

Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Procurement
2.2. Patient Characteristics
2.3. Specimen Processing
2.4. Lipid Extraction
2.5. Internal Standards
2.6. Retention-Time Standards
2.7. Mass Spectrometry Analysis
2.8. Statistical Analysis
3. Results
3.1. Self-Identified Race-Independent and Disease-Specific Analysis of Sphingolipid Metabolism Alterations
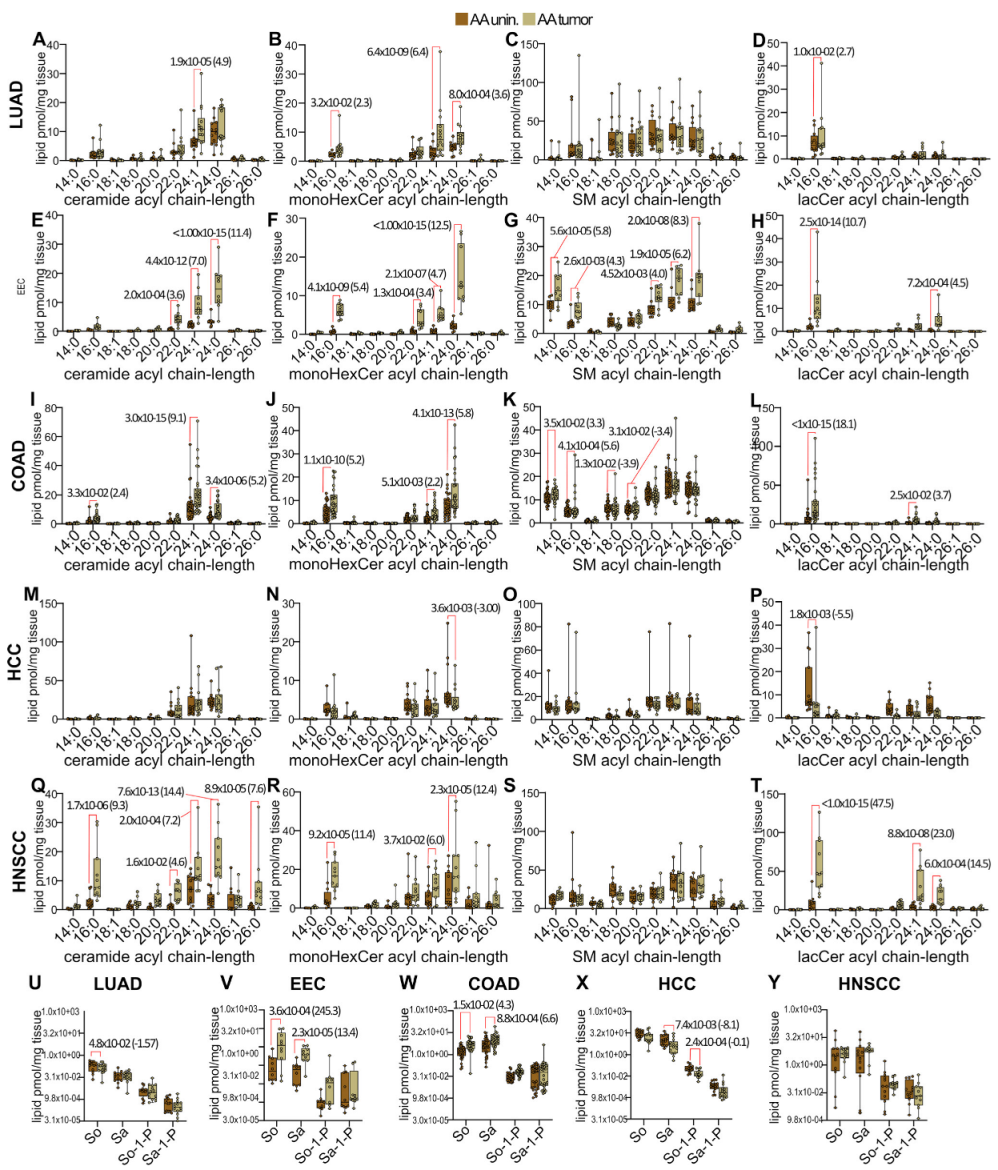
3.2. Disease-Specific Analysis of Sphingolipid Metabolism Alterations in Self-Identified AA Males and Females
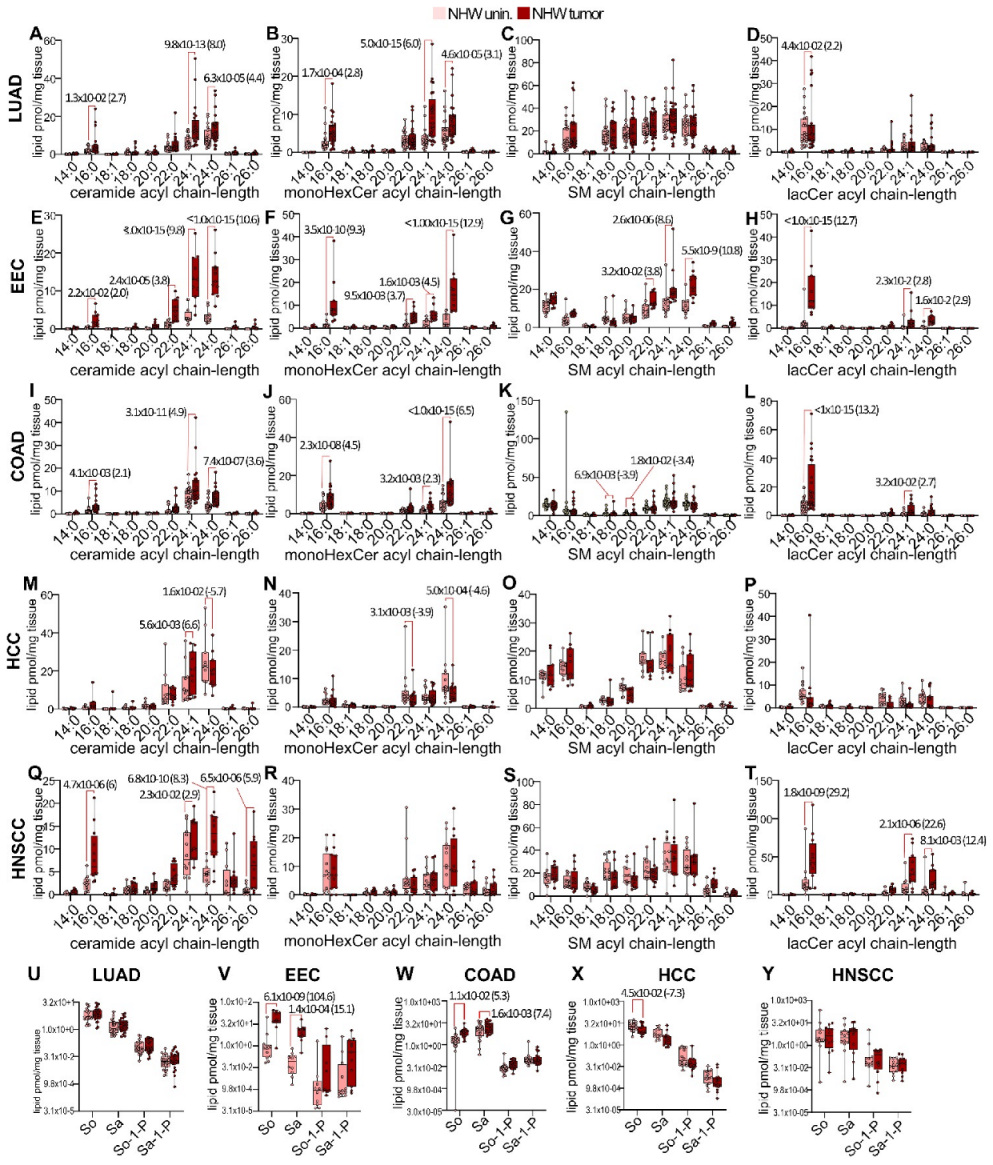
3.3. Disease-Specific Analysis of Sphingolipid Metabolism Alterations in Self-Identified NHW Males and Females
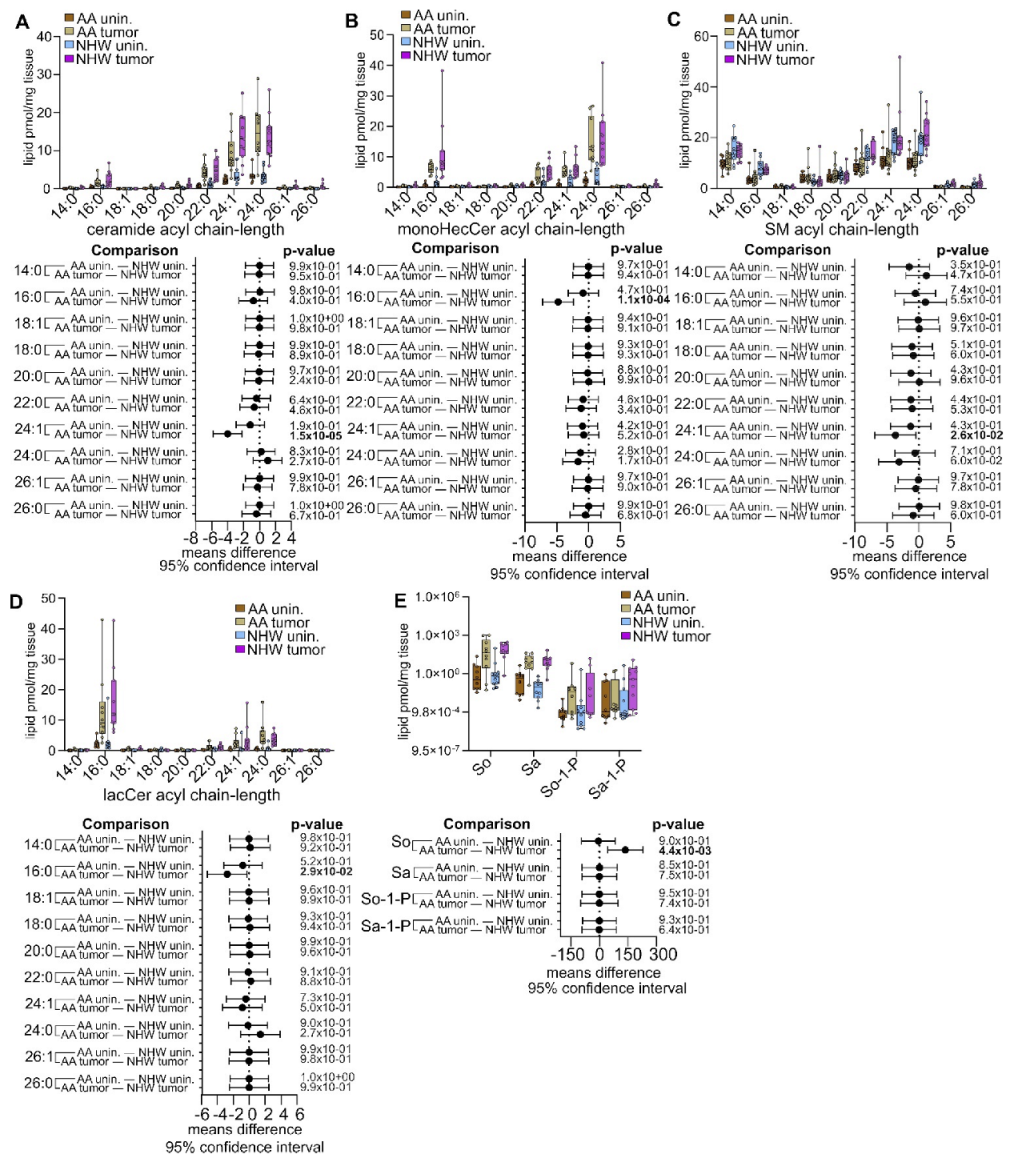
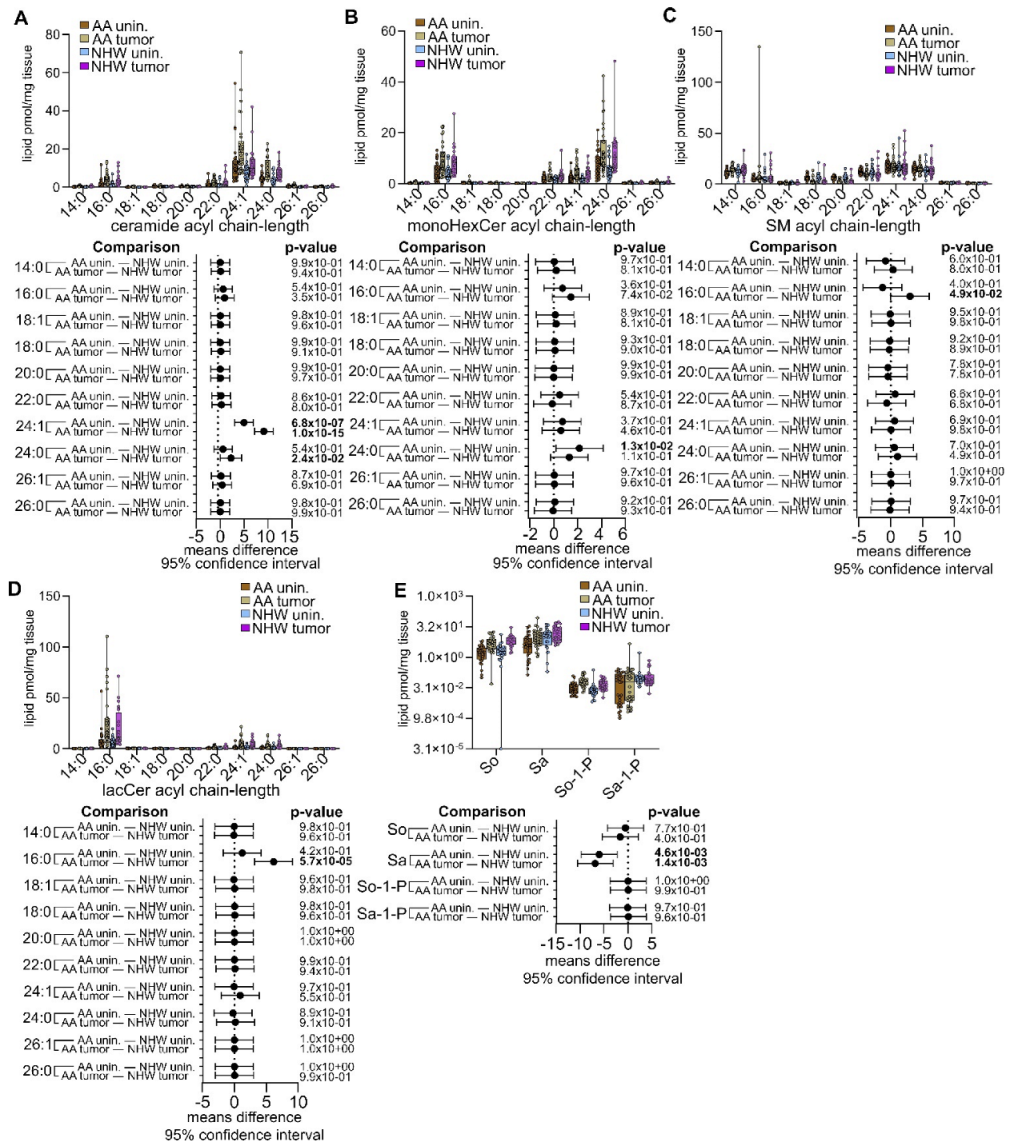
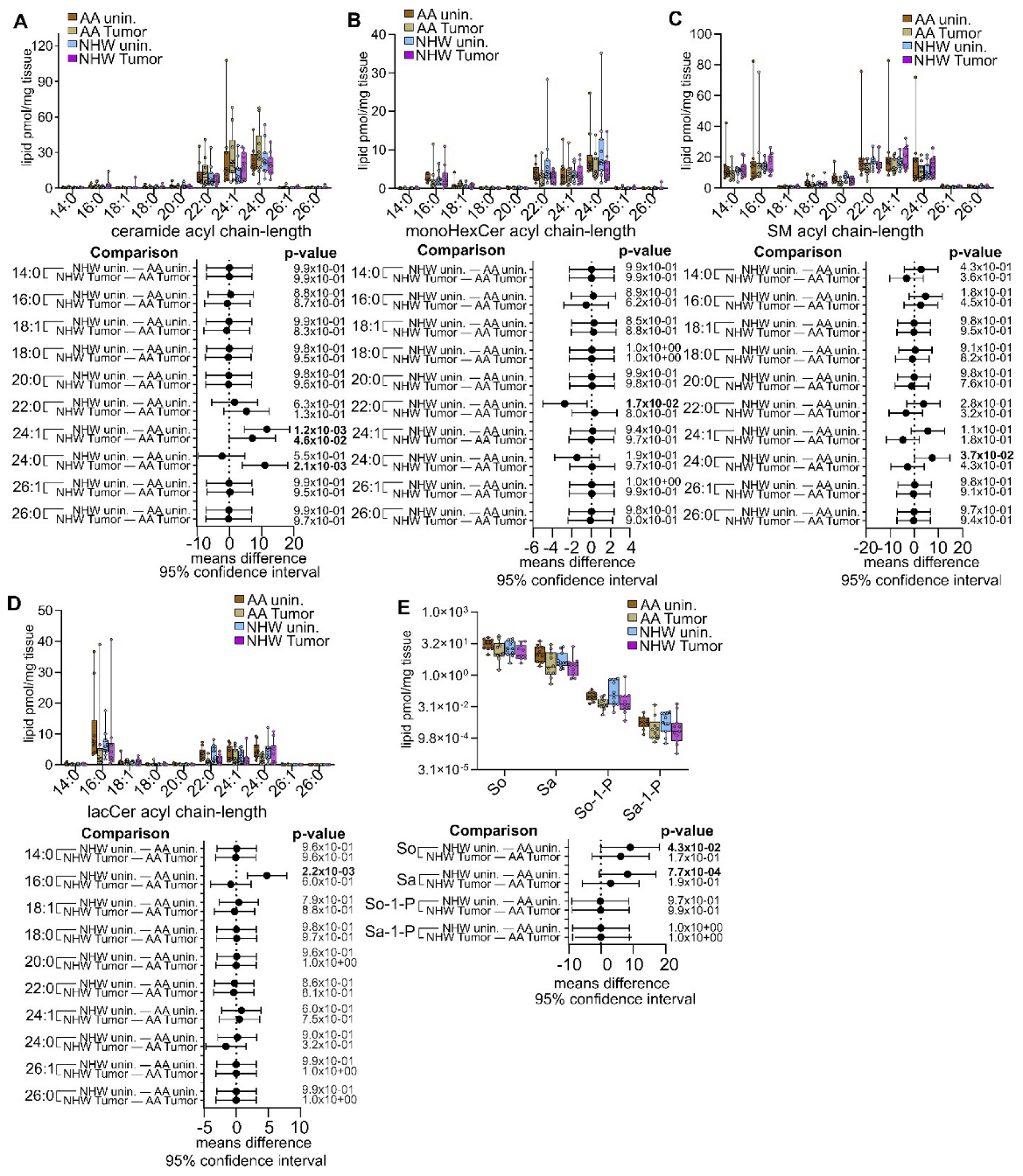
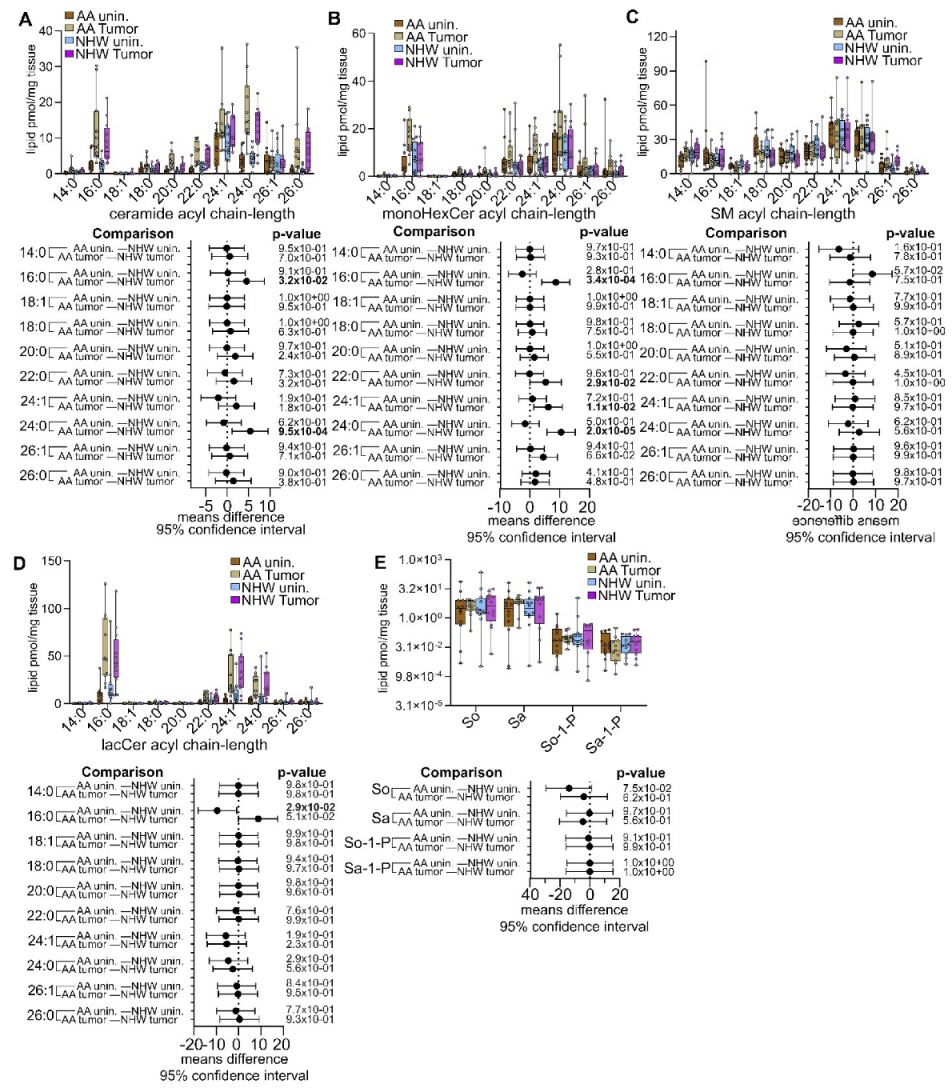
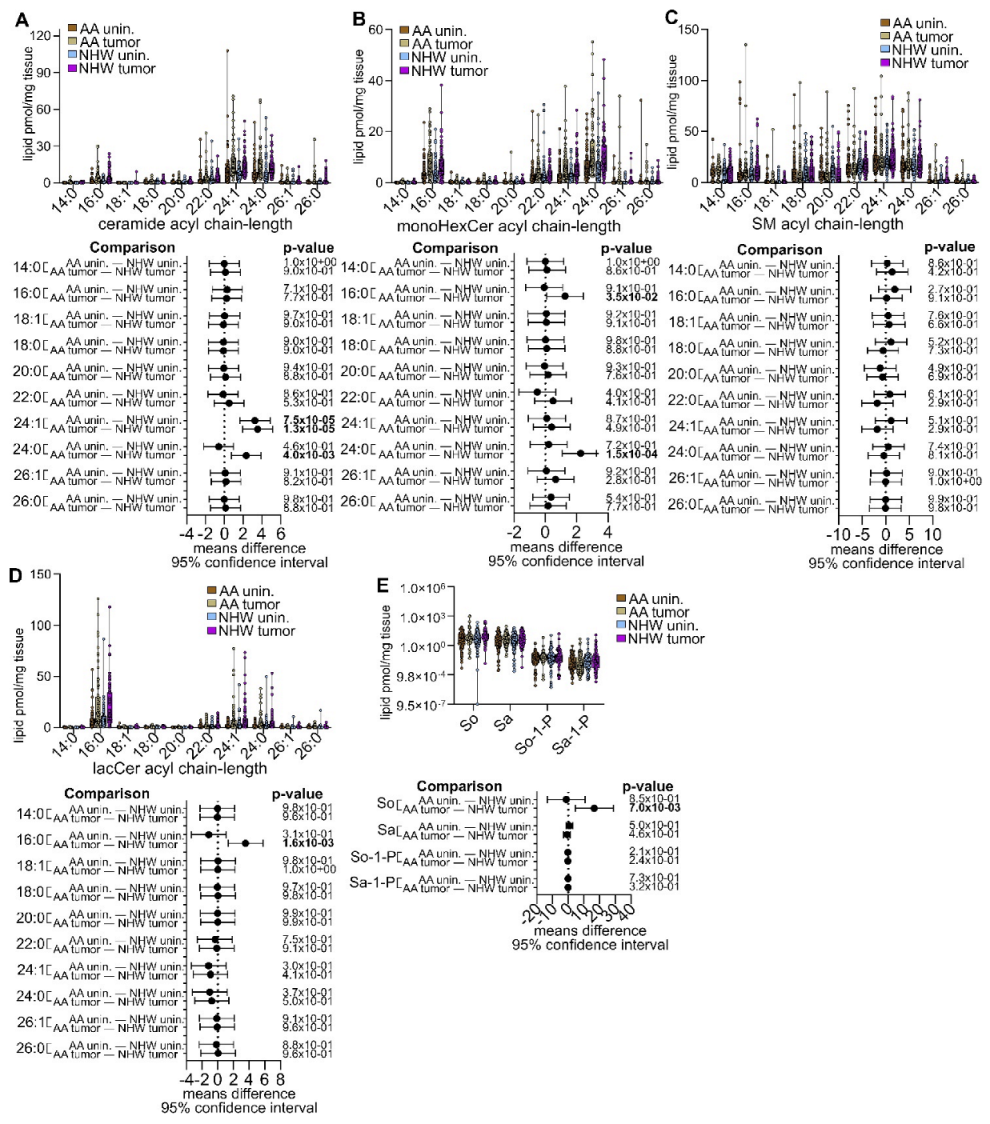
3.4. Disease-Specific Comparisons between Self-Identified AA and NHW with Cancer
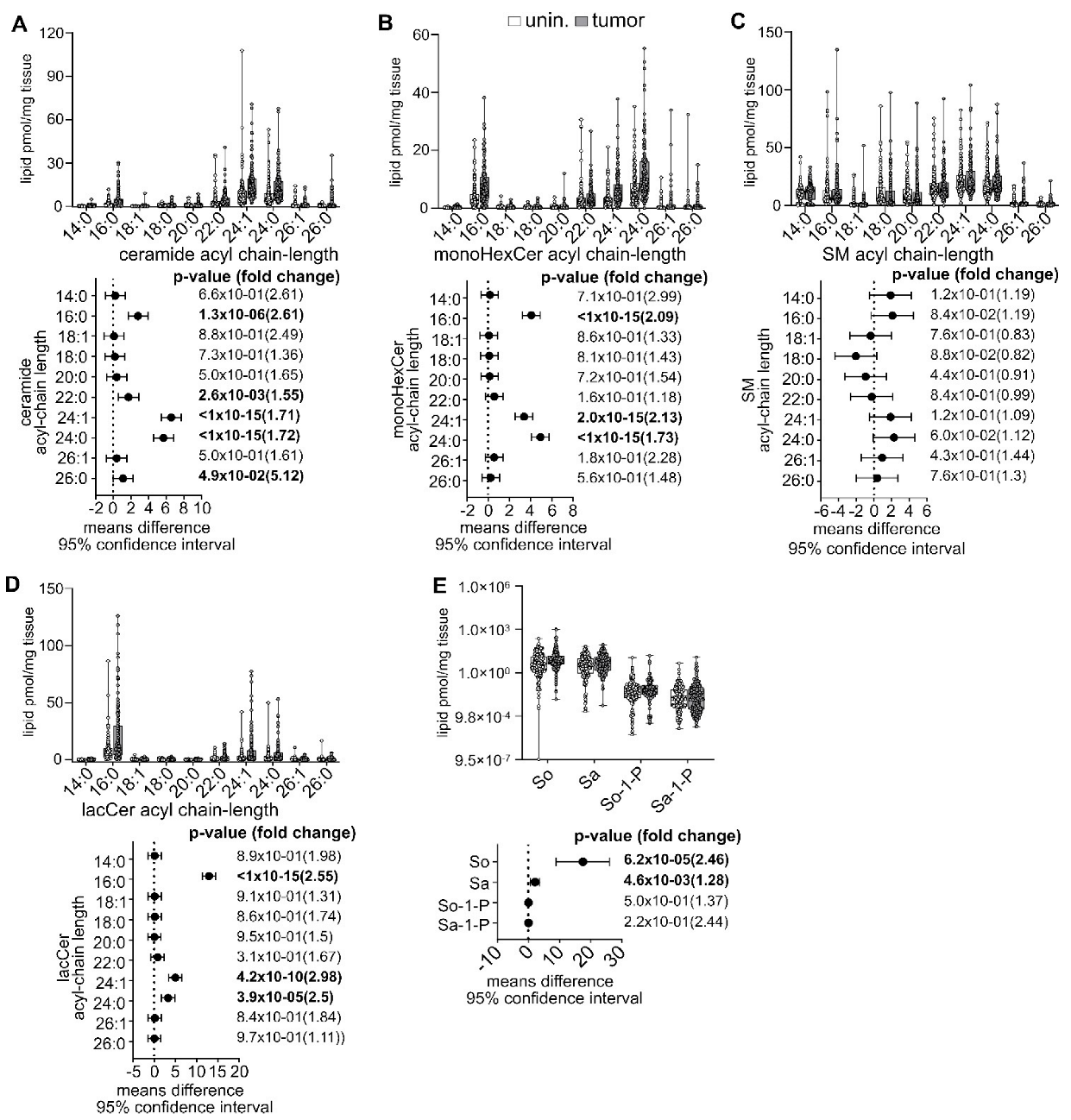
3.5. Pan-Cancer Analysis of Sphingolipid Metabolism Reprogramming
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell Dev. Biol. 2016, 4, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, D.K.; Weissman, J.S. Membranes in Balance: Mechanisms of Sphingolipid Homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, S.; Milstien, S.; Spiegel, S. Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking. J. Biol. Chem. 2017, 292, 3074–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.-G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.M.; Takahashi, Y.; Fox, T.E.; Yun, J.K.; Kester, M.; Wang, H.-G. Sphingosine Kinase 1 Cooperates with Autophagy to Maintain Endocytic Membrane Trafficking. Cell Rep. 2016, 17, 1532–1545. [Google Scholar] [CrossRef] [Green Version]
- Young, M.M.; Wang, H.-G. Sphingolipids as Regulators of Autophagy and Endocytic Trafficking. Adv. Cancer Res. 2018, 140, 27–60. [Google Scholar] [CrossRef]
- Shen, H.; Giordano, F.; Wu, Y.; Chan, J.; Zhu, C.; Milosevic, I.; Wu, X.; Yao, K.; Chen, B.; Baumgart, T.; et al. Coupling between endocytosis and sphingosine kinase 1 recruitment. Nature 2014, 16, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Russo, D.; Parashuraman, S.; D’Angelo, G. Glycosphingolipid–Protein Interaction in Signal Transduction. Int. J. Mol. Sci. 2016, 17, 1732. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, S.; Sandner, J.; Birod, K.; Wobst, I.; Angioni, C.; Ruckhäberle, E.; Kaufmann, M.; Ackermann, H.; Lötsch, J.; Schmidt, H.; et al. Ceramide Synthases and Ceramide Levels Are Increased in Breast Cancer Tissue. Carcinogenesis 2009, 30, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Moro, K.; Kawaguchi, T.; Tsuchida, J.; Gabriel, E.; Qi, Q.; Yan, L.; Wakai, T.; Takabe, K.; Nagahashi, M. Ceramide species are elevated in human breast cancer and are associated with less aggressiveness. Oncotarget 2018, 9, 19874–19890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Tsuchida, J.; Moro, K.; Hasegawa, M.; Tatsuda, K.; Woelfel, I.A.; Takabe, K.; Wakai, T. High levels of sphingolipids in human breast cancer. J. Surg. Res. 2016, 204, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadwal, P.; Dahiya, D.; Shinde, D.; Vaiphei, K.; Math, R.G.H.; Randhawa, V.; Agnihotri, N. LC-HRMS based approach to identify novel sphingolipid biomarkers in breast cancer patients. Sci. Rep. 2020, 10, 4668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakiet, A.; Sikora, K.; Kobiela, J.; Rostkowska, O.; Mika, A.; Sledzinski, T. Alterations in complex lipids in tumor tissue of patients with colorectal cancer. Lipids Health Dis. 2021, 20, 85. [Google Scholar] [CrossRef]
- Rohrbach, T.D.; Boyd, A.E.; Grizzard, P.J.; Spiegel, S.; Allegood, J.; Lima, S. A simple method for sphingolipid analysis of tissues embedded in optimal cutting temperature compound. J. Lipid Res. 2020, 61, 953–967. [Google Scholar] [CrossRef]
- Boyd, A.E.; Allegood, J.; Lima, S. Preparation of Human Tissues Embedded in Optimal Cutting Temperature Compound for Mass Spectrometry Analysis. J. Vis. Exp. 2021, 170, e62552. [Google Scholar]
- Karahatay, S.; Thomas, K.; Koybasi, S.; Senkal, C.E.; ElOjeimy, S.; Liu, X.; Bielawski, J.; Day, T.A.; Gillespie, M.B.; Sinha, D.; et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): Attenuation of C18-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007, 256, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Knapp, P.; Baranowski, M.; Knapp, M.; Zabielski, P.; Błachnio-Zabielska, A.U.; Górski, J. Altered sphingolipid metabolism in human endometrial cancer. Prostaglandins Other Lipid Mediat. 2010, 92, 62–66. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.B.; Hou, J.; Bandaru, V.V.R.; Pezhouh, M.K.; Mannan, A.A.S.R.; Sharma, R. Lactosylceramide Synthase Β-1,4-Galt-V: A Novel Target for the Diagnosis and Therapy of Human Colorectal Cancer. Biochem. Biophys. Res. Commun. 2019, 508, 380–386. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2017, 18, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.Z.; Wang, X.R.; Wang, J.; Xie, C.; Wang, X.X.; Pan, H.D.; Meng, W.Y.; Liang, T.L.; Li, J.X.; Yan, P.Y.; et al. The Key Role of Sphingolipid Metabolism in Cancer: New Therapeutic Targets, Diagnostic and Prognostic Values, and Anti-Tumor Immunotherapy Resistance. Front. Oncol. 2022, 12, 941643. [Google Scholar] [CrossRef]
- Cronin, K.A.; Scott, S.; Firth, A.U.; Sung, H.; Henley, S.J.; Sherman, R.L.; Siegel, R.L.; Anderson, R.N.; Kohler, B.A.; Benard, V.B.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. Cancer 2022, 128, 4251–4284. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, S.; Burke, W.; Sheth, S.S.; Brooks, R.; Cavens, A.; Huber-Keener, K.; Scott, D.M.; Worly, B.; Chelmow, D. Health Disparities in Uterine Cancer: Report from the Uterine Cancer Evidence Review Conference. Obstet. Gynecol. 2022, 139, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Doll, K.M.; Khor, S.; Odem-Davis, K.; He, H.; Wolff, E.M.; Flum, D.R.; Ramsey, S.D.; Goff, B.A. Role of bleeding recognition and evaluation in Black-White disparities in endometrial cancer. Am. J. Obstet. Gynecol. 2018, 219, 593.e1–593.e14. [Google Scholar] [CrossRef]
- Williams, P.A.; Zaidi, S.K.; Sengupta, R. AACR Cancer Disparities Progress Report 2022. Cancer Epidemiol. Biomark. Prev. 2022, 31, 1249–1250. [Google Scholar] [CrossRef]
- Cancer Facts & Figures for African American/Black People 2022–2024; American Cancer Society: Atlanta, GA, USA, 2022.
- Zavala, V.A.; Bracci, P.M.; Carethers, J.M.; Carvajal-Carmona, L.; Coggins, N.B.; Cruz-Correa, M.R.; Davis, M.; de Smith, A.J.; Dutil, J.; Figueiredo, J.C.; et al. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer 2021, 124, 315–332. [Google Scholar] [CrossRef]
- Fatumo, S.; Chikowore, T.; Choudhury, A.; Ayub, M.; Martin, A.R.; Kuchenbaecker, K. A roadmap to increase diversity in genomic studies. Nat. Med. 2022, 28, 243–250. [Google Scholar] [CrossRef]
- Nazha, B.; Mishra, M.; Pentz, R.; Owonikoko, T.K. Enrollment of Racial Minorities in Clinical Trials: Old Problem Assumes New Urgency in the Age of Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 3–10. [Google Scholar] [CrossRef]
- Borrell, L.N.; Elhawary, J.R.; Fuentes-Afflick, E.; Witonsky, J.; Bhakta, N.; Wu, A.H.B.; Bibbins-Domingo, K.; Rodríguez-Santana, J.R.; Lenoir, M.A.; Gavin, J.R., 3rd; et al. Race and Genetic Ancestry in Medicine—A Time for Reckoning with Racism. N. Engl. J. Med. 2021, 384, 474–480. [Google Scholar] [CrossRef]
- Shaner, R.L.; Allegood, J.C.; Park, H.; Wang, E.; Kelly, S.; Haynes, C.A.; Sullards, M.C.; Merrill, A.H., Jr. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 2009, 50, 1692–1707. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, B.; Sun, Z. Spectrum of EGFR aberrations and potential clinical implications: Insights from integrative pan-cancer analysis. Cancer Commun. 2020, 40, 43–59. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zhang, Y.; Gibbons, D.L.; Deneen, B.; Kwiatkowski, D.J.; Ittmann, M.; Creighton, C.J. Pan-Cancer Molecular Classes Transcending Tumor Lineage across 32 Cancer Types, Multiple Data Platforms, and over 10,000 Cases. Clin. Cancer Res. 2018, 24, 2182–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Mitchell, K.A.; Zingone, A.; Bowman, E.; Sinha, N.; Schäffer, A.A.; Lee, J.S.; Ruppin, E.; Ryan, B.M. Higher prevalence of homologous recombination deficiency in tumors from African Americans versus European Americans. Nat. Cancer 2020, 1, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.C.; Adamson, A.W.; Steele, L.; Bailis, A.M.; John, E.M.; Tomlinson, G.; Neuhausen, S.L. Discovery of mutations in homologous recombination genes in African-American women with breast cancer. Fam. Cancer 2018, 17, 187–195. [Google Scholar] [CrossRef]
- Deveaux, A.E.; Allen, T.A.; Al Abo, M.; Qin, X.; Zhang, D.; Patierno, B.M.; Gu, L.; Gray, J.E.; Pecot, C.V.; Dressman, H.K.; et al. RNA splicing and aggregate gene expression differences in lung squamous cell carcinoma between patients of West African and European ancestry. Lung Cancer 2021, 153, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Anbalagan, M.; Baddoo, M.; Chellamuthu, V.K.; Mukhopadhyay, S.; Woods, C.; Jiang, W.; Moroz, K.; Flemington, E.K.; Makridakis, N. Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 2020, 39, 4299–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, K.A.; Nichols, N.; Tang, W.; Walling, J.; Stevenson, H.; Pineda, M.; Stefanescu, R.; Edelman, D.C.; Girvin, A.T.; Zingone, A.; et al. Recurrent PTPRT/JAK2 mutations in lung adenocarcinoma among African Americans. Nat. Commun. 2019, 10, 5735. [Google Scholar] [CrossRef] [Green Version]
- Bauml, J.; Mick, R.; Zhang, Y.; Watt, C.D.; Vachani, A.; Aggarwal, C.; Evans, T.; Langer, C. Frequency of EGFR and KRAS mutations in patients with non small cell lung cancer by racial background: Do disparities exist? Lung Cancer 2013, 81, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Steuer, C.E.; Behera, M.; Berry, L.; Kim, S.; Rossi, M.; Sica, G.; Owonikoko, T.K.; Johnson, B.E.; Kris, M.G.; Bunn, P.A.; et al. Role of race in oncogenic driver prevalence and outcomes in lung adenocarcinoma: Results from the Lung Cancer Mutation Consortium. Cancer 2016, 122, 766–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, S.; Kanteti, R.; Duke-Cohan, J.S.; Loganathan, S.; Liu, W.; Ma, P.C.; Sattler, M.; Singleton, P.A.; Ramnath, N.; Innocenti, F.; et al. Ethnic Differences and Functional Analysis of MET Mutations in Lung Cancer. Clin. Cancer Res. 2009, 15, 5714–5723. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Shen, X.J.; Kim, S.; Araujo-Perez, F.; Galanko, J.A.; Martin, C.F.; Sandler, R.S.; Keku, T.O. Somatic gene mutations in African Americans may predict worse outcomes in colorectal cancer. Cancer Biomark. 2013, 13, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Patel, Y.M.; Park, S.L.; Carmella, S.G.; Paiano, V.; Olvera, N.; Stram, D.O.; Haiman, C.A.; Le Marchand, L.; Hecht, S.S. Metabolites of the Polycyclic Aromatic Hydrocarbon Phenanthrene in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer. PLoS ONE 2016, 11, e0156203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.E.; Park, S.-S.L.; Thompson, E.F.; Wilkens, L.R.; Patel, Y.; Stram, D.O.; Le Marchand, L. Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 2014, 35, 2526–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dator, R.; Villalta, P.W.; Thomson, N.; Jensen, J.; Hatsukami, D.K.; Stepanov, I.; Warth, B.; Balbo, S. Metabolomics Profiles of Smokers from Two Ethnic Groups with Differing Lung Cancer Risk. Chem. Res. Toxicol. 2020, 33, 2087–2098. [Google Scholar] [CrossRef]
- Ross, K.C.; Gubner, N.R.; Tyndale, R.F.; Hawk, L.W.; Lerman, C.; George, T.P.; Cinciripini, P.; Schnoll, R.A.; Benowitz, N.L. Racial differences in the relationship between rate of nicotine metabolism and nicotine intake from cigarette smoking. Pharmacol. Biochem. Behav. 2016, 148, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenoweth, M.J.; Ware, J.J.; Zhu, A.Z.X.; Cole, C.B.; Cox, L.S.; Nollen, N.; Ahluwalia, J.S.; Benowitz, N.L.; Schnoll, R.A.; Hawk, L.W.; et al. Genome-wide association study of a nicotine metabolism biomarker in African American smokers: Impact of chromosome 19 genetic influences. Addiction 2017, 113, 509–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.L.; Tiirikainen, M.I.; Patel, Y.M.; Wilkens, L.R.; Stram, D.O.; Le Marchand, L.; Murphy, S.E. Genetic determinants of CYP2A6 activity across racial/ethnic groups with different risks of lung cancer and effect on their smoking intensity. Carcinogenesis 2016, 37, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.V.; Smith, A.K.; Conneely, K.N.; Chang, Q.; Li, W.; Lazarus, A.; Smith, J.; Almli, L.; Binder, E.B.; Klengel, T.; et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum. Genet. 2013, 132, 1027–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.E.; Park, S.L.; Balbo, S.; Haiman, C.A.; Hatsukami, D.K.; Patel, Y.; Peterson, L.A.; Stepanov, I.; Stram, D.O.; Tretyakova, N.; et al. Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. npj Precis. Oncol. 2018, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Kanaan, Y.M.; Sampey, B.P.; Beyene, D.; Esnakula, A.K.; Naab, T.J.; Ricks-Santi, L.J.; Dasi, S.; Day, A.; Blackman, K.W.; Frederick, W.; et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genom.-Proteom. 2014, 11, 29–294. [Google Scholar]
- Vantaku, V.; Donepudi, S.R.; Piyarathna, D.W.B.; Amara, C.S.; Ambati, C.S.R.; Tang, W.; Putluri, V.; Chandrashekar, D.S.; Varambally, S.; Terris, M.K.; et al. Large-scale profiling of serum metabolites in African American and European American patients with bladder cancer reveals metabolic pathways associated with patient survival. Cancer 2019, 125, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Shen, J.; Moore, S.C.; Ye, Y.; Wu, X.; Esteva, F.J.; Tripathy, D.; Chow, W.H. Breast Cancer Risk in Relation to Plasma Metabolites among Hispanic and African American Women. Breast Cancer Res. Treat. 2019, 176, 687–696. [Google Scholar] [CrossRef]
- Shen, J.; Yan, L.; Liu, S.; Ambrosone, C.B.; Zhao, H. Plasma Metabolomic Profiles in Breast Cancer Patients and Healthy Controls: By Race and Tumor Receptor Subtypes. Transl. Oncol. 2013, 6, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Gohlke, J.H.; Lloyd, S.M.; Basu, S.; Putluri, V.; Vareed, S.K.; Rasaily, U.; Piyarathna, D.W.B.; Fuentes, H.; Rajendiran, T.M.; Dorsey, T.H.; et al. Methionine-Homocysteine Pathway in African-American Prostate Cancer. JNCI Cancer Spectr. 2019, 3, pkz019. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.P.; Haffner, S.M.; Baillargeon, J. Adiposity, the Metabolic Syndrome, and Breast Cancer in African-American and White American Women. Endocr. Rev. 2007, 28, 763–777. [Google Scholar] [CrossRef]
- Aminov, Z.; Haase, R.; Olson, J.R.; Pavuk, M.; Carpenter, D.O. Racial differences in levels of serum lipids and effects of exposure to persistent organic pollutants on lipid levels in residents of Anniston, Alabama. Environ. Int. 2014, 73, 216–223. [Google Scholar] [CrossRef]
- Zhou, X.; Mei, H.; Agee, J.; Brown, T.; Mao, J. Racial differences in distribution of fatty acids in prostate cancer and benign prostatic tissues. Lipids Health Dis. 2019, 18, 189. [Google Scholar] [CrossRef] [Green Version]
- Purwaha, P.; Gu, F.; Piyarathna, D.W.B.; Rajendiran, T.; Ravindran, A.; Omilian, A.R.; Jiralerspong, S.; Das, G.; Morrison, C.; Ambrosone, C.; et al. Unbiased Lipidomic Profiling of Triple-Negative Breast Cancer Tissues Reveals the Association of Sphingomyelin Levels with Patient Disease-Free Survival. Metabolites 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Q.W.C.; Zheng, X.; Ali, Y. Ceramide Acyl Chain Length and Its Relevance to Intracellular Lipid Regulation. Int. J. Mol. Sci. 2022, 23, 9697. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A.H.; Park, W.-J. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Hartmann, D.; Wegner, M.-S.; Wanger, R.A.; Ferreirós, N.; Schreiber, Y.; Lucks, J.; Schiffmann, S.; Geisslinger, G.; Grösch, S. The equilibrium between long and very long chain ceramides is important for the fate of the cell and can be influenced by co-expression of CerS. Int. J. Biochem. Cell Biol. 2013, 45, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Ho, Q.W.C.; Chua, M.; Stelmashenko, O.; Yeo, X.Y.; Muralidharan, S.; Torta, F.; Chew, E.G.Y.; Lian, M.M.; Foo, J.N.; et al. Destabilization of β Cell FIT2 by saturated fatty acids alter lipid droplet numbers and contribute to ER stress and diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2113074119. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, L.; Ubhayasekera, S.J.K.A.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-Induced Impairments of β-Cell Function Are Linked with Generation of Specific Ceramide Species via Acylation of Sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Doddapattar, P.; Radović, B.; Povoden, S.; Kolb, D.; Vujić, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 Ceramide Is Crucial for Triacylglycerol-Induced Apoptosis in Macrophages. Cell Death Dis. 2012, 3, e280. [Google Scholar] [CrossRef] [Green Version]
- Rudd, A.K.; Devaraj, N.K. Traceless synthesis of ceramides in living cells reveals saturation-dependent apoptotic effects. Proc. Natl. Acad. Sci. USA 2018, 115, 7485–7490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Jeffries, K.A.; Esmaeilniakooshkghazi, A.; Szulc, Z.M.; Knagge, K.J.; Kirchner, D.R.; Horita, D.A.; Krupenko, S.A.; Krupenko, N.I. C16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat. Commun. 2018, 9, 4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiban, J.; Perera, M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta 2015, 1848, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Cabot, M.C. Chapter Nine—The Onus of Sphingolipid Enzymes in Cancer Drug Resistance. In Advances in Cancer Research; Chalfant, C.E., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 235–263. [Google Scholar]
- Liu, J.C.; Egleston, B.L.; Blackman, E.; Ragin, C. Racial Survival Disparities in Head and Neck Cancer Clinical Trials. J. Natl. Cancer Inst. 2023, 115, 288–294. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, S.; Chen, C.; Yan, J.; Cai, M.; Zhu, X.; Gu, J. Over-expression of beta-1,4-galactosyltransferase V increases the growth of astrocytoma cell line. J. Exp. Clin. Cancer Res. 2002, 21, 409–414. [Google Scholar]
- Jeong, H.Y.; Park, S.; Kim, H.; Moon, S.; Lee, S.; Lee, S.H.; Kim, S. B3GNT5 is a novel marker correlated with stem-like phenotype and poor clinical outcome in human gliomas. CNS Neurosci. Ther. 2020, 26, 1147–1154. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, B.; Xu, G.; Han, C.; Xing, G. Lncrna Mir4435-2hg Promotes the Progression of Liver Cancer by Upregulating B3gnt5 Expression. Mol. Med. Rep. 2022, 25, 38. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EEC | LUAD | HCC | HNSCC | COAD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA (n = 10) | NHW (n = 12) | AA (Unin., n = 12; Tumor, n = 14) | NHW (Unin., n = 22; Tumor, n = 26) | AA (n = 10) | NHW (n = 11) | AA (Unin., n = 12; Tumor, n = 11) | NHW (n = 12) | AA (n = 30) | NHW (n = 24) | |
| Sex | Female | Male | Male | Male | Male | |||||
| Mean age (SD) | 60.7 (9.1) | 61.3 (14.4) | 60.8 (6.7) | 69.6 (9.1) | 62.2 (5.5) | 64.5 (16.8) | 62 (9.5) | 58.6 (11.6) | 63.5 (13.6) | 65.7 (10.5) |
| Minimum–maximum | 49–79 | 30–82 | 52–75 | 47–83 | 53–73 | 19–78 | 39–75 | 44–78 | 46–94 | 41–86 |
| p-value | 0.9176 | 0.0018 | 0.6901 | 0.4345 | 0.5214 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyd, A.E.; Grizzard, P.J.; Hylton Rorie, K.; Lima, S. Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers 2023, 15, 2238. https://doi.org/10.3390/cancers15082238
Boyd AE, Grizzard PJ, Hylton Rorie K, Lima S. Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers. 2023; 15(8):2238. https://doi.org/10.3390/cancers15082238
Chicago/Turabian StyleBoyd, April E., Pamela J. Grizzard, Katherine Hylton Rorie, and Santiago Lima. 2023. "Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer" Cancers 15, no. 8: 2238. https://doi.org/10.3390/cancers15082238
APA StyleBoyd, A. E., Grizzard, P. J., Hylton Rorie, K., & Lima, S. (2023). Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers, 15(8), 2238. https://doi.org/10.3390/cancers15082238