] [Green Version]
- Hobo, W.; Broen, K.; van der Velden, W.J.F.M.; Greupink-Draaisma, A.; Adisty, N.; Wouters, Y.; Kester, M.; Fredrix, H.; Jansen, J.H.; Reijden, B.v.d.; et al. Association of Disparities in Known Minor Histocompatibility Antigens with Relapse-Free Survival and Graft-versus-Host Disease after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2013, 19, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Spierings, E.; Kim, Y.-H.H.; Hendriks, M.; Borst, E.; Sergeant, R.; Canossi, A.; Oudshoorn, M.; Loiseau, P.; Dolstra, H.; Markiewicz, M.; et al. Multicenter Analyses Demonstrate Significant Clinical Effects of Minor Histocompatibility Antigens on GvHD and GvL after HLA-Matched Related and Unrelated Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2013, 19, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Marijt, W.A.E.; Heemskerk, M.H.M.; Kloosterboer, F.M.; Goulmy, E.; Kester, M.G.D.; van der Hoorn, M.A.W.G.; Luxemburg-Heys, S.A.P.v.; Hoogeboom, M.; Mutis, T.; Drijfhout, J.W.; et al. Hematopoiesis-Restricted Minor Histocompatibility Antigens HA-1- or HA-2-Specific T Cells Can Induce Complete Remissions of Relapsed Leukemia. Proc. Natl. Acad. Sci. USA 2003, 100, 2742–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, B.; Stevanovic, S.; Urbanek, M.; Mitterschiffthaler, A.; Rammensee, H.; Grünewald, K.; Gastl, G.; Nachbaur, D. Induction of HA-1-specific Cytotoxic T-cell Clones Parallels the Therapeutic Effect of Donor Lymphocyte Infusion. Brit. J. Haematol. 2002, 117, 935–939. [Google Scholar] [CrossRef] [PubMed]
- van Balen, P.; Jedema, I.; Loenen, M.M.v.; Boer, R.d.; Egmond, H.v.; Hagedoorn, R.S.; Hoogstaten, C.; Veld, S.A.; Hageman, L.; Liempt, P.v.; et al. HA-1H T-Cell Receptor Gene Transfer to Redirect Virus-Specific T Cells for Treatment of Hematological Malignancies After Allogeneic Stem Cell Transplantation: A Phase 1 Clinical Study. Front. Immunol. 2020, 11, 1804. [Google Scholar] [CrossRef] [PubMed]
- Dossa, R.G.; Cunningham, T.; Sommermeyer, D.; Medina-Rodriguez, I.; Biernacki, M.A.; Foster, K.; Bleakley, M. Development of T-Cell Immunotherapy for Hematopoietic Stem Cell Transplantation Recipients at Risk of Leukemia Relapse. Blood 2018, 131, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Rosskopf, S.; Leitner, J.; Paster, W.; Morton, L.T.; Hagedoorn, R.S.; Steinberger, P.; Heemskerk, M. A Jurkat 76 Based Triple Parameter Reporter System to Evaluate TCR Functions and Adoptive T Cell Strategies. Oncotarget 2018, 9, 17608–17619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishore, A.; Petrek, M. Next-Generation Sequencing Based HLA Typing: Deciphering Immunogenetic Aspects of Sarcoidosis. Front. Genet. 2018, 9, 503. [Google Scholar] [CrossRef] [Green Version]
- Bray, R.A. Flow Cytometry in Human Leukocyte Antigen Testing. Semin. Hematol. 2001, 38, 194–200. [Google Scholar] [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.A.E. IPD-IMGT/HLA Database. Nucleic. Acids. Res. 2019, 48, D948–D955. [Google Scholar] [CrossRef]
- Romaniuk, D.S.; Khmelevskaya, A.A.; Postovskaya, A.M.; Malko, D.B.; Kuzminova, E.P.; Khamaganova, E.G.; Efimov, G.A. Clinically Relevant Minor Histocompatibility Antigens For Russian Patients Undergoing Hematopoietic Stem Cell Transplantation. Medical. Immunol. Russ. 2019, 21, 847–860. [Google Scholar] [CrossRef]
- Wölfl, M.; Greenberg, P.D. Antigen-Specific Activation and Cytokine-Facilitated Expansion of Naive, Human CD8+ T Cells. Nat. Protoc. 2014, 9, 950–966. [Google Scholar] [CrossRef] [Green Version]
- Pogorelyy, M.V.; Elhanati, Y.; Marcou, Q.; Sycheva, A.L.; Komech, E.A.; Nazarov, V.I.; Britanova, O.V.; Chudakov, D.M.; Mamedov, I.Z.; Lebedev, Y.B.; et al. Persisting Fetal Clonotypes Influence the Structure and Overlap of Adult Human T Cell Receptor Repertoires. PLoS Comput. Biol. 2017, 13, e1005572. [Google Scholar] [CrossRef] [Green Version]
- Mamedov, I.Z.; Britanova, O.V.; Zvyagin, I.V.; Turchaninova, M.A.; Bolotin, D.A.; Putintseva, E.V.; Lebedev, Y.B.; Chudakov, D.M. Preparing Unbiased T-Cell Receptor and Antibody CDNA Libraries for the Deep next Generation Sequencing Profiling. Front. Immunol. 2013, 4, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for Comprehensive Adaptive Immunity Profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. VDJtools: Unifying Post-Analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Stauss, H.J.; Morris, E.C. Molecular Immunology Lessons from Therapeutic T-cell Receptor Gene Transfer. Immunology 2010, 129, 170–177. [Google Scholar] [CrossRef]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer. Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, M.L.L.; Luke, G.; Mehrotra, A.; Li, X.; Hughes, L.E.; Gani, D.; Ryan, M.D. Analysis of the Aphthovirus 2A/2B Polyprotein ‘Cleavage’ Mechanism Indicates Not a Proteolytic Reaction, but a Novel Translational Effect: A Putative Ribosomal ‘Skip. ’ J. Gen. Virol. 2001, 82, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Engler, C.; Gruetzner, R.; Werner, S.; Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 2011, 6, e16765. [Google Scholar] [CrossRef]
- Stronen, E.; Abrahamsen, I.W.; Gaudernack, G.; Wälchli, S.; Munthe, E.; Buus, S.; Johansen, F.-E.; Olweus, J. Dendritic Cells Engineered to Express Defined Allo-HLA Peptide Complexes Induce Antigen-specific Cytotoxic T Cells Efficiently Killing Tumour Cells. Scand. J. Immunol. 2009, 69, 319–328. [Google Scholar] [CrossRef]
- Abrahamsen, I.; Stronen, E.; Wälchli, S.; Johansen, J.; Kjellevoll, S.; Kumari, S.; Komada, M.; Gaudernack, G.; Tjonnfjord, G.; Toebes, M.; et al. Targeting B Cell Leukemia with Highly Specific Allogeneic T Cells with a Public Recognition Motif. Leukemia 2010, 24, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, R.M.; Mutis, T.; Wilke, M.; Pool, J.; Schrama, E.; Brand, A.; Goulmy, E. Exclusive TCRVbeta Chain Usage of Ex Vivo Generated Minor Histocompatibility Antigen HA-1 Specific Cytotoxic T Cells: Implications for Monitoring of Immunotherapy of Leukemia by TCRBV Spectratyping. Hematol. J. Off. J. Eur. Haematol. Assoc./EHA 2002, 3, 271–275. [Google Scholar] [CrossRef]
- Yin, Y.; Li, Y.; Mariuzza, R.A. Structural Basis for Self-recognition by Autoimmune T-cell Receptors. Immunol. Rev. 2012, 250, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Campillo-Davo, D.; Flumens, D.; Lion, E. The Quest for the Best: How TCR Affinity, Avidity, and Functional Avidity Affect TCR-Engineered T-Cell Antitumor Responses. Cells 2020, 9, 1720. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA Class I Antigen Downregulation in Human Cancers: T-Cell Immunotherapy Revives an Old Story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.B.; Norberg, S.M.; Sinkoe, A.L.; Adhikary, S.; Meyer, T.J.; Lack, J.B.; Warner, A.C.; Schweitzer, C.; Doran, S.L.; Korrapati, S.; et al. TCR-Engineered T Cells Targeting E7 for Patients with Metastatic HPV-Associated Epithelial Cancers. Nat. Med. 2021, 27, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Theoret, M.R.; Zheng, Z.; Lamers, C.H.J.; Rosenberg, S.A.; Morgan, R.A. Development of Human Anti-Murine T-Cell Receptor Antibodies in Both Responding and Nonresponding Patients Enrolled in TCR Gene Therapy Trials. Clin. Cancer Res. 2010, 16, 5852–5861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, J.P.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming Human T Cell Function and Specificity with Non-Viral Genome Targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Morton, L.T.; Reijmers, R.M.; Wouters, A.K.; Kweekel, C.; Remst, D.F.; Pothast, C.R.; Falkenburg, F.J.; Heemskerk, M.H. Simultaneous Deletion of Endogenous TCRαβ for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Ther. 2019, 28, 64–74. [Google Scholar] [CrossRef]
- Ivica, N.A.; Young, C.M. Tracking the CAR-T Revolution: Analysis of Clinical Trials of CAR-T and TCR-T Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The Therapeutic Landscape for Cells Engineered with Chimeric Antigen Receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y. T Cell Receptor-Engineered T Cells for Leukemia Immunotherapy. Cancer Cell. Int. 2019, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Oppermans, N.; Kueberuwa, G.; Hawkins, R.E.; Bridgeman, J.S. Transgenic T-Cell Receptor Immunotherapy for Cancer: Building on Clinical Success. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520933509. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Zheng, Z.; Lowery, F.J.; Gartner, J.J.; Prickett, T.D.; Robbins, P.F.; Rosenberg, S.A. Direct Identification of Neoantigen-Specific TCRs from Tumor Specimens by High-Throughput Single-Cell Sequencing. J. Immunother. Cancer 2021, 9, e002595. [Google Scholar] [CrossRef] [PubMed]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Koedam, J.; Wermke, M.; Ehninger, A.; Cartellieri, M.; Ehninger, G. Chimeric Antigen Receptor T-Cell Therapy in Acute Myeloid Leukemia. Curr. Opin. Hematol. 2022, 29, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z.; Wei, W.; Li, Y. TCR Engineered T Cells for Solid Tumor Immunotherapy. Exp. Hematol. Oncol. 2022, 11, 38. [Google Scholar] [CrossRef]
- Kozani, P.S.; Kozani, P.S.; Najafabadi, M.A.; Yousefi, F.; Mirarefin, S.M.J.; Rahbarizadeh, F. Recent Advances in Solid Tumor CAR-T Cell Therapy: Driving Tumor Cells from Hero to Zero? Front. Immunol. 2022, 13, 795164. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Wiebel, M.; Jamitzky, S.; Rossig, C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers 2020, 12, 1075. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Reinoso, A.; Nehme-Álvarez, D.; Domínguez-Alonso, C.; Álvarez-Vallina, L. Synthetic TILs: Engineered Tumor-Infiltrating Lymphocytes With Improved Therapeutic Potential. Front. Oncol. 2021, 10, 593848. [Google Scholar] [CrossRef]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells against Solid Tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The Determinants of Tumour Immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Arnaud, M.; Bobisse, S.; Chiffelle, J.; Harari, A. The Promise of Personalized TCR-Based Cellular Immunotherapy for Cancer Patients. Front. Immunol. 2021, 12, 701636. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-Derived HLA-A1–Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3–Directed T Cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Brickner, A.N.G. Mechanisms of Minor Histocompatibility Antigen Immunogenicity. Immunol. Res. 2006, 36, 33–41. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217. [Google Scholar] [CrossRef] [Green Version]
- Jonas, B.A. On the Origin of Relapse in AML. Sci. Transl. Med. 2017, 9, eaan8205. [Google Scholar] [CrossRef]
- Heemskerk, M.H.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.; Willemze, R.; Falkenburg, J.F. Reprogramming of Virus-Specific T Cells into Leukemia-Reactive T Cells Using T Cell Receptor Gene Transfer. J. Exp. Med. 2004, 199, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, M.; van Egmond, H.M.E.; Barnby-Porritt, H.; Hoorn, M.A.W.G.v.d.; Hagedoorn, R.S.; Kester, M.G.D.; Schwabe, N.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Genetic Engineering of Virus-Specific T Cells with T-Cell Receptors Recognizing Minor Histocompatibility Antigens for Clinical Application. Haematologica 2008, 93, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Oostvogels, R.; Lokhorst, H.; Mutis, T. Minor Histocompatibility Ags: Identification Strategies, Clinical Results and Translational Perspectives. Bone Marrow Transplant. 2015, 51, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heemskerk, M.H.; Hoogeboom, M.; de Paus, R.A.; Kester, M.G.; Hoorn, M.A.v.d.; Goulmy, E.; Willemze, R.; Falkenburg, J. Redirection of Antileukemic Reactivity of Peripheral T Lymphocytes Using Gene Transfer of Minor Histocompatibility Antigen HA-2-Specific T-Cell Receptor Complexes Expressing a Conserved Alpha Joining Region. Blood 2003, 102, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Pilunov, A.; Kuchmiy, A.; Sheetikov, S.; Filkin, S.; Romaniuk, D.; Rosov, F.; Efimov, G. Modification of Cytotoxic Lymphocytes with T Cell Receptor Specific for Minor Histocompatibility Antigen ACC-1Y. Mol. Biol. 2019, 53, 402–410. [Google Scholar] [CrossRef]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework Engineering to Produce Dominant T Cell Receptors with Enhanced Antigen-Specific Function. Nat. Commun. 2019, 10, 4451. [Google Scholar] [CrossRef] [Green Version]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody Humanization Methods—A Review and Update. Biotechnol. Genetic. Eng. Rev 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Khan, A.N.; Chowdhury, A.; Karulkar, A.; Jaiswal, A.K.; Banik, A.; Asija, S.; Purwar, R. Immunogenicity of CAR-T Cell Therapeutics: Evidence, Mechanism and Mitigation. Front. Immunol. 2022, 13, 886546. [Google Scholar] [CrossRef]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T Cells in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol. Blood Marrow Transpl. J. Am. Soc. Blood Marrow Transpl. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Van Loenen, M.M.; Boer, R.d.; Amir, A.L.; Hagedoorn, R.S.; Volbeda, G.L.; Willemze, R.; Rood, J.J.v.; Falkenburg, F.J.; Heemskerk, M.H. Mixed T Cell Receptor Dimers Harbor Potentially Harmful Neoreactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 10972–10977. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Dolton, G.; Mian, A.; Ottmann, O.G.; Sewell, A.K. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic T Cells. Blood 2017, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Wachsmann, T.L.A.; Wouters, A.K.; Remst, D.F.G.; Hagedoorn, R.S.; Meeuwsen, M.H.; van Diest, E.; Leusen, J.; Kuball, J.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Comparing CAR and TCR Engineered T Cell Performance as a Function of Tumor Cell Exposure. Oncoimmunology 2022, 11, 2033528. [Google Scholar] [CrossRef]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T Cell Therapy Using Antigen-Specific CD8+ T Cell Clones for the Treatment of Patients with Metastatic Melanoma: In Vivo Persistence, Migration, and Antitumor Effect of Transferred T Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, A.N.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-Reactive CD8+ T Cells Can Mediate Antileukemic Activity and Persist in Post-Transplant Patients. Sci. Transl. Med. 2013, 5, 174ra27. [Google Scholar] [CrossRef] [Green Version]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and Persistence of WT1-Specific T-Cell Receptor Gene−transduced Lymphocytes in Patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Abate-Daga, D.; Hanada, K.; Davis, J.L.; Yang, J.C.; Rosenberg, S.A.; Morgan, R.A. Expression Profiling of TCR-Engineered T Cells Demonstrates Overexpression of Multiple Inhibitory Receptors in Persisting Lymphocytes. Blood 2013, 122, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.B.; Glod, J.; Kaplan, R.N.; Grupp, S.A.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1c259T Cells in Synovial Sarcoma. Cancer. Discov. 2018, 8, CD-17-1417. [Google Scholar] [CrossRef] [Green Version]
- Meij, P.; Jedema, I.; van der Hoorn, M.; Bongaerts, R.; Cox, L.; Wafelman, A.R.; Marijt, E.W.; Willemze, R.; Falkenburg, F.J. Generation and Administration of HA-1-Specific T-Cell Lines for the Treatment of Patients with Relapsed Leukemia after Allogeneic Stem Cell Transplantation: A Pilot Study. Haematologica 2012, 97, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangeli, S.; Bove, C.; Mezzanotte, C.; Camisa, B.; Falcone, L.; Manfredi, F.; Bezzecchi, E.; Khoury, R.E.; Norata, R.; Sanvito, F.; et al. CAR T-Cell Manufacturing from Naive/Stem Memory T-Lymphocytes Enhances Antitumor Responses While Curtailing Cytokine Release Syndrome. J. Clin. Investig. 2022, 132, e150807. [Google Scholar] [CrossRef] [PubMed]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-Long Leukaemia Remissions with Persistence of CD4+ CAR T Cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilunov, A.; Romaniuk, D.S.; Shmelev, A.; Sheetikov, S.; Gabashvili, A.N.; Khmelevskaya, A.; Dianov, D.; Zornikova, K.; Shakirova, N.T.; Vagida, M.; et al. Transgenic HA-1-Specific CD8+ T-Lymphocytes Selectively Target Leukemic Cells. Cancers 2023, 15, 1592. https://doi.org/10.3390/cancers15051592
Pilunov A, Romaniuk DS, Shmelev A, Sheetikov S, Gabashvili AN, Khmelevskaya A, Dianov D, Zornikova K, Shakirova NT, Vagida M, et al. Transgenic HA-1-Specific CD8+ T-Lymphocytes Selectively Target Leukemic Cells. Cancers. 2023; 15(5):1592. https://doi.org/10.3390/cancers15051592
Chicago/Turabian StylePilunov, Artem, Dmitrii S. Romaniuk, Anton Shmelev, Savely Sheetikov, Anna N. Gabashvili, Alexandra Khmelevskaya, Dmitry Dianov, Ksenia Zornikova, Naina T. Shakirova, Murad Vagida, and et al. 2023. "Transgenic HA-1-Specific CD8+ T-Lymphocytes Selectively Target Leukemic Cells" Cancers 15, no. 5: 1592. https://doi.org/10.3390/cancers15051592
APA StylePilunov, A., Romaniuk, D. S., Shmelev, A., Sheetikov, S., Gabashvili, A. N., Khmelevskaya, A., Dianov, D., Zornikova, K., Shakirova, N. T., Vagida, M., Bogolyubova, A., & Efimov, G. A. (2023). Transgenic HA-1-Specific CD8+ T-Lymphocytes Selectively Target Leukemic Cells. Cancers, 15(5), 1592. https://doi.org/10.3390/cancers15051592