
Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Syndecans’ Expression in Breast Cancer Cell Lines and Its Correlation with Tumor Progression
3. Tumor-Associated Stromal Cell Expression of Syndecans
4. Relationship between Syndecans and Epithelial–Mesenchymal Transition
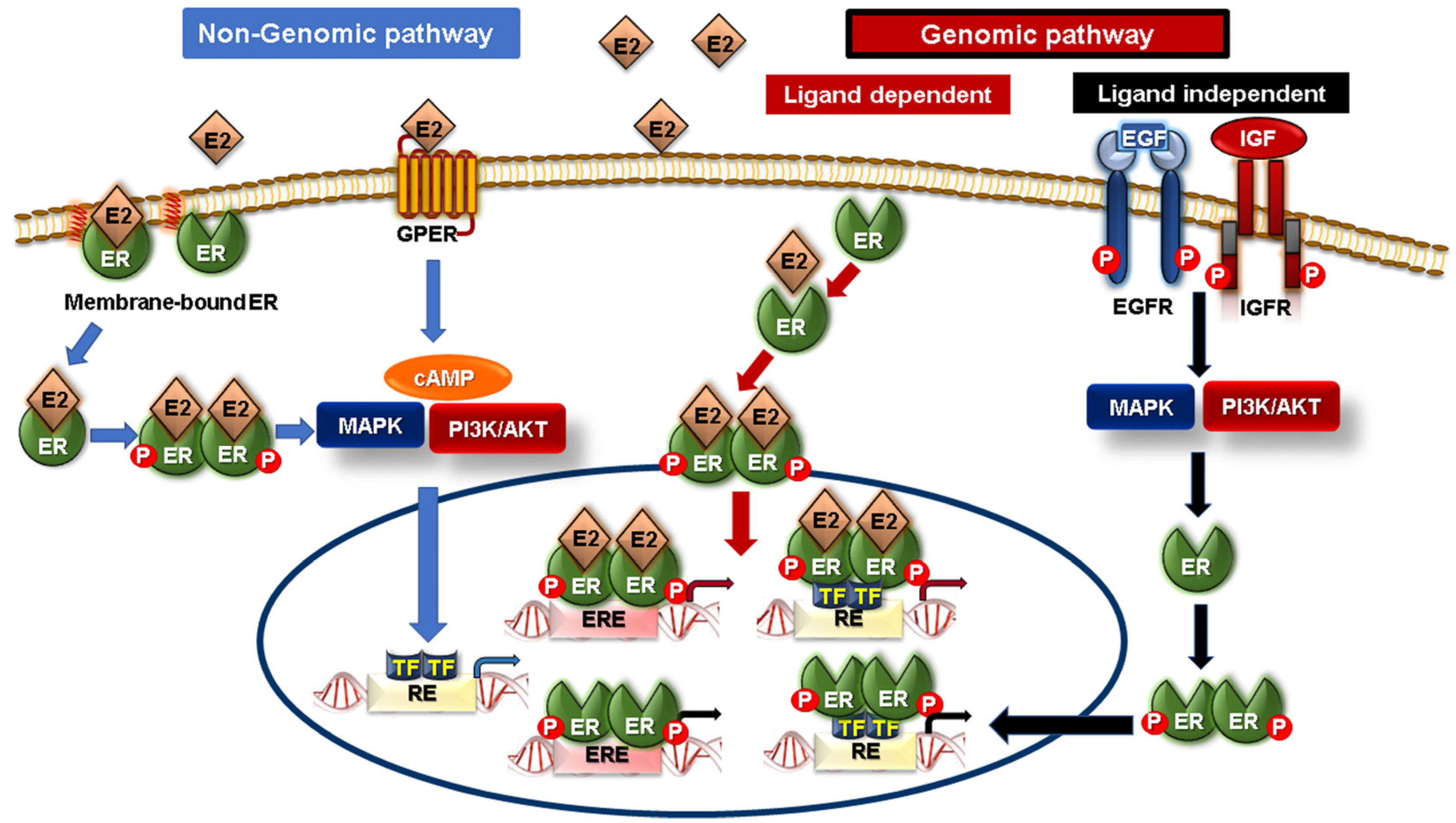
5. Interplay between Syndecans and Nuclear Hormone Receptors in Breast Cancer Progression
5.1. Nuclear Hormone Receptors
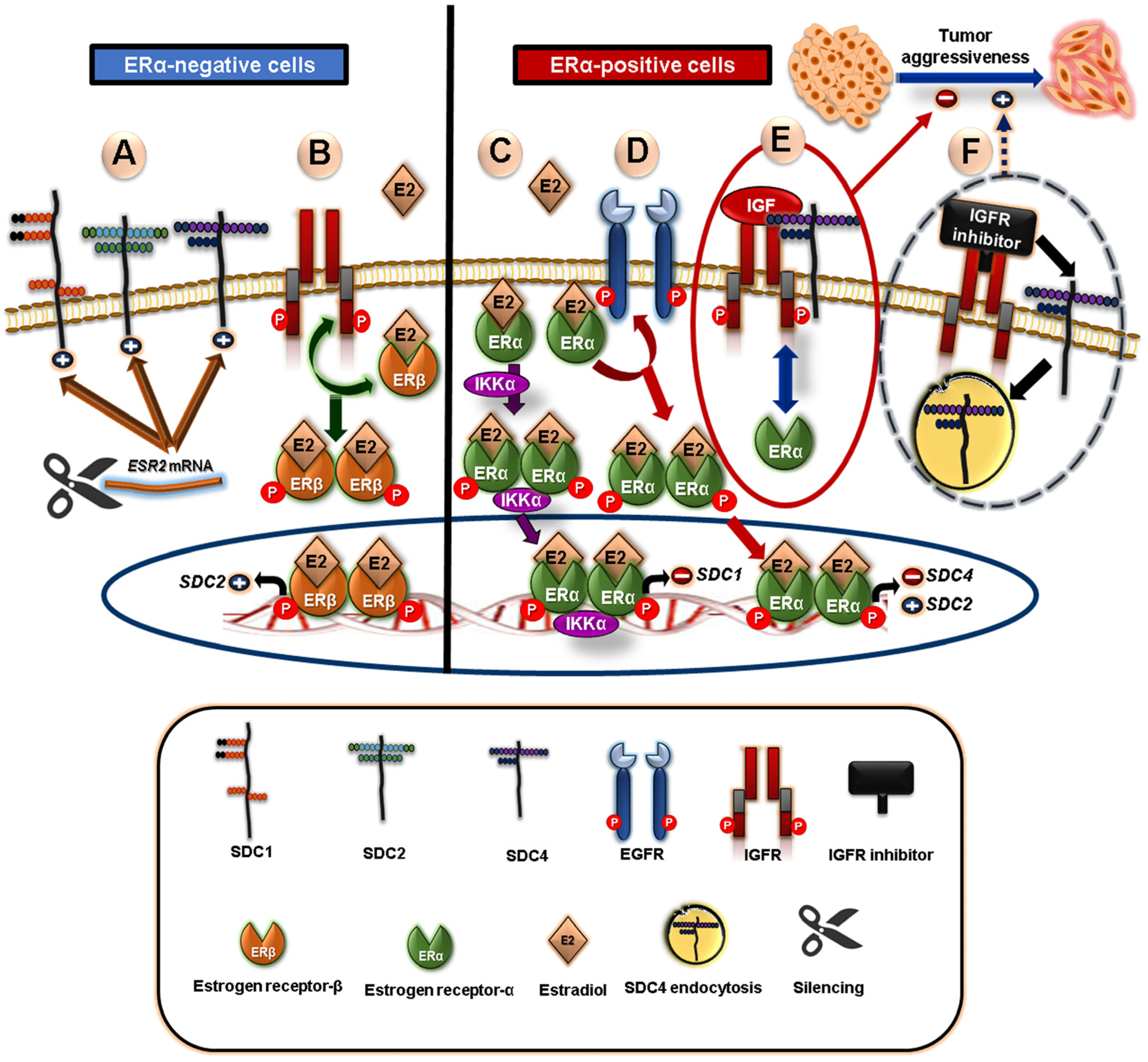
5.2. Crosstalk between Syndecans and Steroid Nuclear Receptors in Breast Cancer
5.3. Crosstalk of Syndecans with PPAR-Gamma “Adopted Orphan Nuclear Receptors”
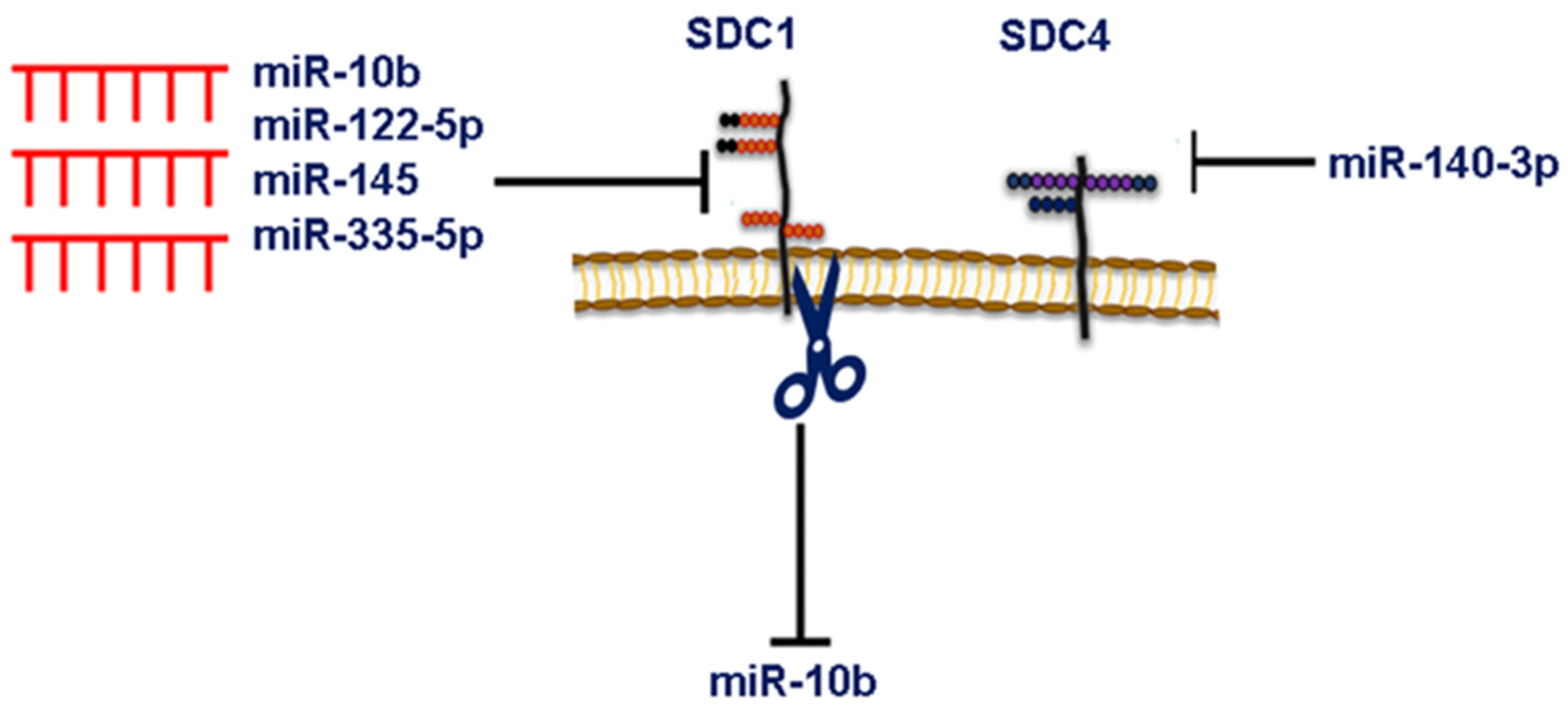
6. MicroRNA-Dependent Syndecan Regulation and Breast Cancer
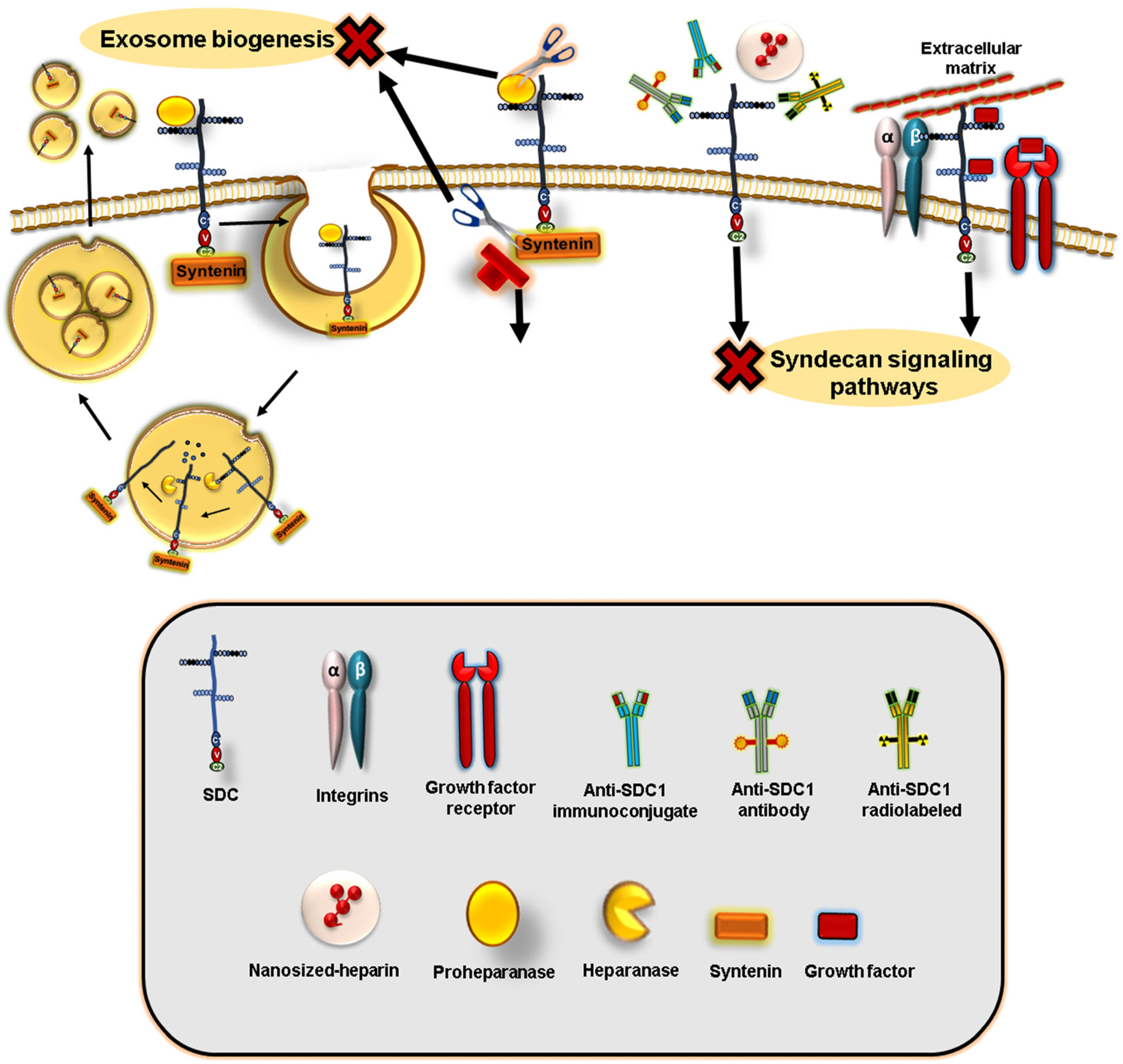
7. Syndecans Regulate Exosome Biogenesis and Composition
8. Syndecans as Targets for Breast Cancer Therapies
8.1. Monoclonal Antibodies
8.2. HS/GAGs Mimetics
8.3. Pharmacological or Peptide Inhibitors
8.4. Exosome Targeting-Based Therapies
8.5. Other SDCs Targeting Therapeutic Modalities
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Couchman, J.R. Syndecans: Proteoglycan Regulators of Cell-Surface Microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–938. [Google Scholar] [CrossRef]
- Afratis, N.A.; Nikitovic, D.; Multhaupt, H.A.B.; Theocharis, A.D.; Couchman, J.R.; Karamanos, N.K. Syndecans—Key Regulators of Cell Signaling and Biological Functions. FEBS J. 2017, 284, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Couchman, J.R.; Chen, L.; Woods, A. Syndecans and Cell Adhesion. Int. Rev. Cytol. 2001, 207, 113–150. [Google Scholar]
- Gopal, S. Syndecans in Inflammation at a Glance. Front. Immunol. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.; Greve, B.; Espinoza-Sánchez, N.A.; Götte, M. Cell-Surface Heparan Sulfate Proteoglycans as Multifunctional Integrators of Signaling in Cancer. Cell. Signal. 2021, 77, 109822. [Google Scholar] [CrossRef]
- Couchman, J.R.; Gopal, S.; Lim, H.C.; Nørgaard, S.; Multhaupt, H.A.B. Fell-Muir Lecture: Syndecans: From Peripheral Coreceptors to Mainstream Regulators of Cell Behaviour. Int. J. Exp. Pathol. 2015, 96, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Manon-Jensen, T.; Itoh, Y.; Couchman, J.R. Proteoglycans in Health and Disease: The Multiple Roles of Syndecan Shedding. FEBS J. 2010, 277, 3876–3889. [Google Scholar] [CrossRef]
- Steinfeld, R.; Van Den Berghe, H.; David, G. Stimulation of Fibroblast Growth Factor Receptor-1 Occupancy and Signaling by Cell Surface-Associated Syndecans and Glypican. J. Cell Biol. 1996, 133, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin Ligands at a Glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O. Integrins. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Bloom, L.; Ingham, K.C.; Hynes, R.O. Fibronectin Regulates Assembly of Actin Filaments and Focal Contacts in Cultured Cells via the Heparin-Binding Site in Repeat III 13. Mol. Biol. Cell 1999, 10, 1521–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, K.J.; Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. Syndecan-1 Regulates αvβ5 Integrin Activity in B82L Fibroblasts. J. Cell Sci. 2006, 119, 2445–2456. [Google Scholar] [CrossRef] [Green Version]
- Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. The Syndecan-1 Ectodomain Regulates αvβ3 Integrin Activity in Human Mammary Carcinoma Cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Tsubota, Y.; Hashimoto, J.; Kariya, Y.; Miyazaki, K. The Short Arm of Laminin Γ2 Chain of Laminin-5 (Laminin-332) Binds Syndecan-1 and Regulates Cellular Adhesion and Migration by Suppressing Phosphorylation of Integrin Β4 Chain. Mol. Biol. Cell 2007, 18, 1621–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hozumi, K.; Suzuki, N.; Nielsen, P.K.; Nomizu, M.; Yamada, Y. Laminin α1 Chain LG4 Module Promotes Cell Attachment through Syndecans and Cell Spreading through Integrin α2β1. J. Biol. Chem. 2006, 281, 32929–32940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, H.; Greve, B.; Pavao, M.S.G.; Kiesel, L.; Ibrahim, S.A.; Götte, M. Syndecan-1 Modulates β-Integrin-Dependent and Interleukin-6-Dependent Functions in Breast Cancer Cell Adhesion, Migration, and Resistance to Irradiation. FEBS J. 2013, 280, 2216–2227. [Google Scholar] [CrossRef]
- Gondelaud, F.; Ricard-Blum, S. Structures and Interactions of Syndecans. FEBS J. 2019, 286, 2994–3007. [Google Scholar] [CrossRef] [Green Version]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brézillon, S.; Götte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, S.S.; Abdel-Mawgood, A.L.; Ibrahim, S.A. EGFR-Dependent Extracellular Matrix Protein Interactions Might Light a Candle in Cell Behavior of Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 766659. [Google Scholar] [CrossRef]
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycan Signaling in Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 6588. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Purushothaman, A.; Stewart, M.D.; Thompson, C.A.; Vlodavsky, I.; Au, J.L.-S.; Sanderson, R.D. The Heparanase/Syndecan-1 Axis in Cancer: Mechanisms and Therapies. FEBS J. 2013, 280, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, J.; Xiao, M.; Zhao, L.; Zhao, Y.; Wang, S.; Gao, S.; Zhuang, Y.; Niu, Y.; Li, S.; et al. Uncovering the Subtype-Specific Molecular Characteristics of Breast Cancer by Multiomics Analysis of Prognosis-Associated Genes, Driver Genes, Signaling Pathways, and Immune Activity. Front. Cell Dev. Biol. 2021, 9, 689028. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated Observation of Breast Tumor Subtypes in Independent Gene Expression Data Sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha (ERα) and Beta (ERβ): Subtype-Selective Ligands and Clinical Potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.; Metzger-Filho, O.; Winer, E.P. The Natural History of Hormone Receptor-Positive Breast Cancer. Oncology 2012, 26, 688–694, 696. [Google Scholar]
- Sotoca Covaleda, A.M.; van den Berg, H.; Vervoort, J.; van der Saag, P.; Ström, A.; Gustafsson, J.-Å.; Rietjens, I.; Murk, A.J. Influence of Cellular ERα/ERβ Ratio on the ERα-Agonist Induced Proliferation of Human T47D Breast Cancer Cells. Toxicol. Sci. 2008, 105, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, M.K.; Movérare, S.; Skrtic, S.; Gao, H.; Dahlman-Wright, K.; Gustafsson, J.-A.; Ohlsson, C. Estrogen Receptor (ER)-β Reduces ERα-Regulated Gene Transcription, Supporting a “Ying Yang” Relationship between ERα and ERβ in Mice. Mol. Endocrinol. 2003, 17, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J. Estrogen and Progesterone in Normal Mammary Gland Development and in Cancer. Horm. Cancer 2011, 2, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Barbareschi, M.; Maisonneuve, P.; Aldovini, D.; Cangi, M.G.; Pecciarini, L.; Angelo Mauri, F.; Veronese, S.; Caffo, O.; Lucenti, A.; Palma, P.D.; et al. High Syndecan-1 Expression in Breast Carcinoma Is Related to an Aggressive Phenotype and to Poorer Prognosis. Cancer 2003, 98, 474–483. [Google Scholar] [CrossRef]
- Leivonen, M.; Lundin, J.; Nordling, S.; von Boguslawski, K.; Haglund, C. Prognostic Value of Syndecan-1 Expression in Breast Cancer. Oncology 2004, 67, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, L.; Sahlin, L.; Jiang, S.; Von Schoultz, B.; Fernstad, R.; Skoog, L.; Von Schoultz, E. Expression of Syndecan-1 in Paired Samples of Normal and Malignant Breast Tissue from Postmenopausal Women. Anticancer Res. 2007, 27, 3045–3050. [Google Scholar]
- Liu, W.; Litwack, E.D.; Stanley, M.J.; Langford, J.K.; Lander, A.D.; Sanderson, R.D. Heparan Sulfate Proteoglycans as Adhesive and Anti-Invasive Molecules. J. Biol. Chem. 1998, 273, 22825–22832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauvais, D.M.; Rapraeger, A.C. Syndecan-1-Mediated Cell Spreading Requires Signaling by αvβ3 Integrins in Human Breast Carcinoma Cells. Exp. Cell Res. 2003, 286, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 Regulates Alphavbeta3 and Alphavbeta5 Integrin Activation during Angiogenesis and Is Blocked by Synstatin, a Novel Peptide Inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Purushothaman, A.; Uyama, T.; Kobayashi, F.; Yamada, S.; Sugahara, K.; Rapraeger, A.C.; Sanderson, R.D. Heparanase-Enhanced Shedding of Syndecan-1 by Myeloma Cells Promotes Endothelial Invasion and Angiogenesis. Blood 2010, 115, 2449–2457. [Google Scholar] [CrossRef] [Green Version]
- Sayyad, M.R.; Puchalapalli, M.; Vergara, N.G.; Wangensteen, S.M.; Moore, M.; Mu, L.; Edwards, C.; Anderson, A.; Kall, S.; Sullivan, M.; et al. Syndecan-1 Facilitates Breast Cancer Metastasis to the Brain. Breast Cancer Res. Treat. 2019, 178, 35–49. [Google Scholar] [CrossRef]
- Nadanaka, S.; Bai, Y.; Kitagawa, H. Cleavage of Syndecan-1 Promotes the Proliferation of the Basal-Like Breast Cancer Cell Line BT-549 Via Akt SUMOylation. Front. Cell Dev. Biol. 2021, 9, 659428. [Google Scholar] [CrossRef]
- Sun, M.; Gomes, S.; Chen, P.; Frankenberger, C.A.; Sankarasharma, D.; Chung, C.H.; Chada, K.K.; Rosner, M.R. RKIP and HMGA2 Regulate Breast Tumor Survival and Metastasis through Lysyl Oxidase and Syndecan-2. Oncogene 2014, 33, 3528–3537. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.C.; Couchman, J.R. Syndecan-2 Regulation of Morphology in Breast Carcinoma Cells Is Dependent on RhoGTPases. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Stavik, B.; Louch, W.; Carlson, C.R.; Sletten, M.; Ruf, W.; Skretting, G.; Sandset, P.M.; Iversen, N. Syndecan-3 and TFPI Colocalize on the Surface of Endothelial-, Smooth Muscle-, and Cancer Cells. PLoS ONE 2015, 10, e0117404. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.; Efing, J.; Kiesel, L.; Bendas, G.; Götte, M. The Tissue Factor Pathway in Cancer: Overview and Role of Heparan Sulfate Proteoglycans. Cancers 2023, 15, 1524. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-S.; Pandey, V.; Wu, W.-Y.; Ye, S.; Zhu, T.; Lobie, P.E. Prognostic Significance of the Expression of GFRα1, GFRα3 and Syndecan-3, Proteins Binding ARTEMIN, in Mammary Carcinoma. BMC Cancer 2013, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller-Pinter, A.; Bottka, S.; Timar, J.; Kulka, J.; Katona, R.; Dux, L.; Deak, F.; Szilak, L. Syndecan-4 Promotes Cytokinesis in a Phosphorylation-Dependent Manner. Cell. Mol. Life Sci. 2010, 67, 1881–1894. [Google Scholar] [CrossRef]
- Onyeisi, J.O.S.; Greve, B.; Espinoza-Sánchez, N.A.; Kiesel, L.; Lopes, C.C.; Götte, M. MicroRNA-140-3p Modulates Invasiveness, Motility, and Extracellular Matrix Adhesion of Breast Cancer Cells by Targeting Syndecan-4. J. Cell. Biochem. 2021, 122, 1491–1505. [Google Scholar] [CrossRef]
- Mundhenke, C.; Meyer, K.; Drew, S.; Friedl, A. Heparan Sulfate Proteoglycans as Regulators of Fibroblast Growth Factor-2 Receptor Binding in Breast Carcinomas. Am. J. Pathol. 2002, 160, 185–194. [Google Scholar] [CrossRef]
- Vijaya Kumar, A.; Brézillon, S.; Untereiner, V.; Sockalingum, G.D.; Kumar Katakam, S.; Mohamed, H.T.; Kemper, B.; Greve, B.; Mohr, B.; Ibrahim, S.A.; et al. HS2ST1-dependent Signaling Pathways Determine Breast Cancer Cell Viability, Matrix Interactions, and Invasive Behavior. Cancer Sci. 2020, 111, 2907–2922. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-Associated Stromal Cells as Key Contributors to the Tumor Microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Tchou, J.; Conejo-Garcia, J. Targeting the Tumor Stroma as a Novel Treatment Strategy for Breast Cancer. Adv. Pharmacol. 2012, 65, 45–61. [Google Scholar]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J.; Yokota, S.J.; Bankert, R.B. Characterization of Human Lung Tumor-Associated Fibroblasts and Their Ability to Modulate the Activation of Tumor-Associated T Cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef] [Green Version]
- Rodemann, H.P.; Müller, G.A. Characterization of Human Renal Fibroblasts in Health and Disease: II. In Vitro Growth, Differentiation, and Collagen Synthesis of Fibroblasts From Kidneys With Interstitial Fibrosis. Am. J. Kidney Dis. 1991, 17, 684–686. [Google Scholar] [CrossRef]
- Xing, F. Cancer Associated Fibroblasts (CAFs) in Tumor Microenvironment. Front. Biosci. 2010, 15, 166. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Alexander, C.M.; Friedl, A. Induction of Syndecan-1 Expression in Stromal Fibroblasts Promotes Proliferation of Human Breast Cancer Cells. Cancer Res. 2004, 64, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.J.; Stanley, M.W.; Sanderson, R.D.; Zera, R. Syndecan-1 Expression Is Induced in the Stroma of Infiltrating Breast Carcinoma. Am. J. Clin. Pathol. 1999, 112, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Loftus, P.G.; Watson, L.; Deedigan, L.M.; Camarillo-Retamosa, E.; Dwyer, R.M.; O’Flynn, L.; Alagesan, S.; Griffin, M.; O’Brien, T.; Kerin, M.J.; et al. Targeting Stromal Cell Syndecan-2 Reduces Breast Tumour Growth, Metastasis and Limits Immune Evasion. Int. J. Cancer 2021, 148, 1245–1259. [Google Scholar] [CrossRef]
- Prieto-Fernández, E.; Egia-Mendikute, L.; Bosch, A.; García del Río, A.; Jimenez-Lasheras, B.; Antoñana-Vildosola, A.; Lee, S.Y.; Palazon, A. Hypoxia Promotes Syndecan-3 Expression in the Tumor Microenvironment. Front. Immunol. 2020, 11, 586977. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic Domain Interactions of Syndecan-1 and Syndecan-4 with A6β4 Integrin Mediate Human Epidermal Growth Factor Receptor (HER1 and HER2)-Dependent Motility and Survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, H.; Li, X.; Pan, H.; Li, Z.; Wang, J.; Zheng, Z. TNF- α and TGF- β 1 Regulate Syndecan-4 Expression in Nucleus Pulposus Cells: Role of the Mitogen-Activated Protein Kinase and NF- κ B Pathways. Connect. Tissue Res. 2015, 56, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Weinberg, R.A. Linking EMT Programmes to Normal and Neoplastic Epithelial Stem Cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, C.; Zimmermann, C.C.; Espinoza-Sanchez, N.A.; Greve, B.; Schmidt, A.; Kiesel, L.; von Wahlde, M.; Götte, M. The Heparan Sulphate Proteoglycan Syndecan-1 (CD138) Regulates Tumour Progression in a 3D Model of Ductal Carcinoma in Situ of the Breast. IUBMB Life 2022, 74, 955–968. [Google Scholar] [CrossRef]
- Vitale, D.; Kumar Katakam, S.; Greve, B.; Jang, B.; Oh, E.-S.; Alaniz, L.; Götte, M. Proteoglycans and Glycosaminoglycans as Regulators of Cancer Stem Cell Function and Therapeutic Resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 Is a Novel Molecular Marker for Triple Negative Inflammatory Breast Cancer and Modulates the Cancer Stem Cell Phenotype via the IL-6/STAT3, Notch and EGFR Signaling Pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Vargas, H.; Ouzounova, M.; Le Calvez-Kelm, F.; Lambert, M.-P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome Analysis Reveals Jak-STAT Pathway Deregulation in Putative Breast Cancer Stem Cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Liu, C.; Zhu, G.; Wang, K.; Yang, Y.; Wang, C. Relationship between SDC1 and Cadherin Signalling Activation in Cancer. Pathol.-Res. Pract. 2020, 216, 152756. [Google Scholar] [CrossRef]
- Mytilinaiou, M.; Nikitovic, D.; Berdiaki, A.; Kostouras, A.; Papoutsidakis, A.; Tsatsakis, A.M.; Tzanakakis, G.N. Emerging Roles of Syndecan 2 in Epithelial and Mesenchymal Cancer Progression. IUBMB Life 2017, 69, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Ramani, V.C.; Sanderson, R.D. Shed Syndecan-1 Translocates to the Nucleus of Cells Delivering Growth Factors and Inhibiting Histone Acetylation. J. Biol. Chem. 2015, 290, 941–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmári, T.; Mundt, F.; Kumar-Singh, A.; Möbus, L.; Ötvös, R.; Hjerpe, A.; Dobra, K. Molecular Targets and Signaling Pathways Regulated by Nuclear Translocation of Syndecan-1. BMC Cell Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Singh, A.; Parniewska, M.M.; Giotopoulou, N.; Javadi, J.; Sun, W.; Szatmári, T.; Dobra, K.; Hjerpe, A.; Fuxe, J. Nuclear Syndecan-1 Regulates Epithelial-Mesenchymal Plasticity in Tumor Cells. Biology 2021, 10, 521. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. SnapShot: Nuclear Receptors I. Cell 2010, 142, 822–822.e1. [Google Scholar] [CrossRef] [Green Version]
- Garattini, E.; Bolis, M.; Gianni’, M.; Paroni, G.; Fratelli, M.; Terao, M. Lipid-Sensors, Enigmatic-Orphan and Orphan Nuclear Receptors as Therapeutic Targets in Breast-Cancer. Oncotarget 2016, 7, 42661–42682. [Google Scholar] [CrossRef] [Green Version]
- Sonoda, J.; Pei, L.; Evans, R.M. Nuclear Receptors: Decoding Metabolic Disease. FEBS Lett. 2008, 582, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Lee, J.X.T.; Wahli, W.; Tan, N.S. Exploiting Vulnerabilities of Cancer by Targeting Nuclear Receptors of Stromal Cells in Tumor Microenvironment. Mol. Cancer 2019, 18, 51. [Google Scholar] [CrossRef]
- Doan, T.B.; Graham, J.D.; Clarke, C.L. Emerging Functional Roles of Nuclear Receptors in Breast Cancer. J. Mol. Endocrinol. 2017, 58, R169–R190. [Google Scholar] [CrossRef]
- CORDERA, F.; JORDAN, V. Steroid Receptors and Their Role in the Biology and Control of Breast Cancer Growth. Semin. Oncol. 2006, 33, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Harrell, J.C.; Korach, K.S. Lessons in Estrogen Biology from Knockout and Transgenic Animals. Annu. Rev. Physiol. 2005, 67, 285–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korach, K.S.; Couse, J.F.; Curtis, S.W.; Washburn, T.F.; Lindzey, J.; Kimbro, K.S.; Eddy, E.M.; Migliaccio, S.; Snedeker, S.M.; Lubahn, D.B.; et al. Estrogen Receptor Gene Disruption: Molecular Characterization and Experimental and Clinical Phenotypes. Recent Prog. Horm. Res. 1996, 51, 159–186; discussion 186–188. [Google Scholar] [PubMed]
- Lydon, J.P.; DeMayo, F.J.; Funk, C.R.; Mani, S.K.; Hughes, A.R.; Montgomery, C.A.; Shyamala, G.; Conneely, O.M.; O’Malley, B.W. Mice Lacking Progesterone Receptor Exhibit Pleiotropic Reproductive Abnormalities. Genes Dev. 1995, 9, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, C.W.; Silberstein, G.B.; Strickland, P. Direct Action of 17 Beta-Estradiol on Mouse Mammary Ducts Analyzed by Sustained Release Implants and Steroid Autoradiography. Cancer Res. 1987, 47, 6052–6057. [Google Scholar]
- Tiemann, K.; Weigel, M.T.; Alkatout, I.; Wenners, A.S.; Mundhenke, H.; Schäfer, F.W.; Bauer, M.; Schem, C.; Maass, N.; Jonat, W.; et al. Significance of Syndecan-1 Expression in Ductal Carcinoma in Situ of the Breast. Anticancer Res. 2014, 34, 3607–3616. [Google Scholar]
- Baba, F.; Swartz, K.; van Buren, R.; Eickhoff, J.; Zhang, Y.; Wolberg, W.; Friedl, A. Syndecan-1 and Syndecan-4 Are Overexpressed in an Estrogen Receptor-Negative, Highly Proliferative Breast Carcinoma Subtype. Breast Cancer Res. Treat. 2006, 98, 91–98. [Google Scholar] [CrossRef]
- Lendorf, M.E.; Manon-Jensen, T.; Kronqvist, P.; Multhaupt, H.A.B.; Couchman, J.R. Syndecan-1 and Syndecan-4 Are Independent Indicators in Breast Carcinoma. J. Histochem. Cytochem. 2011, 59, 615–629. [Google Scholar] [CrossRef] [Green Version]
- Onyeisi, J.O.S.; Lopes, C.C.; Götte, M. Role of Syndecan-4 in Breast Cancer Pathophysiology. Am. J. Physiol. Cell Physiol. 2022, 323, C1345–C1354. [Google Scholar] [CrossRef]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Tsonis, A.I.; Afratis, N.; Gialeli, C.; Ellina, M.-I.; Piperigkou, Z.; Skandalis, S.S.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Evaluation of the Coordinated Actions of Estrogen Receptors with Epidermal Growth Factor Receptor and Insulin-like Growth Factor Receptor in the Expression of Cell Surface Heparan Sulfate Proteoglycans and Cell Motility in Breast Cancer Cells. FEBS J. 2013, 280, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Luo, Y.; Zhang, L.; Cai, Q.; Pan, X. Known and Emerging Factors Modulating Estrogenic Effects of Endocrine-Disrupting Chemicals. Environ. Rev. 2014, 22, 87–98. [Google Scholar] [CrossRef]
- Khatpe, A.; Adebayo, A.; Herodotou, C.; Kumar, B.; Nakshatri, H. Nexus between PI3K/AKT and Estrogen Receptor Signaling in Breast Cancer. Cancers 2021, 13, 369. [Google Scholar] [CrossRef]
- Lipovka, Y.; Konhilas, J.P. The Complex Nature of Oestrogen Signalling in Breast Cancer: Enemy or Ally? Biosci. Rep. 2016, 36, e00352. [Google Scholar] [CrossRef] [Green Version]
- Słowikowski, B.K.; Lianeri, M.; Jagodziński, P.P. Exploring Estrogenic Activity in Lung Cancer. Mol. Biol. Rep. 2017, 44, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kousidou, O.C.; Berdiaki, A.; Kletsas, D.; Zafiropoulos, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Estradiol-Estrogen Receptor: A Key Interplay of the Expression of Syndecan-2 and Metalloproteinase-9 in Breast Cancer Cells. Mol. Oncol. 2008, 2, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Afratis, N.A.; Bouris, P.; Skandalis, S.S.; Multhaupt, H.A.; Couchman, J.R.; Theocharis, A.D.; Karamanos, N.K. IGF-IR Cooperates with ERα to Inhibit Breast Cancer Cell Aggressiveness by Regulating the Expression and Localisation of ECM Molecules. Sci. Rep. 2017, 7, 40138. [Google Scholar] [CrossRef] [Green Version]
- Fleurot, E.; Goudin, C.; Hanoux, V.; Bonnamy, P.-J.; Levallet, J. Estrogen Receptor α Regulates the Expression of Syndecan-1 in Human Breast Carcinoma Cells. Endocr. Relat. Cancer 2019, 26, 615–628. [Google Scholar] [CrossRef]
- Park, K.-J.; Krishnan, V.; O’Malley, B.W.; Yamamoto, Y.; Gaynor, R.B. Formation of an IKKα-Dependent Transcription Complex Is Required for Estrogen Receptor-Mediated Gene Activation. Mol. Cell 2005, 18, 71–82. [Google Scholar] [CrossRef]
- Weitsman, G.E.; Li, L.; Skliris, G.P.; Davie, J.R.; Ung, K.; Niu, Y.; Curtis-Snell, L.; Tomes, L.; Watson, P.H.; Murphy, L.C. Estrogen Receptor-α Phosphorylated at Ser118 Is Present at the Promoters of Estrogen-Regulated Genes and Is Not Altered Due to HER-2 Overexpression. Cancer Res. 2006, 66, 10162–10170. [Google Scholar] [CrossRef] [Green Version]
- Piperigkou, Z.; Bouris, P.; Onisto, M.; Franchi, M.; Kletsas, D.; Theocharis, A.D.; Karamanos, N.K. Estrogen Receptor Beta Modulates Breast Cancer Cells Functional Properties, Signaling and Expression of Matrix Molecules. Matrix Biol. 2016, 56, 4–23. [Google Scholar] [CrossRef]
- Chen, M.; Yang, Y.; Xu, K.; Li, L.; Huang, J.; Qiu, F. Androgen Receptor in Breast Cancer: From Bench to Bedside. Front. Endocrinol. 2020, 11, 573. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Bonafè, M.; Bravaccini, S. Androgen Receptor in Breast Cancer: A Wolf in Sheep’s Clothing? A Lesson from Prostate Cancer. Semin. Cancer Biol. 2020, 60, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, S.K.; Diamandis, E.P. Androgen Receptor: A Promising Therapeutic Target in Breast Cancer. Crit. Rev. Clin. Lab. Sci. 2019, 56, 200–223. [Google Scholar] [CrossRef]
- Leppä, S.; Mali, M.; Miettinen, H.M.; Jalkanen, M. Syndecan Expression Regulates Cell Morphology and Growth of Mouse Mammary Epithelial Tumor Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Leppä, S.; Härkönen, P.; Jalkanen, M. Steroid-Induced Epithelial-Fibroblastic Conversion Associated with Syndecan Suppression in S115 Mouse Mammary Tumor Cells. Cell Regul. 1991, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viklund, L.; Vorontsova, N.; Henttinen, T.; Salmivirta, M. Syndecan-1 Regulates FGF8b Responses in S115 Mammary Carcinoma Cells. Growth Factors 2006, 24, 151–157. [Google Scholar] [CrossRef]
- Hofling, M.; Ma, L.; Sahlin, L.; Haglund, C.; Nordling, S.; von Schoultz, B.; Cline, J.M. Expression of the Androgen Receptor and Syndecan-1 in Breast Tissue during Different Hormonal Treatments in Cynomolgus Monkeys. Climacteric 2009, 12, 72–79. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications—A Review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Hong, O.-Y.; Youn, H.J.; Jang, H.-Y.; Jung, S.H.; Noh, E.-M.; Chae, H.S.; Jeong, Y.-J.; Kim, W.; Kim, C.-H.; Kim, J.-S. Troglitazone Inhibits Matrix Metalloproteinase-9 Expression and Invasion of Breast Cancer Cell through a Peroxisome Proliferator-Activated Receptor γ-Dependent Mechanism. J. Breast Cancer 2018, 21, 28. [Google Scholar] [CrossRef]
- Rovito, D.; Giordano, C.; Plastina, P.; Barone, I.; De Amicis, F.; Mauro, L.; Rizza, P.; Lanzino, M.; Catalano, S.; Bonofiglio, D.; et al. Omega-3 DHA- and EPA–Dopamine Conjugates Induce PPARγ-Dependent Breast Cancer Cell Death through Autophagy and Apoptosis. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 2185–2195. [Google Scholar] [CrossRef]
- Rovito, D.; Gionfriddo, G.; Barone, I.; Giordano, C.; Grande, F.; De Amicis, F.; Lanzino, M.; Catalano, S.; Andò, S.; Bonofiglio, D. Ligand-Activated PPARγ Downregulates CXCR4 Gene Expression through a Novel Identified PPAR Response Element and Inhibits Breast Cancer Progression. Oncotarget 2016, 7, 65109–65124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome Proliferator-Activated Receptor γ–Mediated Up-Regulation of Syndecan-1 by n-3 Fatty Acids Promotes Apoptosis of Human Breast Cancer Cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pon, C.K.; Firth, S.M.; Baxter, R.C. Involvement of Insulin-like Growth Factor Binding Protein-3 in Peroxisome Proliferator-Activated Receptor Gamma-Mediated Inhibition of Breast Cancer Cell Growth. Mol. Cell. Endocrinol. 2015, 399, 354–361. [Google Scholar] [CrossRef]
- Abduljabbar, R.; Negm, O.H.; Lai, C.-F.; Jerjees, D.A.; Al-Kaabi, M.; Hamed, M.R.; Tighe, P.J.; Buluwela, L.; Mukherjee, A.; Green, A.R.; et al. Clinical and Biological Significance of Glucocorticoid Receptor (GR) Expression in Breast Cancer. Breast Cancer Res. Treat. 2015, 150, 335–346. [Google Scholar] [CrossRef]
- Carter, J.C.; Church, F.C. Obesity and Breast Cancer: The Roles of Peroxisome Proliferator-Activated Receptor-γ and Plasminogen Activator Inhibitor-1. PPAR Res. 2009, 2009, 345320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The Role of PPARγ Ligands in Breast Cancer: From Basic Research to Clinical Studies. Cancers 2020, 12, 2623. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative Cobinding of Synthetic and Natural Ligands to the Nuclear Receptor PPARγ. eLife 2018, 7, e43320. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hu, Y.; Gu, Z.; Owens, R.T.; Chen, Y.Q.; Edwards, I.J. Omega-3 Fatty Acids Induce Apoptosis in Human Breast Cancer Cells and Mouse Mammary Tissue through Syndecan-1 Inhibition of the MEK-Erk Pathway. Carcinogenesis 2011, 32, 1518–1524. [Google Scholar] [CrossRef] [Green Version]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.-I.; et al. Syndecan-1 Is Required to Maintain Intradermal Fat and Prevent Cold Stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef] [Green Version]
- D’Esposito, V.; Ambrosio, M.R.; Giuliano, M.; Cabaro, S.; Miele, C.; Beguinot, F.; Formisano, P. Mammary Adipose Tissue Control of Breast Cancer Progression: Impact of Obesity and Diabetes. Front. Oncol. 2020, 10, 1554. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Loedige, I.; Filipowicz, W. The Widespread Regulation of MicroRNA Biogenesis, Function and Decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.A.; Hassan, H.; Götte, M. MicroRNA-Dependent Targeting of the Extracellular Matrix as a Mechanism of Regulating Cell Behavior. Biochim. Biophys. Acta 2014, 1840, 2609–2620. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Tzaferi, K.; Makrokanis, G.; Cheli, K.; Karamanos, N.K. The MicroRNA-Cell Surface Proteoglycan Axis in Cancer Progression. Am. J. Physiol. Cell Physiol. 2022, 322, C825–C832. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.A.; Hassan, H.; Götte, M. MicroRNA Regulation of Proteoglycan Function in Cancer. FEBS J. 2014, 281, 5009–5022. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Piperigkou, Z.; Franchi, M.; Götte, M.; Karamanos, N.K. Estrogen Receptor Beta as Epigenetic Mediator of MiR-10b and MiR-145 in Mammary Cancer. Matrix Biol. 2017, 64, 94–111. [Google Scholar] [CrossRef]
- Zolota, V.; Tzelepi, V.; Piperigkou, Z.; Kourea, H.; Papakonstantinou, E.; Argentou, Μ.-I.; Karamanos, N.K. Epigenetic Alterations in Triple-Negative Breast Cancer-The Critical Role of Extracellular Matrix. Cancers 2021, 13, 713. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Fahim, S.A.; Abdullah, M.S.; Espinoza-Sánchez, N.A.; Hassan, H.; Ibrahim, A.M.; Ahmed, S.H.; Shakir, G.; Badawy, M.A.; Zakhary, N.I.; Greve, B.; et al. Inflammatory Breast Carcinoma: Elevated MicroRNA MiR-181b-5p and Reduced MiR-200b-3p, MiR-200c-3p, and MiR-203a-3p Expression as Potential Biomarkers with Diagnostic Value. Biomolecules 2020, 10, 1059. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Espinoza-Sánchez, N.A.; El-Damen, A.; Fahim, S.A.; Badawy, M.A.; Greve, B.; El-Shinawi, M.; Götte, M.; Ibrahim, S.A. Small Extracellular Vesicle-Encapsulated MiR-181b-5p, MiR-222-3p and Let-7a-5p: Next Generation Plasma Biopsy-Based Diagnostic Biomarkers for Inflammatory Breast Cancer. PLoS ONE 2021, 16, e0250642. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Yip, G.W.; Stock, C.; Pan, J.-W.; Neubauer, C.; Poeter, M.; Pupjalis, D.; Koo, C.Y.; Kelsch, R.; Schüle, R.; et al. Targeting of Syndecan-1 by MicroRNA MiR-10b Promotes Breast Cancer Cell Motility and Invasiveness via a Rho-GTPase- and E-Cadherin-Dependent Mechanism. Int. J. Cancer 2012, 131, E884–E896. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Götte, M.; Mohr, C.; Koo, C.-Y.; Stock, C.; Vaske, A.-K.; Viola, M.; Ibrahim, S.A.; Peddibhotla, S.; Teng, Y.H.-F.; Low, J.-Y.; et al. MiR-145-Dependent Targeting of Junctional Adhesion Molecule A and Modulation of Fascin Expression Are Associated with Reduced Breast Cancer Cell Motility and Invasiveness. Oncogene 2010, 29, 6569–6580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperigkou, Z.; Franchi, M.; Riethmüller, C.; Götte, M.; Karamanos, N.K. MiR-200b Restrains EMT and Aggressiveness and Regulates Matrix Composition Depending on ER Status and Signaling in Mammary Cancer. Matrix Biol. Plus 2020, 6–7, 100024. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, S.S.; Ibrahim, S.A.; Abdel-Mawgood, A.L. Cell Behavior of Non-Small Cell Lung Cancer Is at EGFR and MicroRNAs Hands. Int. J. Mol. Sci. 2021, 22, 124969. [Google Scholar] [CrossRef]
- Jiang, F.; Wu, C.; Wang, M.; Wei, K.; Wang, J. Identification of Novel Cell Glycolysis Related Gene Signature Predicting Survival in Patients with Breast Cancer. Sci. Rep. 2021, 11, 3986. [Google Scholar] [CrossRef] [PubMed]
- Valla, S.; Hassan, N.; Vitale, D.L.; Madanes, D.; Spinelli, F.M.; Teixeira, F.C.O.B.; Greve, B.; Espinoza-Sánchez, N.A.; Cristina, C.; Alaniz, L.; et al. Syndecan-1 Depletion Has a Differential Impact on Hyaluronic Acid Metabolism and Tumor Cell Behavior in Luminal and Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5874. [Google Scholar] [CrossRef]
- Uen, Y.; Wang, J.-W.; Wang, C.; Jhang, Y.; Chung, J.-Y.; Tseng, T.; Sheu, M.; Lee, S. Mining of Potential MicroRNAs with Clinical Correlation—Regulation of Syndecan-1 Expression by MiR-122-5p Altered Mobility of Breast Cancer Cells and Possible Correlation with Liver Injury. Oncotarget 2018, 9, 28165–28175. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Ma, Y.; Ma, Y.; Liu, P.; Hou, L.; Xu, Z.; Jiang, J.; Shen, Y.; Cao, Y.; Zhao, Y. MiR-335-5p Targets SDC1 to Regulate the Progression of Breast Cancer. Crit. Rev. Eukaryot. Gene Expr. 2022, 32, 21–31. [Google Scholar] [CrossRef]
- Grootjans, J.J.; Zimmermann, P.; Reekmans, G.; Smets, A.; Degeest, G.; Dürr, J.; David, G. Syntenin, a PDZ Protein That Binds Syndecan Cytoplasmic Domains. Proc. Natl. Acad. Sci. USA 1997, 94, 13683–13688. [Google Scholar] [CrossRef] [Green Version]
- Pu, T.; Shen, M.; Li, S.; Yang, L.; Gao, H.; Xiao, L.; Zhong, X.; Zheng, H.; Liu, Y.; Ye, F.; et al. Repression of MiR-135b-5p Promotes Metastasis of Early-Stage Breast Cancer by Regulating Downstream Target SDCBP. Lab. Investig. 2019, 99, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Lai, F.; Florio, T.; Caviglioli, G. Exosomes and Extracellular Vesicles as Emerging Theranostic Platforms in Cancer Research. Cells 2020, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Lorenc, T.; Chrzanowski, J.; Olejarz, W. Current Perspectives on Clinical Use of Exosomes as a Personalized Contrast Media and Theranostics. Cancers 2020, 12, 3386. [Google Scholar] [CrossRef]
- Gould, G.W.; Lippincott-Schwartz, J. New Roles for Endosomes: From Vesicular Carriers to Multi-Purpose Platforms. Nat. Rev. Mol. Cell Biol. 2009, 10, 287–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan–Syntenin–ALIX Regulates the Biogenesis of Exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Friand, V.; David, G.; Zimmermann, P. Syntenin and Syndecan in the Biogenesis of Exosomes. Biol. Cell 2015, 107, 331–341. [Google Scholar] [CrossRef]
- Zimmermann, P.; Zhang, Z.; Degeest, G.; Mortier, E.; Leenaerts, I.; Coomans, C.; Schulz, J.; N’Kuli, F.; Courtoy, P.J.; David, G. Syndecan Recyling Is Controlled by Syntenin-PIP2 Interaction and Arf6. Dev. Cell 2005, 9, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX Exosome Biogenesis and Budding into Multivesicular Bodies Are Controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [Green Version]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase Activates the Syndecan-Syntenin-ALIX Exosome Pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, F.C.O.B.; Götte, M. Involvement of Syndecan-1 and Heparanase in Cancer and Inflammation. Adv. Exp. Med. Biol. 2020, 1221, 97–135. [Google Scholar] [CrossRef]
- Yip, G.W.; Smollich, M.; Götte, M. Therapeutic Value of Glycosaminoglycans in Cancer. Mol. Cancer Ther. 2006, 5, 2139–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theocharis, A.D.; Skandalis, S.S.; Neill, T.; Multhaupt, H.A.B.; Hubo, M.; Frey, H.; Gopal, S.; Gomes, A.; Afratis, N.; Lim, H.C.; et al. Insights into the Key Roles of Proteoglycans in Breast Cancer Biology and Translational Medicine. Biochim. Biophys. Acta 2015, 1855, 276–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbouri, D.; Afratis, N.; Gialeli, C.; Vynios, D.H.; Theocharis, A.D.; Karamanos, N.K. Syndecans as Modulators and Potential Pharmacological Targets in Cancer Progression. Front. Oncol. 2014, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, K.; Herbener, P.; Zuber, C.; Häder, T.; Bernöster, K.; Uherek, C.; Schüttrumpf, J. Activity of Indatuximab Ravtansine against Triple-Negative Breast Cancer in Preclinical Tumor Models. Pharm. Res. 2018, 35, 118. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, C.; Ruellan, A.L.; Bernardeau, K.; Kraeber-Bodéré, F.; Gouard, S.; Loussouarn, D.; Saï-Maurel, C.; Faivre-Chauvet, A.; Wijdenes, J.; Barbet, J.; et al. Syndecan-1 Antigen, a Promising New Target for Triple-Negative Breast Cancer Immuno-PET and Radioimmunotherapy. A Preclinical Study on MDA-MB-468 Xenograft Tumors. EJNMMI Res. 2011, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, R.; Sahay, D.; Houssin, A.; Machuca-Gayet, I.; Peyruchaud, O. Autotaxin-β Interaction with the Cell Surface via Syndecan-4 Impacts on Cancer Cell Proliferation and Metastasis. Oncotarget 2018, 9, 33170–33185. [Google Scholar] [CrossRef] [Green Version]
- Onyeisi, J.O.S.; Castanho de Almeida Pernambuco Filho, P.; de Araujo Lopes, S.; Nader, H.B.; Lopes, C.C. Heparan Sulfate Proteoglycans as Trastuzumab Targets in Anoikis-Resistant Endothelial Cells. J. Cell. Biochem. 2019, 120, 13826–13840. [Google Scholar] [CrossRef]
- van den Born, J.; van den Heuvel, L.P.W.J.; Bakker, M.A.H.; Veerkamp, J.H.; Assmann, K.J.M.; Berden, J.H.M. A Monoclonal Antibody against GBM Heparan Sulfate Induces an Acute Selective Proteinuria in Rats. Kidney Int. 1992, 41, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Dam, G.B.t; Kurup, S.; van de Westerlo, E.M.A.; Versteeg, E.M.M.; Lindahl, U.; Spillmann, D.; van Kuppevelt, T.H. 3-O-Sulfated Oligosaccharide Structures Are Recognized by Anti-Heparan Sulfate Antibody HS4C3. J. Biol. Chem. 2006, 281, 4654–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieboldt, R.; Läubli, H. Glycosaminoglycans in Cancer Therapy. Am. J. Physiol. Physiol. 2022, 322, C1187–C1200. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Abrahams, J.P.; Skinner, R.; Petitou, M.; Pike, R.N.; Carrell, R.W. The Anticoagulant Activation of Antithrombin by Heparin. Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [Google Scholar] [CrossRef] [Green Version]
- Läubli, H.; Borsig, L. Heparins Attenuate Cancer Metastasis: Are Selectins the Link? Cancer Investig. 2009, 27, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, I.-K.; Hursting, M.J. When Heparins Promote Thrombosis. Circulation 2005, 111, 2671–2683. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.H.; Viera, M.; Yip, G.W.; Bay, B.H. Theranostic Applications of Glycosaminoglycans in Metastatic Renal Cell Carcinoma. Cancers 2022, 15, 266. [Google Scholar] [CrossRef]
- Huang, X.; Reye, G.; Momot, K.I.; Blick, T.; Lloyd, T.; Tilley, W.D.; Hickey, T.E.; Snell, C.E.; Okolicsanyi, R.K.; Haupt, L.M.; et al. Heparanase Promotes Syndecan-1 Expression to Mediate Fibrillar Collagen and Mammographic Density in Human Breast Tissue Cultured Ex Vivo. Front. Cell Dev. Biol. 2020, 8, 599. [Google Scholar] [CrossRef]
- Zhang, L.; Ngo, J.A.; Wetzel, M.D.; Marchetti, D. Heparanase Mediates a Novel Mechanism in Lapatinib-Resistant Brain Metastatic Breast Cancer. Neoplasia 2015, 17, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a Chemically Modified Heparin, Inhibits Myeloma Growth and Angiogenesis via Disruption of the Heparanase/Syndecan-1 Axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Taher, F.A.; Ibrahim, S.A.; El-Aziz, A.A.; Abou El-Nour, M.F.; El-Sheikh, M.A.; El-Husseiny, N.; Mohamed, M.M. Anti-Proliferative Effect of Chitosan Nanoparticles (Extracted from Crayfish Procambarus Clarkii, Crustacea: Cambaridae) against MDA-MB-231 and SK-BR-3 Human Breast Cancer Cell Lines. Int. J. Biol. Macromol. 2019, 126, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Karamanou, K.; Afratis, N.A.; Bouris, P.; Gialeli, C.; Belmiro, C.L.R.; Pavão, M.S.G.; Vynios, D.H.; Tsatsakis, A.M. Biochemical and Toxicological Evaluation of Nano-Heparins in Cell Functional Properties, Proteasome Activation and Expression of Key Matrix Molecules. Toxicol. Lett. 2016, 240, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Dedes, P.G.; Gialeli, C.; Tsonis, A.I.; Kanakis, I.; Theocharis, A.D.; Kletsas, D.; Tzanakakis, G.N.; Karamanos, N.K. Expression of Matrix Macromolecules and Functional Properties of Breast Cancer Cells Are Modulated by the Bisphosphonate Zoledronic Acid. Biochim. Biophys. Acta 2012, 1820, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Blaine, S.A.; Qiao, D.; Friedl, A. Membrane Type 1 Matrix Metalloproteinase–Mediated Stromal Syndecan-1 Shedding Stimulates Breast Carcinoma Cell Proliferation. Cancer Res. 2008, 68, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Pasqualon, T.; Pruessmeyer, J.; Weidenfeld, S.; Babendreyer, A.; Groth, E.; Schumacher, J.; Schwarz, N.; Denecke, B.; Jahr, H.; Zimmermann, P.; et al. A Transmembrane C-Terminal Fragment of Syndecan-1 Is Generated by the Metalloproteinase ADAM17 and Promotes Lung Epithelial Tumor Cell Migration and Lung Metastasis Formation. Cell. Mol. Life Sci. 2015, 72, 3783–3801. [Google Scholar] [CrossRef]
- Ramirez Williams, L.; Brüggemann, K.; Hubert, M.; Achmad, N.; Kiesel, L.; Schäfer, S.D.; Greve, B.; Götte, M. γ-Secretase Inhibition Affects Viability, Apoptosis, and the Stem Cell Phenotype of Endometriotic Cells. Acta Obstet. Gynecol. Scand. 2019, 98, 1565–1574. [Google Scholar] [CrossRef] [Green Version]
- Malavaki, C.J.; Roussidis, A.E.; Gialeli, C.; Kletsas, D.; Tsegenidis, T.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Imatinib as a Key Inhibitor of the Platelet-Derived Growth Factor Receptor Mediated Expression of Cell Surface Heparan Sulfate Proteoglycans and Functional Properties of Breast Cancer Cells. FEBS J. 2013, 280, 2477–2489. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Nelson, S.E.; Adams, K.M.; Stueven, N.A.; Jung, O.; Rapraeger, A.C. Plasma Membrane Proteoglycans Syndecan-2 and Syndecan-4 Engage with EGFR and RON Kinase to Sustain Carcinoma Cell Cycle Progression. J. Biol. Chem. 2022, 298, 102029. [Google Scholar] [CrossRef]
- Leblanc, R.; Kashyap, R.; Barral, K.; Egea-Jimenez, A.L.; Kovalskyy, D.; Feracci, M.; Garcia, M.; Derviaux, C.; Betzi, S.; Ghossoub, R.; et al. Pharmacological Inhibition of Syntenin PDZ2 Domain Impairs Breast Cancer Cell Activities and Exosome Loading with Syndecan and EpCAM Cargo. J. Extracell. Vesicles 2020, 10, e12039. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Hoffer, L.; Leblanc, R.; Benmansour, F.; Feracci, M.; Derviaux, C.; Egea-Jimenez, A.L.; Roche, P.; Zimmermann, P.; Morelli, X.; et al. Fragment-Based Drug Design Targeting Syntenin PDZ2 Domain Involved in Exosomal Release and Tumour Spread. Eur. J. Med. Chem. 2021, 223, 113601. [Google Scholar] [CrossRef]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase Regulates Secretion, Composition, and Function of Tumor Cell-Derived Exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [Green Version]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.-P.; Belting, M. Cancer Cell Exosomes Depend on Cell-Surface Heparan Sulfate Proteoglycans for Their Internalization and Functional Activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [Green Version]
- Parimon, T.; Brauer, R.; Schlesinger, S.Y.; Xie, T.; Jiang, D.; Ge, L.; Huang, Y.; Birkland, T.P.; Parks, W.C.; Habiel, D.M.; et al. Syndecan-1 Controls Lung Tumorigenesis by Regulating MiRNAs Packaged in Exosomes. Am. J. Pathol. 2018, 188, 1094–1103. [Google Scholar] [CrossRef] [Green Version]
- Tavianatou, A.-G.; Piperigkou, Z.; Barbera, C.; Beninatto, R.; Masola, V.; Caon, I.; Onisto, M.; Franchi, M.; Galesso, D.; Karamanos, N.K. Molecular Size-Dependent Specificity of Hyaluronan on Functional Properties, Morphology and Matrix Composition of Mammary Cancer Cells. Matrix Biol. Plus 2019, 3, 100008. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, S.; Ying, H.; Yao, W. Targeting Syndecan-1: New Opportunities in Cancer Therapy. Am. J. Physiol. Cell Physiol. 2022, 323, C29–C45. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, T.; Deshmukh, S. PH-Responsive Antibodies for Therapeutic Applications. J. Biomed. Sci. 2021, 28, 11. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced Tumor-Targeting Selectivity by Modulating Bispecific Antibody Binding Affinity and Format Valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef]
- Cerbelli, B.; Pisano, A.; Pignataro, M.G.; Pernazza, A.; Botticelli, A.; Carosi, M.; Costarelli, L.; Allegretti, M.; D’Amati, G.; Cordone, I. Overexpression in Metastatic Breast Cancer Supports Syndecan-1 as a Marker of Invasiveness and Poor Prognosis. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 Years’ Follow-up of Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Early Breast Cancer: Final Analysis of the HERceptin Adjuvant (HERA) Trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motta, J.M.; Hassan, H.; Ibrahim, S.A. Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma. Cancers 2023, 15, 1794. https://doi.org/10.3390/cancers15061794
Motta JM, Hassan H, Ibrahim SA. Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma. Cancers. 2023; 15(6):1794. https://doi.org/10.3390/cancers15061794
Chicago/Turabian StyleMotta, Juliana Maria, Hebatallah Hassan, and Sherif Abdelaziz Ibrahim. 2023. "Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma" Cancers 15, no. 6: 1794. https://doi.org/10.3390/cancers15061794
APA StyleMotta, J. M., Hassan, H., & Ibrahim, S. A. (2023). Revisiting the Syndecans: Master Signaling Regulators with Prognostic and Targetable Therapeutic Values in Breast Carcinoma. Cancers, 15(6), 1794. https://doi.org/10.3390/cancers15061794