

The Role of NK Cells in EBV Infection and Related Diseases: Current Understanding and Hints for Novel Therapies

Abstract
:Simple Summary
Abstract
1. Introduction
2. EBV-Related Lymphoproliferative and Malignant Diseases
3. Complex EBV Infection in the Context of Immunodeficiency
4. Role of NK Cells in Primary Immunodeficiencies Predisposing to EBV-Induced Diseases
5. Modulation and Function of NK Cells during EBV Infection
5.1. NK Cells in the Context of Primary EBV Infection
5.2. In Vitro Activation/Expansion of NK Cells Triggered by EBV-Infected Cells
5.3. NK Cells in EBV-Related Lymphoproliferative Diseases and Cancer
6. NK/EBV-Infected Cell Interactions
6.1. NK Cell Recognition of HLA-I Molecules on EBV+ Cell Targets
6.2. NKG2A/HLA-E Interaction in NK Cell-Mediated Killing of EBV+ Cell Targets
6.3. NKG2D/NKG2DLs and DNAM-1/DNAM-1Ls Axes Modulated by EBV
6.4. NK Cell-Mediated ADCC against EBV+ Cells
7. Current Treatments for EBV-Induced Diseases and Therapeutic Applications of NK Cells
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Mace, E.M. Human Natural Killer Cells: Form, Function, and Development. J. Allergy Clin. Immunol. 2023, 151, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK Cell Self Tolerance, Responsiveness and Missing Self Recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljunggren, H.G.; Kärre, K. In Search of the ‘Missing Self’: MHC Molecules and NK Cell Recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Ardolino, M.; Santoni, A.; Cerboni, C. NKG2D and DNAM-1 Activating Receptors and their Ligands in NK-T Cell Interactions: Role in the NK Cell-Mediated Negative Regulation of T Cell Responses. Front. Immunol. 2013, 3, 408. [Google Scholar] [CrossRef] [Green Version]
- Di Vito, C.; Mikulak, J.; Mavilio, D. On the Way to Become a Natural Killer Cell. Front. Immunol. 2019, 10, 1812. [Google Scholar] [CrossRef]
- Björkström, N.K.; Ljunggren, H.; Sandberg, J.K. CD56 Negative NK Cells: Origin, Function, and Role in Chronic Viral Disease. Trends Immunol. 2010, 31, 401–406. [Google Scholar] [CrossRef]
- Brillantes, M.; Beaulieu, A.M. Memory and Memory-Like NK Cell Responses to Microbial Pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 102. [Google Scholar] [CrossRef]
- Stary, V.; Stary, G. NK Cell-Mediated Recall Responses: Memory-Like, Adaptive, Or Antigen-Specific? Front. Cell. Infect. Microbiol. 2020, 10, 208. [Google Scholar] [CrossRef]
- Orange, J.S. Natural Killer Cell Deficiency. J. Allergy Clin. Immunol. 2013, 132, 515–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, E.M.; Orange, J.S. Emerging Insights into Human Health and NK Cell Biology from the Study of NK Cell Deficiencies. Immunol. Rev. 2019, 287, 202–225. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, C. Latency and Lytic Replication in Epstein-Barr Virus-Associated Oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein-Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; M Bode, A.; Cao, Y. Epstein-Barr Virus Lytic Reactivation Regulation and its Pathogenic Role in Carcinogenesis. Int. J. Biol. Sci. 2016, 12, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Bu, G.; Xie, C.; Kang, Y.; Zeng, M.; Sun, C. How EBV Infects: The Tropism and Underlying Molecular Mechanism for Viral Infection. Viruses 2022, 14, 2372. [Google Scholar] [CrossRef] [PubMed]
- Rezk, S.A.; Zhao, X.; Weiss, L.M. Epstein-Barr Virus (EBV)-Associated Lymphoid Proliferations, a 2018 Update. Hum. Pathol. 2018, 79, 18–41. [Google Scholar] [CrossRef]
- Callan, M.F.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct Visualization of Antigen-Specific CD8+ T Cells during the Primary Immune Response to Epstein-Barr Virus in Vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The Immunology of Epstein-Barr Virus-Induced Disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Chijioke, O.; Landtwing, V.; Münz, C. NK Cell Influence on the Outcome of Primary Epstein-Barr Virus Infection. Front. Immunol. 2016, 7, 323. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Epstein-Barr Virus-Specific Immune Control by Innate Lymphocytes. Front. Immunol. 2017, 8, 1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balfour, H.H.J.; Dunmire, S.K.; Hogquist, K.A. Infectious Mononucleosis. Clin. Transl. Immunol. 2015, 4, e33. [Google Scholar] [CrossRef]
- El-Mallawany, N.K.; Curry, C.V.; Allen, C.E. Haemophagocytic Lymphohistiocytosis and Epstein-Barr Virus: A Complex Relationship with Diverse Origins, Expression and Outcomes. Br. J. Haematol. 2022, 196, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. A Common Mechanism Links Epstein-Barr Virus Infections and Autoimmune Diseases. J. Med. Virol. 2023, 95, e28363. [Google Scholar] [CrossRef] [PubMed]
- Worth, A.J.J.; Houldcroft, C.J.; Booth, C. Severe Epstein-Barr Virus Infection in Primary Immunodeficiency and the Normal Host. Br. J. Haematol. 2016, 175, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Dharnidharka, V.R.; Webster, A.C.; Martinez, O.M.; Preiksaitis, J.K.; Leblond, V.; Choquet, S. Post-Transplant Lymphoproliferative Disorders. Nat. Rev. Dis. Primers 2016, 2, 15088. [Google Scholar] [CrossRef]
- Dierickx, D.; Habermann, T.M. Post-Transplantation Lymphoproliferative Disorders in Adults. N. Engl. J. Med. 2018, 378, 549–562. [Google Scholar] [CrossRef]
- Vos, A.C.W.; Bakkal, N.; Minnee, R.C.; Casparie, M.K.; de Jong, D.J.; Dijkstra, G.; Stokkers, P.; van Bodegraven, A.A.; Pierik, M.; van der Woude, C.J.; et al. Risk of Malignant Lymphoma in Patients with Inflammatory Bowel Diseases: A Dutch Nationwide Study. Inflamm. Bowel Dis. 2011, 17, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Arakawa, F.; Kiyasu, J.; Sato, K.; Miyoshi, H.; Niino, D.; Kimura, Y.; Takeuchi, M.; Yoshida, M.; Ishibashi, Y.; et al. Methotrexate/iatrogenic Lymphoproliferative Disorders in Rheumatoid Arthritis: Histology, Epstein-Barr Virus, and Clonality are Important Predictors of Disease Progression and Regression. Eur. J. Haematol. 2013, 91, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Cesarman, E.; Spina, M.; Gloghini, A.; Schulz, T.F. HIV-Associated Lymphomas and Gamma-Herpesviruses. Blood 2009, 113, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Modification of EBV-Associated Pathologies and Immune Control by Coinfections. Front. Oncol. 2021, 11, 756480. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Primary Immunodeficiencies Associated with EBV Disease. Curr. Top. Microbiol. Immunol. 2015, 390, 241–265. [Google Scholar]
- Latour, S.; Winter, S. Inherited Immunodeficiencies with High Predisposition to Epstein-Barr Virus-Driven Lymphoproliferative Diseases. Front. Immunol. 2018, 9, 1103. [Google Scholar] [CrossRef]
- Tangye, S.G.; Latour, S. Primary Immunodeficiencies Reveal the Molecular Requirements for Effective Host Defense Against EBV Infection. Blood 2020, 135, 644–655. [Google Scholar] [CrossRef]
- Latour, S.; Fischer, A. Signaling Pathways Involved in the T-Cell-Mediated Immunity Against Epstein-Barr Virus: Lessons from Genetic Diseases. Immunol. Rev. 2019, 291, 174–189. [Google Scholar] [CrossRef]
- Tangye, S.G. Genetic Susceptibility to EBV Infection: Insights from Inborn Errors of Immunity. Hum. Genet. 2020, 139, 885–901. [Google Scholar] [CrossRef]
- Cohen, J.I.; Dropulic, L.; Hsu, A.P.; Zerbe, C.S.; Krogmann, T.; Dowdell, K.; Hornung, R.L.; Lovell, J.; Hardy, N.; Hickstein, D.; et al. Association of GATA2 Deficiency with Severe Primary Epstein-Barr Virus (EBV) Infection and EBV-Associated Cancers. Clin. Infect. Dis. 2016, 63, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 Deficiency in Patients with Growth Retardation, Adrenal Insufficiency, and Natural Killer Cell Deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grier, J.T.; Forbes, L.R.; Monaco-Shawver, L.; Oshinsky, J.; Atkinson, T.P.; Moody, C.; Pandey, R.; Campbell, K.S.; Orange, J.S. Human Immunodeficiency-Causing Mutation Defines CD16 in Spontaneous NK Cell Cytotoxicity. J. Clin. Investig. 2012, 122, 3769–3780. [Google Scholar] [CrossRef] [PubMed]
- Arjunaraja, S.; Angelus, P.; Su, H.C.; Snow, A.L. Impaired Control of Epstein-Barr Virus Infection in B-Cell Expansion with NF-κB and T-Cell Anergy Disease. Front. Immunol. 2018, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Suarez, F.; Mahlaoui, N.; Canioni, D.; Andriamanga, C.; Dubois d’Enghien, C.; Brousse, N.; Jais, J.; Fischer, A.; Hermine, O.; Stoppa-Lyonnet, D. Incidence, Presentation, and Prognosis of Malignancies in Ataxia-Telangiectasia: A Report from the French National Registry of Primary Immune Deficiencies. J. Clin. Oncol. 2015, 33, 202–208. [Google Scholar] [CrossRef]
- Du, S.; Scuderi, R.; Malicki, D.M.; Willert, J.; Bastian, J.; Weidner, N. Hodgkin’s and Non-Hodgkin’s Lymphomas Occurring in Two Brothers with Wiskott-Aldrich Syndrome and Review of the Literature. Pediatr. Dev. Pathol. 2011, 14, 64–70. [Google Scholar] [CrossRef]
- Chaigne-Delalande, B.; Li, F.; O’Connor, G.M.; Lukacs, M.J.; Jiang, P.; Zheng, L.; Shatzer, A.; Biancalana, M.; Pittaluga, S.; Matthews, H.F.; et al. Mg2+ Regulates Cytotoxic Functions of NK and CD8 T Cells in Chronic EBV Infection through NKG2D. Science 2013, 341, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Abolhassani, H.; Edwards, E.S.J.; Ikinciogullari, A.; Jing, H.; Borte, S.; Buggert, M.; Du, L.; Matsuda-Lennikov, M.; Romano, R.; Caridha, R.; et al. Combined Immunodeficiency and Epstein-Barr Virus-Induced B Cell Malignancy in Humans with Inherited CD70 Deficiency. J. Exp. Med. 2017, 214, 91–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, T.; Magg, T.; Laschinger, M.; Rohlfs, M.; Linhares, N.D.; Puchalka, J.; Weisser, T.; Fehlner, K.; Mautner, J.; Walz, C.; et al. A Human Immunodeficiency Syndrome Caused by Mutations in CARMIL2. Nat. Commun. 2017, 8, 14209. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, S.D.; Price, S.; Zou, J.; Hunsberger, S.; Brofferio, A.; Matthews, H.; Similuk, M.; Rosenzweig, S.D.; Su, H.C.; Cohen, J.I.; et al. A Double-Blind, Placebo-Controlled, Crossover Study of Magnesium Supplementation in Patients with XMEN Disease. J. Clin. Immunol. 2022, 42, 108–118. [Google Scholar] [CrossRef]
- Orange, J.S.; Mace, E.M.; French, A.R.; Yokoyama, W.M.; Fehniger, T.A.; Cooper, M.A. Comment on: Evidence of Innate Lymphoid Cell Redundancy in Humans. Nat. Immunol. 2018, 19, 788–789. [Google Scholar] [CrossRef]
- Vivier, E.; Vély, F.; Fischer, A. Reply to ‘Comment on: Evidence of Innate Lymphoid Cell Redundancy in Humans’. Nat. Immunol. 2018, 19, 789–790. [Google Scholar] [CrossRef]
- Vély, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of Innate Lymphoid Cell Redundancy in Humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamili, Q.U.A.; Seeborg, F.O.; Saxena, K.; Nicholas, S.K.; Banerjee, P.P.; Angelo, L.S.; Mace, E.M.; Forbes, L.R.; Martinez, C.; Wright, T.S.; et al. Severe Cutaneous Human Papillomavirus Infection Associated with Natural Killer Cell Deficiency Following Stem Cell Transplantation for Severe Combined Immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 1451–1453.e1 . [Google Scholar] [CrossRef] [Green Version]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and Immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Schlums, H.; Jung, M.; Han, H.; Theorell, J.; Bigley, V.; Chiang, S.C.C.; Allan, D.S.J.; Davidson-Moncada, J.K.; Dickinson, R.E.; Holmes, T.D.; et al. Adaptive NK Cells can Persist in Patients with GATA2 Mutation Depleted of Stem and Progenitor Cells. Blood 2017, 129, 1927–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.; McAulay, K.; Macsween, K.F.; Gallacher, N.J.; Higgins, C.D.; Harrison, N.; Swerdlow, A.J.; Crawford, D.H. The Immune Response to Primary EBV Infection: A Role for Natural Killer Cells. Br. J. Haematol. 2005, 129, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Balfour, H.H.J.; Odumade, O.A.; Schmeling, D.O.; Mullan, B.D.; Ed, J.A.; Knight, J.A.; Vezina, H.E.; Thomas, W.; Hogquist, K.A. Behavioral, Virologic, and Immunologic Factors Associated with Acquisition and Severity of Primary Epstein-Barr Virus Infection in University Students. J. Infect. Dis. 2013, 207, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Azzi, T.; Lünemann, A.; Murer, A.; Ueda, S.; Béziat, V.; Malmberg, K.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for Early-Differentiated Natural Killer Cells in Infectious Mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef] [Green Version]
- Béziat, V.; Descours, B.; Parizot, C.; Debré, P.; Vieillard, V. NK Cell Terminal Differentiation: Correlated Stepwise Decrease of NKG2A and Acquisition of KIRs. PLoS ONE 2010, 5, e11966. [Google Scholar] [CrossRef]
- Björkström, N.K.; Riese, P.; Heuts, F.; Andersson, S.; Fauriat, C.; Ivarsson, M.A.; Björklund, A.T.; Flodström-Tullberg, M.; Michaëlsson, J.; Rottenberg, M.E.; et al. Expression Patterns of NKG2A, KIR, and CD57 Define a Process of CD56dim NK-Cell Differentiation Uncoupled from NK-Cell Education. Blood 2010, 116, 3853–3864. [Google Scholar] [CrossRef] [Green Version]
- Chijioke, O.; Müller, A.; Feederle, R.; Barros, M.H.M.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human Natural Killer Cells Prevent Infectious Mononucleosis Features by Targeting Lytic Epstein-Barr Virus Infection. Cell. Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Botet, M.; Vilches, C.; Redondo-Pachón, D.; Muntasell, A.; Pupuleku, A.; Yélamos, J.; Pascual, J.; Crespo, M. Dual Role of Natural Killer Cells on Graft Rejection and Control of Cytomegalovirus Infection in Renal Transplantation. Front. Immunol. 2017, 8, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, D.W.; Balfour, H.H.J.; Dunmire, S.K.; Schmeling, D.O.; Hogquist, K.A.; Lanier, L.L. Cutting Edge: NKG2C(Hi)CD57+ NK Cells Respond Specifically to Acute Infection with Cytomegalovirus and Not Epstein-Barr Virus. J. Immunol. 2014, 192, 4492–4496. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Brilot, F.; Arrey, F.; Bougras, G.; Thomas, D.; Muller, W.A.; Münz, C. Tonsilar NK Cells Restrict B Cell Transformation by the Epstein-Barr Virus Via IFN-Gamma. PLoS Pathog. 2008, 4, e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lünemann, A.; Vanoaica, L.D.; Azzi, T.; Nadal, D.; Münz, C. A Distinct Subpopulation of Human NK Cells Restricts B Cell Transformation by EBV. J. Immunol. 2013, 191, 4989–4995. [Google Scholar] [CrossRef] [Green Version]
- Jud, A.; Kotur, M.; Berger, C.; Gysin, C.; Nadal, D.; Lünemann, A. Tonsillar CD56brightNKG2A+ NK Cells Restrict Primary Epstein-Barr Virus Infection in B Cells Via IFN-γ. Oncotarget 2017, 8, 6130–6141. [Google Scholar] [CrossRef] [Green Version]
- Hatton, O.; Strauss-Albee, D.M.; Zhao, N.Q.; Haggadone, M.D.; Pelpola, J.S.; Krams, S.M.; Martinez, O.M.; Blish, C.A. NKG2A-Expressing Natural Killer Cells Dominate the Response to Autologous Lymphoblastoid Cells Infected with Epstein-Barr Virus. Front. Immunol. 2016, 7, 607. [Google Scholar] [CrossRef] [Green Version]
- Granzin, M.; Soltenborn, S.; Müller, S.; Kollet, J.; Berg, M.; Cerwenka, A.; Childs, R.W.; Huppert, V. Fully Automated Expansion and Activation of Clinical-Grade Natural Killer Cells for Adoptive Immunotherapy. Cytotherapy 2015, 17, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Djaoud, Z.; Guethlein, L.A.; Horowitz, A.; Azzi, T.; Nemat-Gorgani, N.; Olive, D.; Nadal, D.; Norman, P.J.; Münz, C.; Parham, P. Two Alternate Strategies for Innate Immunity to Epstein-Barr Virus: One using NK Cells and the Other NK Cells and γδ T Cells. J. Exp. Med. 2017, 214, 1827–1841. [Google Scholar] [CrossRef] [Green Version]
- Wiesmayr, S.; Webber, S.A.; Macedo, C.; Popescu, I.; Smith, L.; Luce, J.; Metes, D. Decreased NKp46 and NKG2D and Elevated PD-1 are Associated with Altered NK-Cell Function in Pediatric Transplant Patients with PTLD. Eur. J. Immunol. 2012, 42, 541–550. [Google Scholar] [CrossRef]
- Nakid-Cordero, C.; Choquet, S.; Gauthier, N.; Balegroune, N.; Tarantino, N.; Morel, V.; Arzouk, N.; Burrel, S.; Rousseau, G.; Charlotte, F.; et al. Distinct Immunopathological Mechanisms of EBV-Positive and EBV-Negative Posttransplant Lymphoproliferative Disorders. Am. J. Transplant. 2021, 21, 2846–2863. [Google Scholar] [CrossRef] [PubMed]
- Forconi, C.S.; Cosgrove, C.P.; Saikumar-Lakshmi, P.; Nixon, C.E.; Foley, J.; Ong’echa, J.M.; Otieno, J.A.; Alter, G.; Münz, C.; Moormann, A.M. Poorly Cytotoxic Terminally Differentiated CD56(Neg)CD16(Pos) NK Cells Accumulate in Kenyan Children with Burkitt Lymphomas. Blood Adv. 2018, 2, 1101–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pánisová, E.; Lünemann, A.; Bürgler, S.; Kotur, M.; Lazarovici, J.; Danu, A.; Kaulfuss, M.; Mietz, J.; Chijioke, O.; Münz, C.; et al. Reduced Frequency of Cytotoxic CD56(Dim) CD16(+) NK Cells Leads to Impaired Antibody-Dependent Degranulation in EBV-Positive Classical Hodgkin Lymphoma. Cancer Immunol. Immunother. 2022, 71, 13–24. [Google Scholar] [CrossRef]
- Png, Y.T.; Yang, A.Z.Y.; Lee, M.Y.; Chua, M.J.M.; Lim, C.M. The Role of NK Cells in EBV Infection and EBV-Associated NPC. Viruses 2021, 13, 300. [Google Scholar] [CrossRef]
- Horst, D.; van Leeuwen, D.; Croft, N.P.; Garstka, M.A.; Hislop, A.D.; Kremmer, E.; Rickinson, A.B.; Wiertz, E.J.H.J.; Ressing, M.E. Specific Targeting of the EBV Lytic Phase Protein BNLF2a to the Transporter Associated with Antigen Processing Results in Impairment of HLA Class I-Restricted Antigen Presentation. J. Immunol. 2009, 182, 2313–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampé, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B Lymphocytes by Cellular and Epstein-Barr Virus-Encoded Interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.H.J.; Ressing, M.E. Host Shutoff during Productive Epstein-Barr Virus Infection is Mediated by BGLF5 and may Contribute to Immune Evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.; Hislop, A.D.; Rowe, M. The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2015, 90, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The Switch from Latent to Productive Infection in Epstein-Barr Virus-Infected B Cells is Associated with Sensitization to NK Cell Killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, M.; Zentz, C.; Mayr, C.; Wimmer, R.; Hammerschmidt, W.; Zeidler, R.; Moosmann, A. Conditional Immortalization of Human B Cells by CD40 Ligation. PLoS ONE 2008, 3, e1464. [Google Scholar] [CrossRef] [Green Version]
- Tudor, C.S.; Dawson, C.W.; Eckhardt, J.; Niedobitek, G.; Büttner, A.C.; Seliger, B.; Hartmann, A.; Buettner, M. C-Myc and EBV-LMP1: Two Opposing Regulators of the HLA Class I Antigen Presentation Machinery in Epithelial Cells. Br. J. Cancer 2012, 106, 1980–1988. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.R.; Quinn, L.L.; Rowe, M.; Zuo, J. Induction of the Lytic Cycle Sensitizes Epstein-Barr Virus-Infected B Cells to NK Cell Killing that is Counteracted by Virus-Mediated NK Cell Evasion Mechanisms in the Late Lytic Cycle. J. Virol. 2015, 90, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr Virus microRNAs Reduce Immune Surveillance by Virus-Specific CD8+ T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, J.; Gwozdowicz, S.; Graczyk-Pol, E.; Mika-Witkowska, R.; Rogatko-Koros, M.; Nestorowicz, K.; Szlendak, U.; Malinowska, A.; Kaczmarek, B.; Nasilowska-Adamska, B.; et al. Epstein-Barr Virus Infections are Strongly Dependent on Activating and Inhibitory KIR-HLA Pairs After T-Cell Replate Unrelated Hematopoietic Stem Cell Transplantation, the Principles, and Method of Pairing Analysis. HLA 2019, 94 (Suppl. 2), 40–48. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.J.W.; Stowell, J.; Sergeant, R.; Altmann, D.M.; Long, E.O.; Boyton, R.J. KIR2DL3 and KIR2DL1 show Similar Impact on Licensing of Human NK Cells. Eur. J. Immunol. 2016, 46, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheent, K.S.; Jamil, K.M.; Cassidy, S.; Liu, M.; Mbiribindi, B.; Mulder, A.; Claas, F.H.J.; Purbhoo, M.A.; Khakoo, S.I. Synergistic Inhibition of Natural Killer Cells by the Nonsignaling Molecule CD94. Proc. Natl. Acad. Sci. USA 2013, 110, 16981–16986. [Google Scholar] [CrossRef] [Green Version]
- Mahaweni, N.M.; Ehlers, F.A.I.; Sarkar, S.; Janssen, J.W.H.; Tilanus, M.G.J.; Bos, G.M.J.; Wieten, L. NKG2A Expression is Not Per Se Detrimental for the Anti-Multiple Myeloma Activity of Activated Natural Killer Cells in an in Vitro System Mimicking the Tumor Microenvironment. Front. Immunol. 2018, 9, 1415. [Google Scholar] [CrossRef]
- Griffin, B.D.; Gram, A.M.; Mulder, A.; Van Leeuwen, D.; Claas, F.H.J.; Wang, F.; Ressing, M.E.; Wiertz, E. EBV BILF1 Evolved to Downregulate Cell Surface Display of a Wide Range of HLA Class I Molecules through their Cytoplasmic Tail. J. Immunol. 2013, 190, 1672–1684. [Google Scholar] [CrossRef] [Green Version]
- Ulbrecht, M.; Modrow, S.; Srivastava, R.; Peterson, P.A.; Weiss, E.H. Interaction of HLA-E with Peptides and the Peptide Transporter in Vitro: Implications for its Function in Antigen Presentation. J. Immunol. 1998, 160, 4375–4385. [Google Scholar] [CrossRef]
- Mbiribindi, B.; Pena, J.K.; Arvedson, M.P.; Moreno Romero, C.; McCarthy, S.R.; Hatton, O.L.; Esquivel, C.O.; Martinez, O.M.; Krams, S.M. Epstein-Barr Virus Peptides Derived from Latent Cycle Proteins Alter NKG2A + NK Cell Effector Function. Sci. Rep. 2020, 10, 19973. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D Receptor and its Ligands in Host Defense. Cancer. Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, A.; Shibuya, K. DNAM-1 Versus TIGIT: Competitive Roles in Tumor Immunity and Inflammatory Responses. Int. Immunol. 2021, 33, 687–692. [Google Scholar] [CrossRef]
- Baugh, R.; Khalique, H.; Seymour, L.W. Convergent Evolution by Cancer and Viruses in Evading the NKG2D Immune Response. Cancers 2020, 12, 3827. [Google Scholar] [CrossRef]
- Cifaldi, L.; Doria, M.; Cotugno, N.; Zicari, S.; Cancrini, C.; Palma, P.; Rossi, P. DNAM-1 Activating Receptor and its Ligands: How do Viruses Affect the NK Cell-Mediated Immune Surveillance during the various Phases of Infection? Int. J. Mol. Sci. 2019, 20, 3715. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major Histocompatibility Complex Class I-Related Chain A and UL16-Binding Protein Expression on Tumor Cell Lines of Different Histotypes: Analysis of Tumor Susceptibility to NKG2D-Dependent Natural Killer Cell Cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse Herpesvirus microRNAs Target the Stress-Induced Immune Ligand MICB to Escape Recognition by Natural Killer Cells. Cell. Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Rancan, C.; Schirrmann, L.; Hüls, C.; Zeidler, R.; Moosmann, A. Latent Membrane Protein LMP2A Impairs Recognition of EBV-Infected Cells by CD8+ T Cells. PLoS Pathog. 2015, 11, e1004906. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cao, W.; Xi, X.; Ma, C.; Cui, L.; He, W. The NKG2D Ligand ULBP4 Binds to TCRgamma9/delta2 and Induces Cytotoxicity to Tumor Cells through both TCRgammadelta and NKG2D. Blood 2009, 114, 310–317. [Google Scholar] [CrossRef]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Hömig-Hölzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune Surveillance and Therapy of Lymphomas Driven by Epstein-Barr Virus Protein LMP1 in a Mouse Model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Banerjee, S. Downregulation of HLA-ABC Expression through Promoter Hypermethylation and Downmodulation of MIC-A/B Surface Expression in LMP2A-Positive Epithelial Carcinoma Cell Lines. Sci. Rep. 2020, 10, 5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westhoff Smith, D.; Chakravorty, A.; Hayes, M.; Hammerschmidt, W.; Sugden, B. The Epstein-Barr Virus Oncogene EBNA1 Suppresses Natural Killer Cell Responses and Apoptosis Early After Infection of Peripheral B Cells. mBio 2021, 12, e02243-21. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Chen, S.; Zhang, M.; Chan, J.Y.; Gao, W. Epstein-Barr Virus-Encoded microRNA BART7 Downregulates Major Histocompatibility Complex Class I Chain-Related Peptide A and Reduces the Cytotoxicity of Natural Killer Cells to Nasopharyngeal Carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef] [Green Version]
- Cerboni, C.; Fionda, C.; Soriani, A.; Zingoni, A.; Doria, M.; Cippitelli, M.; Santoni, A. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front. Immunol. 2014, 4, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Montañés, M.; Alari-Pahissa, E.; Sintes, J.; Martínez-Rodríguez, J.E.; Muntasell, A.; López-Botet, M. Antibody-Dependent NK Cell Activation Differentially Targets EBV-Infected Cells in Lytic Cycle and Bystander B Lymphocytes Bound to Viral Antigen-Containing Particles. J. Immunol. 2017, 199, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsten, C.B.; Bartsch, Y.C.; Shin, S.A.; Slein, M.D.; Heller, H.M.; Kolandaivelu, K.; Middeldorp, J.M.; Alter, G.; Julg, B. Evolution of Functional Antibodies Following Acute Epstein-Barr Virus Infection. PLoS Pathog. 2022, 18, e1010738. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel Therapeutics for Epstein⁻Barr Virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef] [Green Version]
- van Zyl, D.G.; Mautner, J.; Delecluse, H. Progress in EBV Vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr Virus-Related Post-Transplant Lymphoproliferative Disease (EBV-PTLD) in the Setting of Allogeneic Stem Cell Transplantation: A Comprehensive Review from Pathogenesis to Forthcoming Treatment Modalities. Bone Marrow Transpl. 2020, 55, 25–39. [Google Scholar] [CrossRef]
- Kim, S.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr Virus–Positive and –Negative Relapsed Or Refractory Non-Hodgkin Lymphomas. Cancer. Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Pan, X.; Chen, C.; Niu, T.; Shuai, X.; Wang, J.; Chen, X.; Liu, J.; Guo, Y.; Xie, L.; et al. Nivolumab Treatment of relapsed/refractory Epstein-Barr Virus-Associated Hemophagocytic Lymphohistiocytosis in Adults. Blood 2020, 135, 826–833. [Google Scholar] [CrossRef]
- Smith, C.; McGrath, M.; Neller, M.A.; Matthews, K.K.; Crooks, P.; Le Texier, L.; Panizza, B.; Porceddu, S.; Khanna, R. Complete Response to PD-1 Blockade Following EBV-Specific T-Cell Therapy in Metastatic Nasopharyngeal Carcinoma. NPJ Precis. Oncol. 2021, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.M.; Xu, M.L.; Yu, H.; Fletcher, C.D.M.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 Expression is Characteristic of a Subset of Aggressive B-Cell Lymphomas and Virus-Associated Malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, L.P.; Bollard, C.M.; Keller, M.D. Adoptive T Cell Therapy for Epstein-Barr Virus Complications in Patients with Primary Immunodeficiency Disorders. Front. Immunol. 2018, 9, 556. [Google Scholar] [CrossRef]
- Li, W.; Duan, X.; Chen, X.; Zhan, M.; Peng, H.; Meng, Y.; Li, X.; Li, X.; Pang, G.; Dou, X. Immunotherapeutic Approaches in EBV-Associated Nasopharyngeal Carcinoma. Front. Immunol. 2023, 13, 1079515. [Google Scholar] [CrossRef]
- Lupo, K.B.; Matosevic, S. Natural Killer Cells as Allogeneic Effectors in Adoptive Cancer Immunotherapy. Cancers (Basel) 2019, 11, 769. [Google Scholar] [CrossRef] [Green Version]
- Heipertz, E.L.; Zynda, E.R.; Stav-Noraas, T.E.; Hungler, A.D.; Boucher, S.E.; Kaur, N.; Vemuri, M.C. Current Perspectives on “Off-the-Shelf” Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021, 12, 732135. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wolterink, K.; Roel, G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric Antigen Receptor Natural Killer (CAR-NK) Cell Design and Engineering for Cancer Therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Lim, C.M.; Liou, A.; Poon, M.; Koh, L.P.; Tan, L.K.; Loh, K.S.; Petersson, B.F.; Ting, E.; Campana, D.; Goh, B.C.; et al. Phase I Study of Expanded Natural Killer Cells in Combination with Cetuximab for recurrent/metastatic Nasopharyngeal Carcinoma. Cancer Immunol. Immunother. 2022, 71, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Dorothea, M.; Hui, K.F.; Chiang, A.K.S. Lytic Induction Therapy Against Epstein-Barr Virus-Associated Malignancies: Past, Present, and Future. Cancers 2020, 12, 2142. [Google Scholar] [CrossRef]
- Cifaldi, L.; Locatelli, F.; Marasco, E.; Moretta, L.; Pistoia, V. Boosting Natural Killer Cell-Based Immunotherapy with Anticancer Drugs: A Perspective. Trends Mol. Med. 2017, 23, 1156–1175. [Google Scholar] [CrossRef] [PubMed]


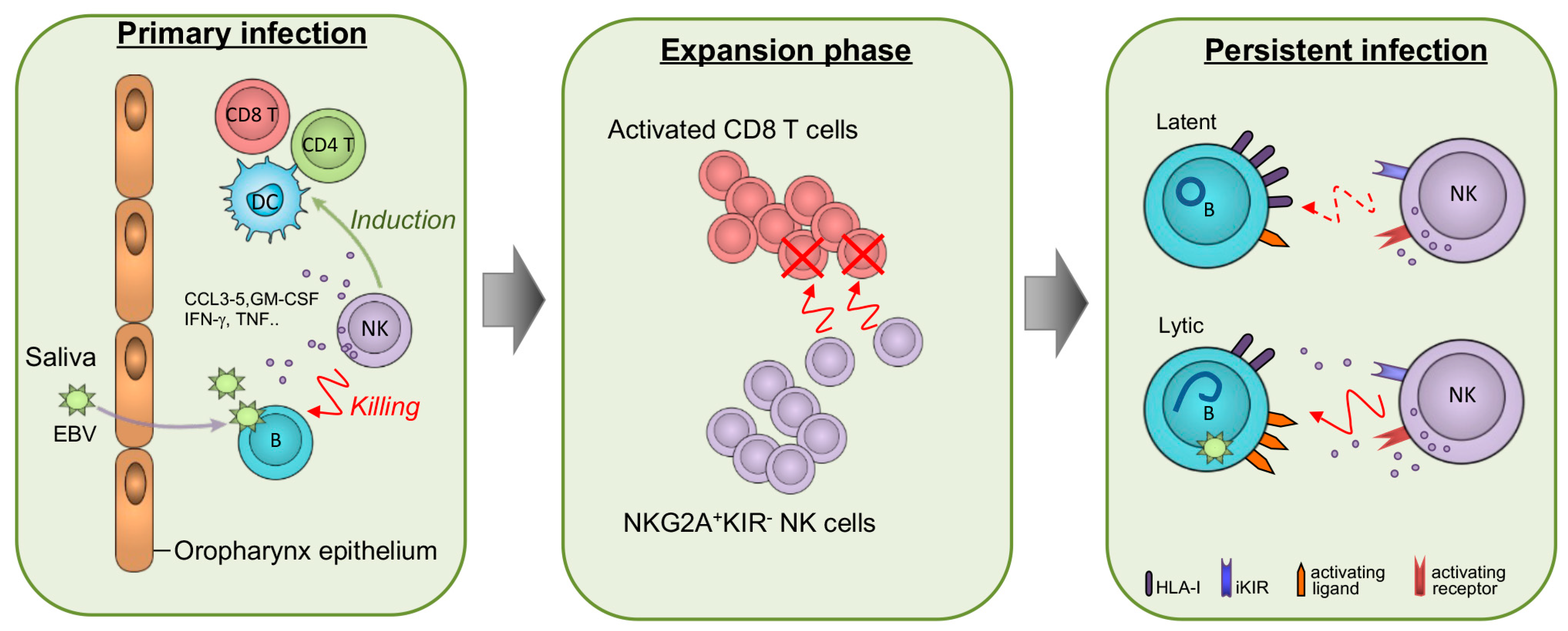{kind=link}
| B-cell target |
|---|
| Chronic active EBV (CAEBV) of B cells |
| Burkitt’s lymphoma (BL) |
| Hodgkin’s lymphoma (HL) |
| Diffuse large B-cell lymphoma (DLBCL) |
| Plasmablastic lymphoma |
| Lymphomatoid granulomatosis |
| Post-transplant lymphoproliferative disorders (PTLDs) |
| NK or T-cell target |
| CAEBV of NK or T cells |
| NK or T-cell lymphoma |
| Epithelial cell target |
| Nasopharyngeal carcinoma (NPC) |
| Gastric carcinoma (GC) |
| Smooth muscle cell target |
| Leiomyosarcoma |
| Cell System | EBV Effect on Activating Ligands for NK Cells | Impact on Immune Killing | Ref. |
|---|---|---|---|
| Daudi, Raji (EBV+ BL) | High ULBP1 expression (not MICA, ULBP2-3) in Daudi but not in Raji cells | Killing of Daudi but not Raji cells by activated primary NK cells was mediated by NKG2D/ULBP1 interaction (reduced by ULBP1 or NKG2D blocking Ab) | [96] |
| AKBM (EBV+ BL) | CD48 but not NKG2DL or DNAM-1L were expressed; ULBP1 and CD112 were induced upon lytic cycle induction (while CD48 was maintained) | Killing by NK cells (activated primary NK, NKL, and DEL NK) was low against latent AKBM (1–20%) but high against lytic AKBM (20–60%); NKG2D and DNAM-1 contributed to NKL lysis of lytic AKBM (reduced by ULBP1 or CD112 blocking Ab) | [80] |
| AKBM, LCL | Not tested in AKBM LCLs expressed MICA and CD48; upon lytic cycle activation, MICA and CD48 were up-modulated and CD155 and CD112 were induced | Cytotoxic degranulation of CD56dimNKG2A+KIR− subset of activated NK cells was low against latent AKBM and LCLs but high against lytic AKBM | [58] |
| AKBM, LCL | Not tested in AKBM LCLs, either latent or lytic cycle-induced, did not express MICA, MICB, ULBP2, CD112, and CD155 | NKL-mediated lysis was low/absent against latent (BZLF1−BcLF1−) or late lytic (BZLF1+BcLF1+) but high against early lytic (BZLF1+BcLF1−) AKBM or LCL cells; NKG2D and, to a smaller extent, DNAM-1 (not NKp46) contributed to killing of lytic AKBM while killing of lytic LCL was mediated by DNAM-1 (not NKG2D or NKp46 by blocking with receptor-specific Ab) | [83] |
| LCL | High CD48 expression | Lysis by autologous NK cells was low (5%) in part involving NKG2D but not 2B4 or NKG2A (as determined by Ab block); CD56dimNKG2A+ cells were more cytotoxic than CD56dimNKG2A− cells | [67] |
| LCL (721.221) | High MICB expression (despite translational repression by miR-BART2-5p), low MICA levels | NK cell lysis was enhanced upon MICB up-modulation by transduction with anti-miR-BART2-5p ‘sponge’ | [97] |
| LCL | High MICB expression, low MICA and ULBP4 levels because of LMP2A-mediated downregulation | NKG2D contributed to recognition by EBV-specific CD8 T cells (in part reduced via NKG2D Ab block); NK lysis not tested | [98] |
| LCL from XMEN patients | Unspecified NKG2DL expression (NKG2D-Fc staining) | Impaired killing by autologous NK or CD8 T cells unless NKG2D expression was restored | [46] |
| Murine LMP1+ B-cell lymphoma | Rae-1 (murine ULBP ortholog) was induced | NKG2D contributed to NK cell lysis (in part reduced via NKG2D Ab block) of LMP1+ lymphoma cells in vitro; treatment with NKG2D-Fc reduced tumor growth in transgenic LMP1+ mice | [100] |
| EBV+ PTLD | Unspecified NKG2DL expression (NKG2D-Fc staining) | Not tested | [100] |
| B cells infected with EBV | ULBP4 was induced by EBV infection | ULBP4 mediated killing by γδ T cells (halved by ULBP4 Ab block); NK lysis not tested | [99] |
| B cells infected with EBV | ULBP1 and ULBP5 mRNA levels were reduced by EBNA1 | NK cells killed more efficiently and in an NKG2D-dependent manner (reduced via NKG2D Ab block) targets infected with EBNA1-deficient EBV as compared with wt virus | [102] |
| BZLF1+ DG75 (EBV− BL) | BZLF1 expression in DG75 cells induced expression of ULBP2 at the transcriptional level (not MICA, MICB, or CD155) | NKL cells killed efficiently BZLF1+ULBP2+ DG75 but not control DG75 cells | [83] |
| miR-BART7+ EBV− NPC | MICA downregulated via translational repression by miR-BART7 | Lysis by the NK92 NK cell line against miR-BART7+ cells was reduced as compared to untreated cells | [103] |
| LMP2+ EBV− GC | LMP2 expression down-modulated MICA and MICB despite increased mRNA levels | NK lysis not tested | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desimio, M.G.; Covino, D.A.; Rivalta, B.; Cancrini, C.; Doria, M. The Role of NK Cells in EBV Infection and Related Diseases: Current Understanding and Hints for Novel Therapies. Cancers 2023, 15, 1914. https://doi.org/10.3390/cancers15061914
Desimio MG, Covino DA, Rivalta B, Cancrini C, Doria M. The Role of NK Cells in EBV Infection and Related Diseases: Current Understanding and Hints for Novel Therapies. Cancers. 2023; 15(6):1914. https://doi.org/10.3390/cancers15061914
Chicago/Turabian StyleDesimio, Maria G., Daniela A. Covino, Beatrice Rivalta, Caterina Cancrini, and Margherita Doria. 2023. "The Role of NK Cells in EBV Infection and Related Diseases: Current Understanding and Hints for Novel Therapies" Cancers 15, no. 6: 1914. https://doi.org/10.3390/cancers15061914
APA StyleDesimio, M. G., Covino, D. A., Rivalta, B., Cancrini, C., & Doria, M. (2023). The Role of NK Cells in EBV Infection and Related Diseases: Current Understanding and Hints for Novel Therapies. Cancers, 15(6), 1914. https://doi.org/10.3390/cancers15061914