
Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell-Based Phenotypic Assays
2.3. Mouse Study
2.4. Western Blotting
2.5. Data Analysis
2.5.1. DNA Methylation Analysis
2.5.2. Transcriptome Analysis
2.5.3. Integrative Data Analysis
2.6. Statistical Analysis
2.7. Data Availability
3. Results
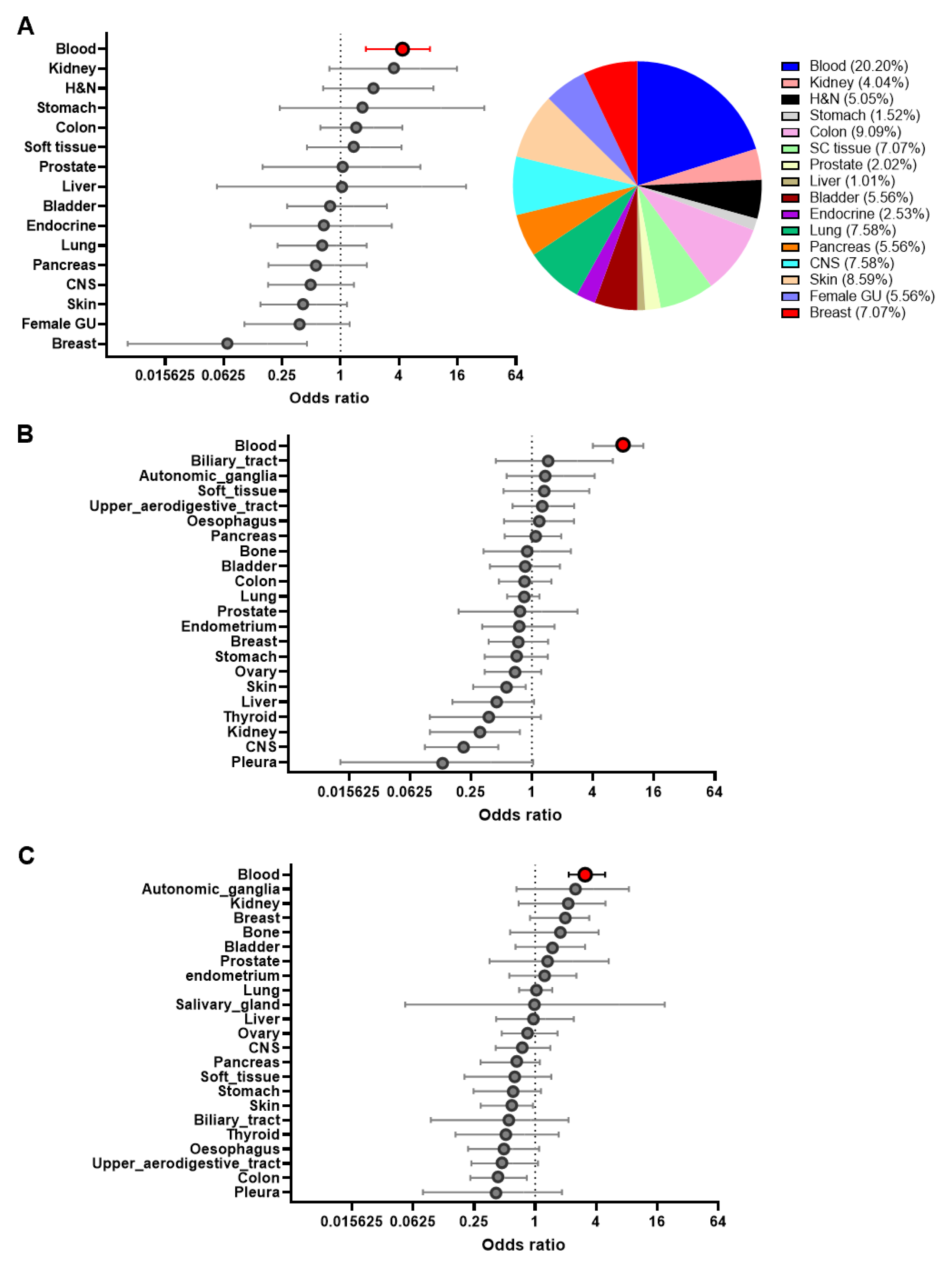
3.1. Sensitivity Profiling of NTX-301 in 199 CCLs
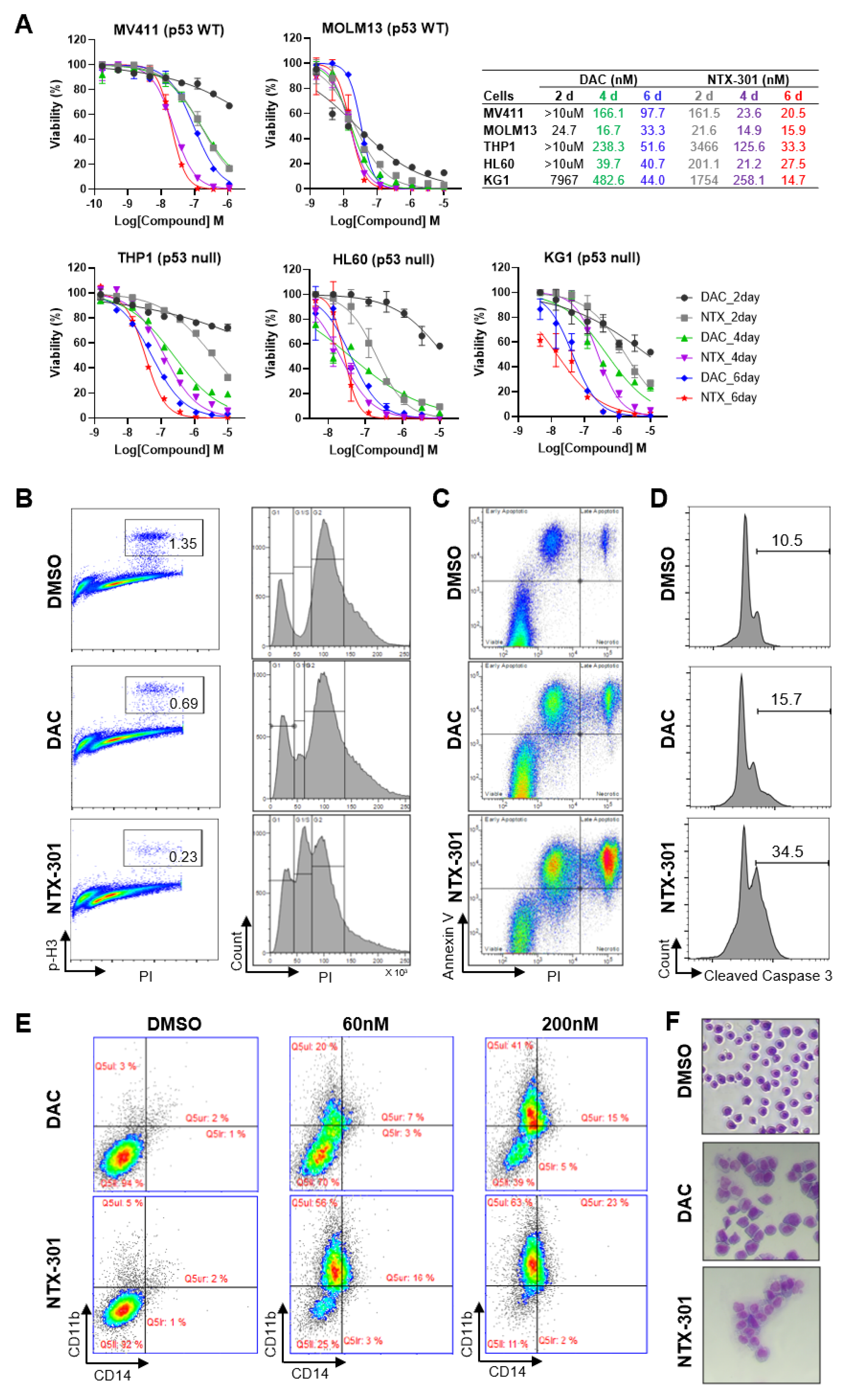
3.2. Cellular Phenotypes Associated with the Antileukemic Activity of NTX-301
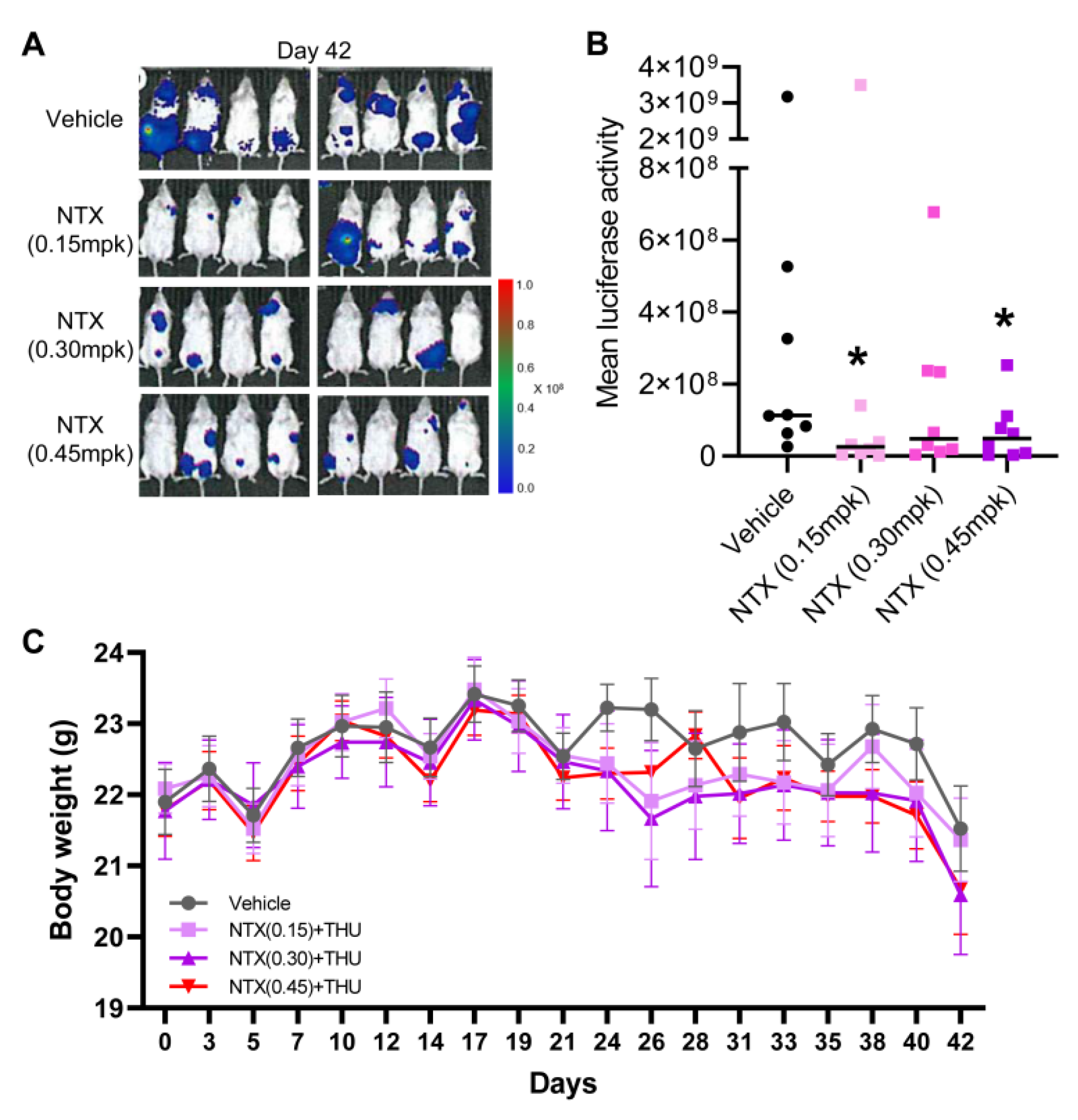
3.3. Antitumor Efficacy of NTX-301 in a Preclinical Model of AML
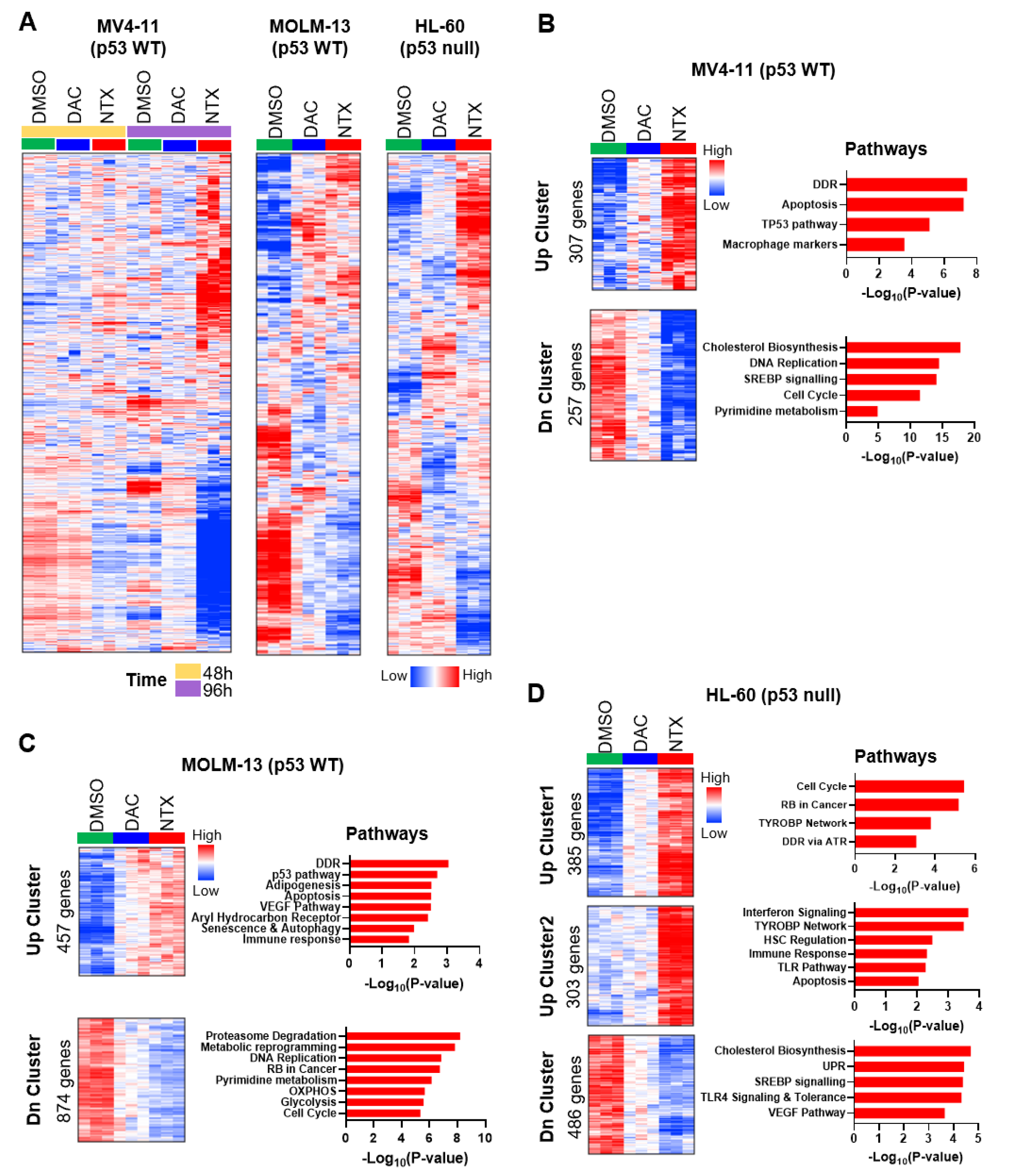
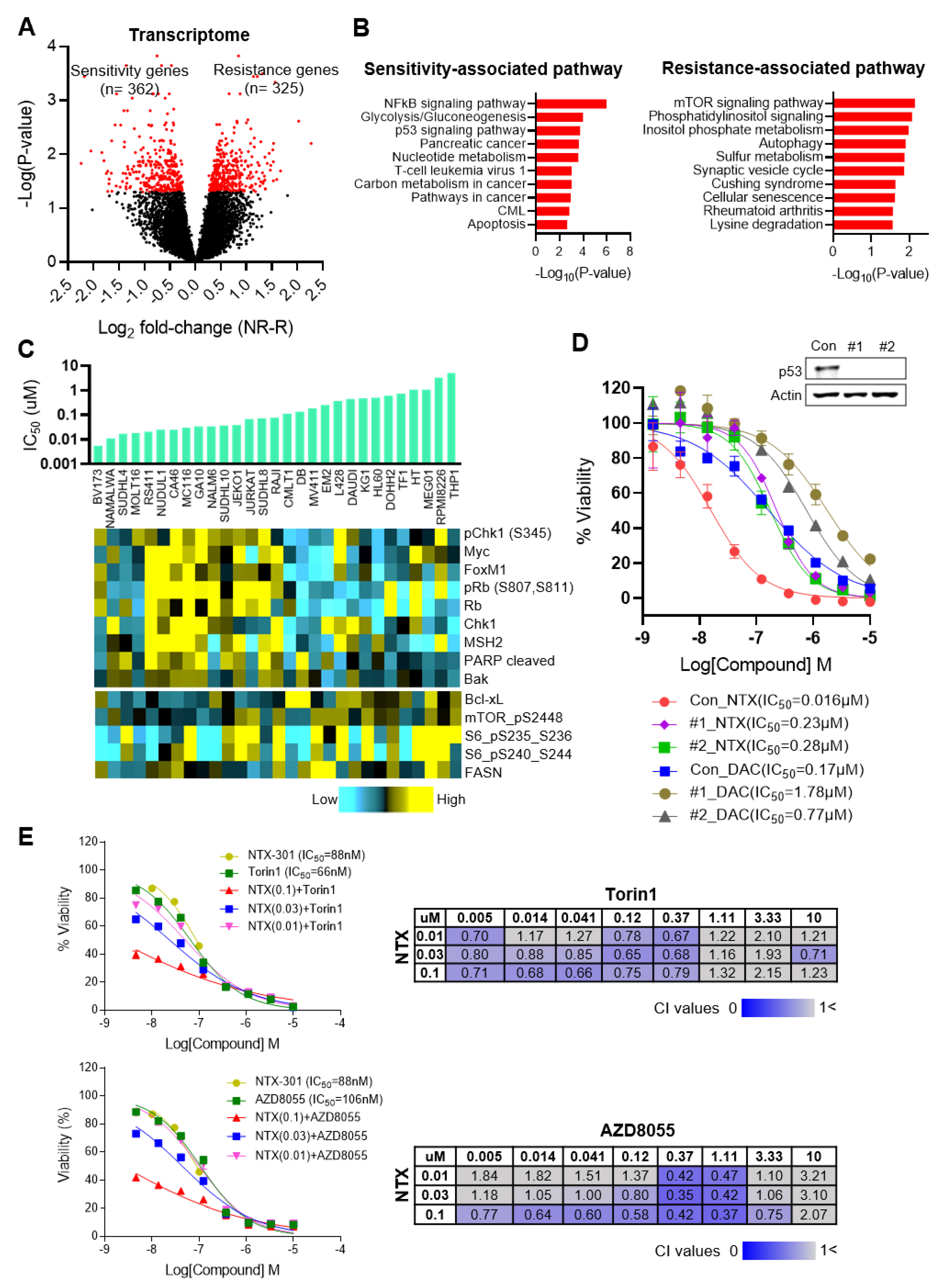
3.4. Transcriptome Analyses Cataloged the MoAs of NTX-301
3.5. Integrative Data Analyses Identified Transcriptional Events Associated with Sensitivity to NTX-301
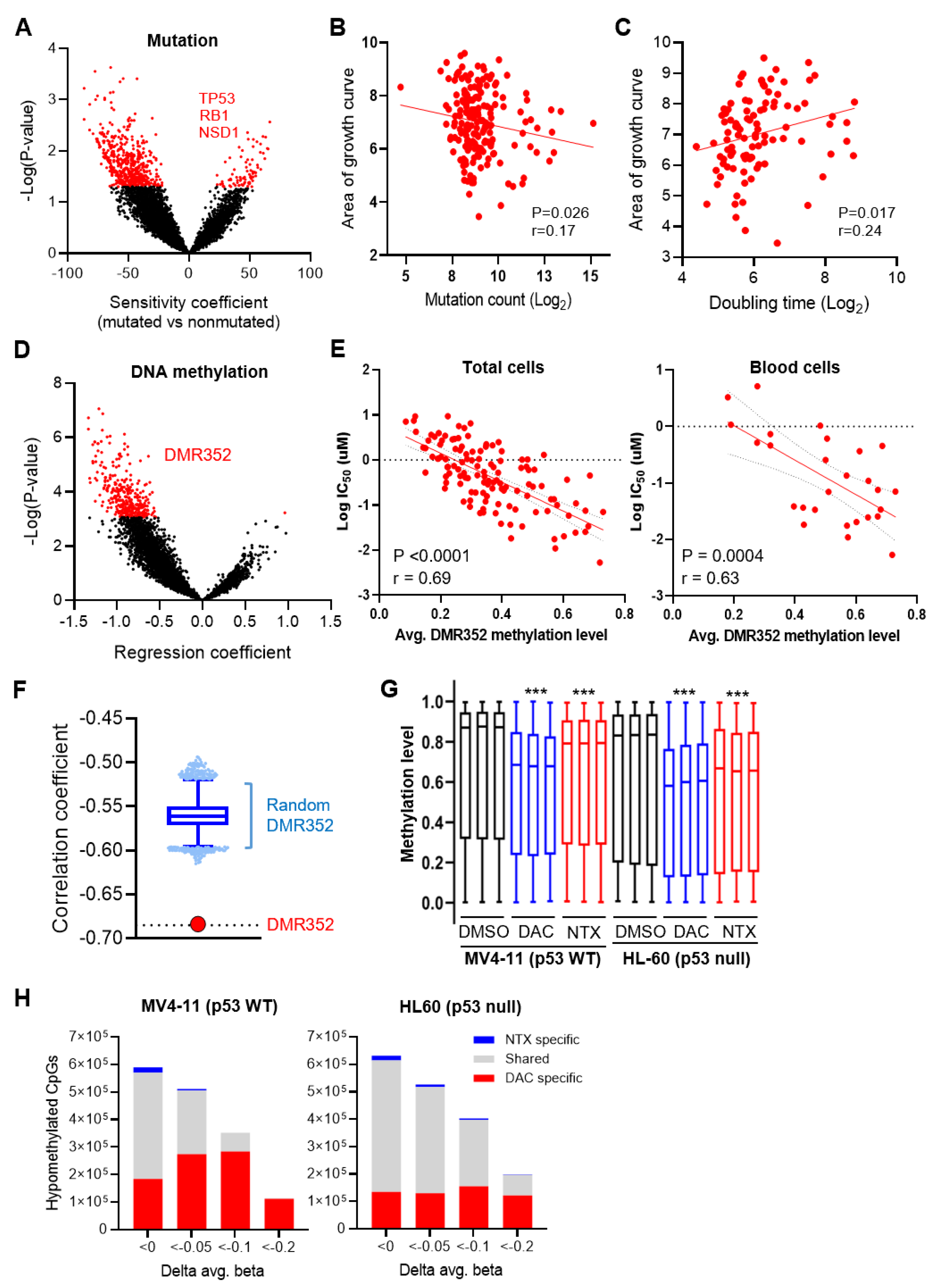
3.6. Integrative Data Analyses Identified Mutation Events That Are Associated with Sensitivity to NTX-301
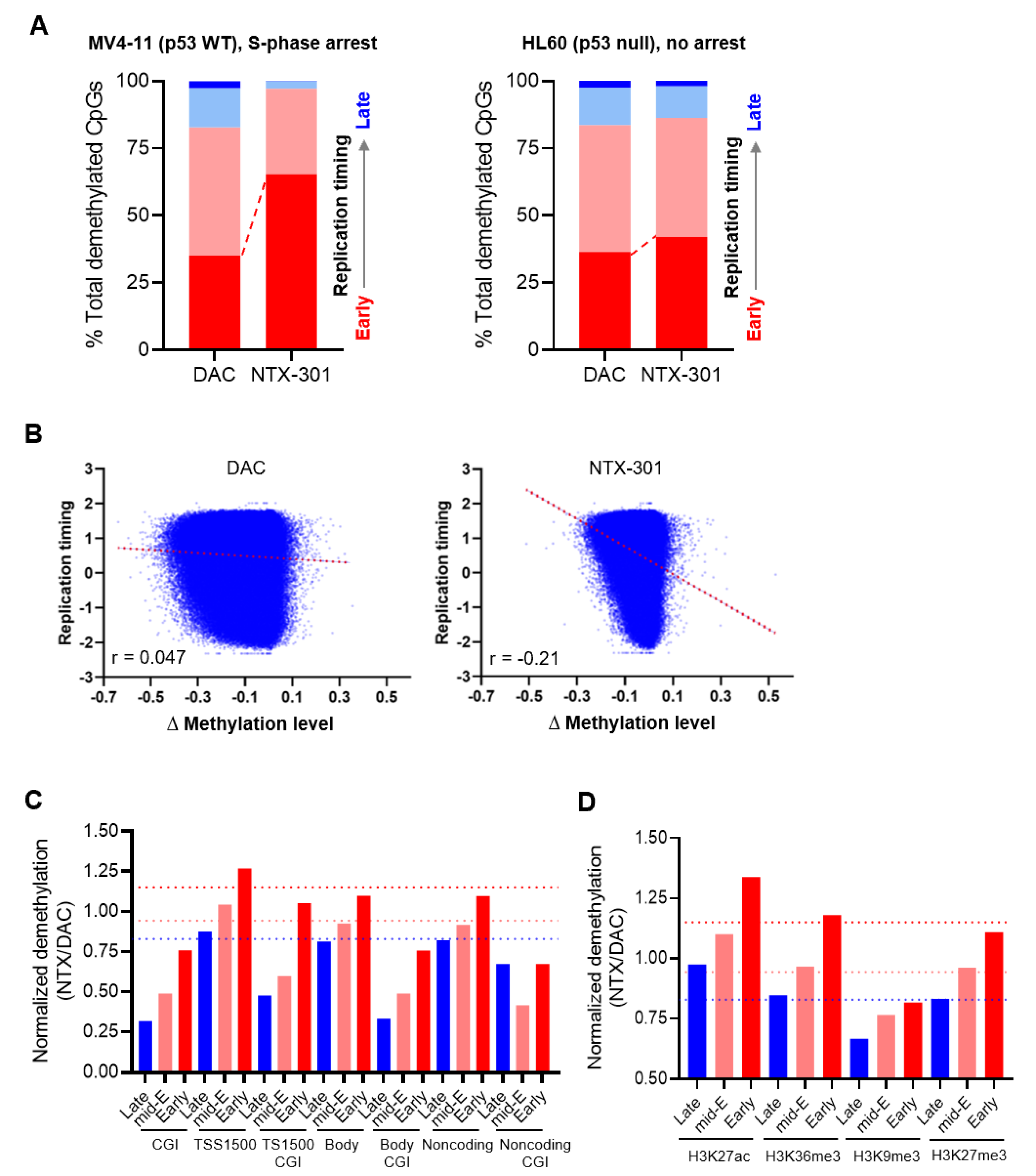
3.7. Integrative Data Analyses Identified Epigenomic Events That Are Associated with Sensitivity to NTX-301
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, X.; Li, M.; Guo, C.; Wu, Y.; Zhao, L.; Shi, Q.; Song, J.; Song, B. Therapeutic Targeting of Cancer: Epigenetic Homeostasis. Front. Oncol. 2021, 11, 747022. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. NeuropsychoPharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.P.; Gehring, M. Principles of Epigenetic Homeostasis Shared Between Flowering Plants and Mammals. Trends Genet. 2020, 36, 751–763. [Google Scholar] [CrossRef]
- Schoofs, T.; Berdel, W.E.; Muller-Tidow, C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 2014, 28, 1–14. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, P.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef]
- Parker, W.B.; Thottassery, J.V. 5-Aza-4′-thio-2′-deoxycytidine, a New Orally Bioavailable Nontoxic “Best-in-Class”: DNA Methyltransferase 1-Depleting Agent in Clinical Development. J. Pharmacol. Exp. Ther. 2021, 379, 211–222. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Yoo, D.; Chun, Y.; Go, A.; Cho, K.J.; Choi, D.; Jung, M.E.; Lee, H.Y.; Boohaker, R.J.; Lee, J.S.; et al. The preclinical efficacy of the novel hypomethylating agent NTX-301 as a monotherapy and in combination with venetoclax in acute myeloid leukemia. Blood Cancer J. 2022, 12, 57. [Google Scholar] [CrossRef]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4’-thio-2’-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer ChemoTher. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Ebrahem, Q.; Negrotto, S.; Mahfouz, R.Z.; Link, K.A.; Hu, Z.; Gu, X.; Advani, A.; Kalaycio, M.; Sobecks, R.; et al. p53 independent epigenetic-differentiation treatment in xenotransplant models of acute myeloid leukemia. Leukemia 2011, 25, 1739–1750. [Google Scholar] [CrossRef] [Green Version]
- Santini, V.; Ossenkoppele, G.J. Hypomethylating agents in the treatment of acute myeloid leukemia: A guide to optimal use. Crit. Rev. Oncol. Hematol. 2019, 140, 1–7. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e924. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, L.; Alvarez-Errico, D.; Esteller, M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020, 41, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell. Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Skibbe, J.R.; Hu, C.; Dong, L.; Ferchen, K.; Su, R.; Li, C.; Huang, H.; Weng, H.; Huang, H.; et al. ALOX5 exhibits anti-tumor and drug-sensitizing effects in MLL-rearranged leukemia. Sci. Rep. 2017, 7, 1853. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Yang, L.; Liu, X.J.; Zhan, Y.; Pan, Y.X.; Wang, X.Z.; Luo, J.M. Association between methylation of tumor suppressor gene SOCS1 and acute myeloid leukemia. Oncol. Rep. 2018, 40, 1008–1016. [Google Scholar] [CrossRef]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012, 19, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 2021, 35, 1023–1036. [Google Scholar] [CrossRef]
- Oellerich, T.; Schneider, C.; Thomas, D.; Knecht, K.M.; Buzovetsky, O.; Kaderali, L.; Schliemann, C.; Bohnenberger, H.; Angenendt, L.; Hartmann, W.; et al. Selective inactivation of hypomethylating agents by SAMHD1 provides a rationale for therapeutic stratification in AML. Nat. Commun. 2019, 10, 3475. [Google Scholar] [CrossRef]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Choufani, S.; Cytrynbaum, C.; Chung, B.H.; Turinsky, A.L.; Grafodatskaya, D.; Chen, Y.A.; Cohen, A.S.; Dupuis, L.; Butcher, D.T.; Siu, M.T.; et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat. Commun. 2015, 6, 10207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, P.; Frankhouser, D.; Murphy, M.; Tam, H.H.; Rodriguez, B.; Curfman, J.; Trimarchi, M.; Geyer, S.; Wu, Y.Z.; Whitman, S.P.; et al. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood 2012, 120, 2466–2474. [Google Scholar] [CrossRef] [PubMed]
- Chilakala, S.; Feng, Y.; Li, L.; Mahfouz, R.; Quteba, E.; Saunthararajah, Y.; Xu, Y. Tracking Decitabine Incorporation into Malignant Myeloid Cell DNA in vitro and in vivo by LC-MS/MS with Enzymatic Digestion. Sci. Rep. 2019, 9, 4558. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Baris, A.; Aladjem, M.I. Replication timing and nuclear structure. Curr. Opin. Cell Biol. 2018, 52, 43–50. [Google Scholar] [CrossRef]
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Orta, M.L.; Calderon-Montano, J.M.; Dominguez, I.; Pastor, N.; Burgos-Moron, E.; Lopez-Lazaro, M.; Cortes, F.; Mateos, S.; Helleday, T. 5-Aza-2’-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Hastak, K.; Paul, R.K.; Agarwal, M.K.; Thakur, V.S.; Amin, A.R.; Agrawal, S.; Sramkoski, R.M.; Jacobberger, J.W.; Jackson, M.W.; Stark, G.R.; et al. DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc. Natl. Acad. Sci. USA 2008, 105, 6314–6319. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, C.C.; Davalos, A.; Martin-Sanchez, C.; de la Pena, G.; Fernandez-Hernando, C.; Lasuncion, M.A. Cholesterol starvation induces differentiation of human leukemia HL-60 cells. Cancer Res. 2007, 67, 3379–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.Y.; Appelbaum, F.R.; Willman, C.L.; Zager, R.A.; Banker, D.E. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003, 101, 3628–3634. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Wu, X.; Niculite, C.; Gilca, M.; Petrusca, D.; Rogozea, A.; Rice, S.; Guo, B.; Griffin, S.; Calin, G.A.; et al. Classic and targeted anti-leukaemic agents interfere with the cholesterol biogenesis metagene in acute myeloid leukaemia: Therapeutic implications. J. Cell. Mol. Med. 2020, 24, 7378–7392. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, B.; Yoo, D.; Chun, Y.; Go, A.; Kim, J.Y.; Lee, H.Y.; Boohaker, R.J.; Cho, K.-J.; Ahn, S.; Lee, J.S.; et al. Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301. Cancers 2023, 15, 1737. https://doi.org/10.3390/cancers15061737
Lim B, Yoo D, Chun Y, Go A, Kim JY, Lee HY, Boohaker RJ, Cho K-J, Ahn S, Lee JS, et al. Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301. Cancers. 2023; 15(6):1737. https://doi.org/10.3390/cancers15061737
Chicago/Turabian StyleLim, Byungho, Dabin Yoo, Younghwa Chun, Areum Go, Ji Yeon Kim, Ha Young Lee, Rebecca J. Boohaker, Kyung-Jin Cho, Sunjoo Ahn, Jin Soo Lee, and et al. 2023. "Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301" Cancers 15, no. 6: 1737. https://doi.org/10.3390/cancers15061737
APA StyleLim, B., Yoo, D., Chun, Y., Go, A., Kim, J. Y., Lee, H. Y., Boohaker, R. J., Cho, K. -J., Ahn, S., Lee, J. S., Jung, D., & Choi, G. (2023). Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301. Cancers, 15(6), 1737. https://doi.org/10.3390/cancers15061737