

STAT3 Inhibition Attenuates MYC Expression by Modulating Co-Activator Recruitment and Suppresses Medulloblastoma Tumor Growth by Augmenting Cisplatin Efficacy In Vivo
 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. MTT Assay
2.3. Apoptotic Assay and Cell Cycle Analysis
2.4. Western Blot Analysis
2.5. RNA-seq and Quantitative PCR (qPCR)
2.6. Cell Migration Assays
2.7. Colony Assay
2.8. MB-Tumorsphere Assay
2.9. Chromatin Immunoprecipitation (ChIP)-qPCR Assay
2.10. In Vivo Animal Xenograft
- (A)
- Subcutaneous:
- (B)
- Intracranial:
2.11. Immunofluorescence (IF) and Immunohistochemistry (IHC)
2.12. Statistical Analysis
3. Results
3.1. Doxycycline-Inducible STAT3 shRNA Decreases Endogenous STAT3 and STAT3 Targets
3.2. STAT3 KD Affects Cell Viability, Apoptosis, Cell Growth and Migration
3.3. Effect of STAT3 KD on MB Sphere Formation
3.4. STAT3 KD Attenuates Target Gene Expression by Affecting Coactivators and RNA Pol II Recruitment
3.5. STAT3 KD Increases Sensitivity of MB Cells to Chemotherapy
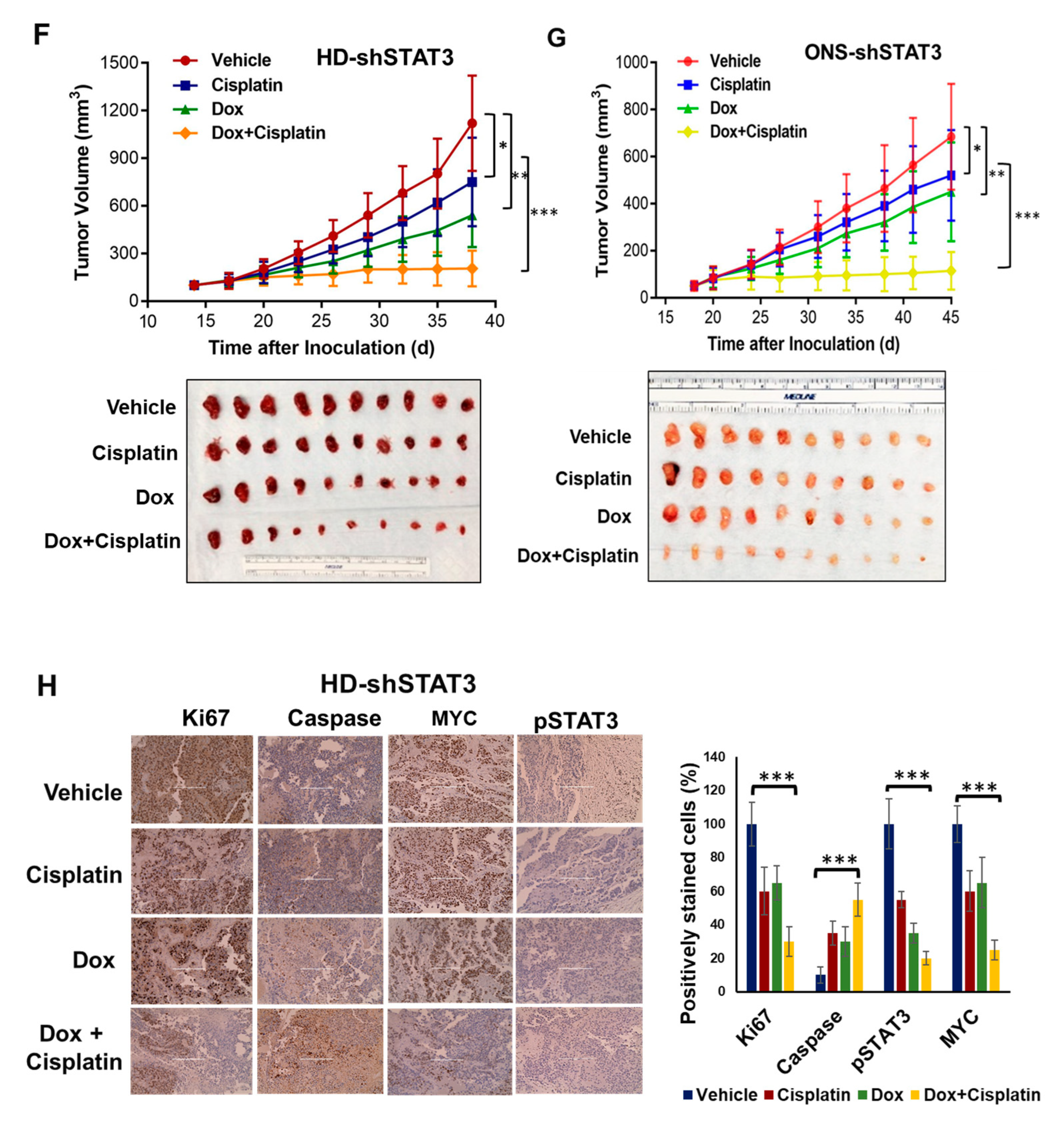
3.6. STAT3 Genetic Inhibition In Vivo Suppresses MB Tumor Growth and Sensitizes MB Tumor to Cisplatin
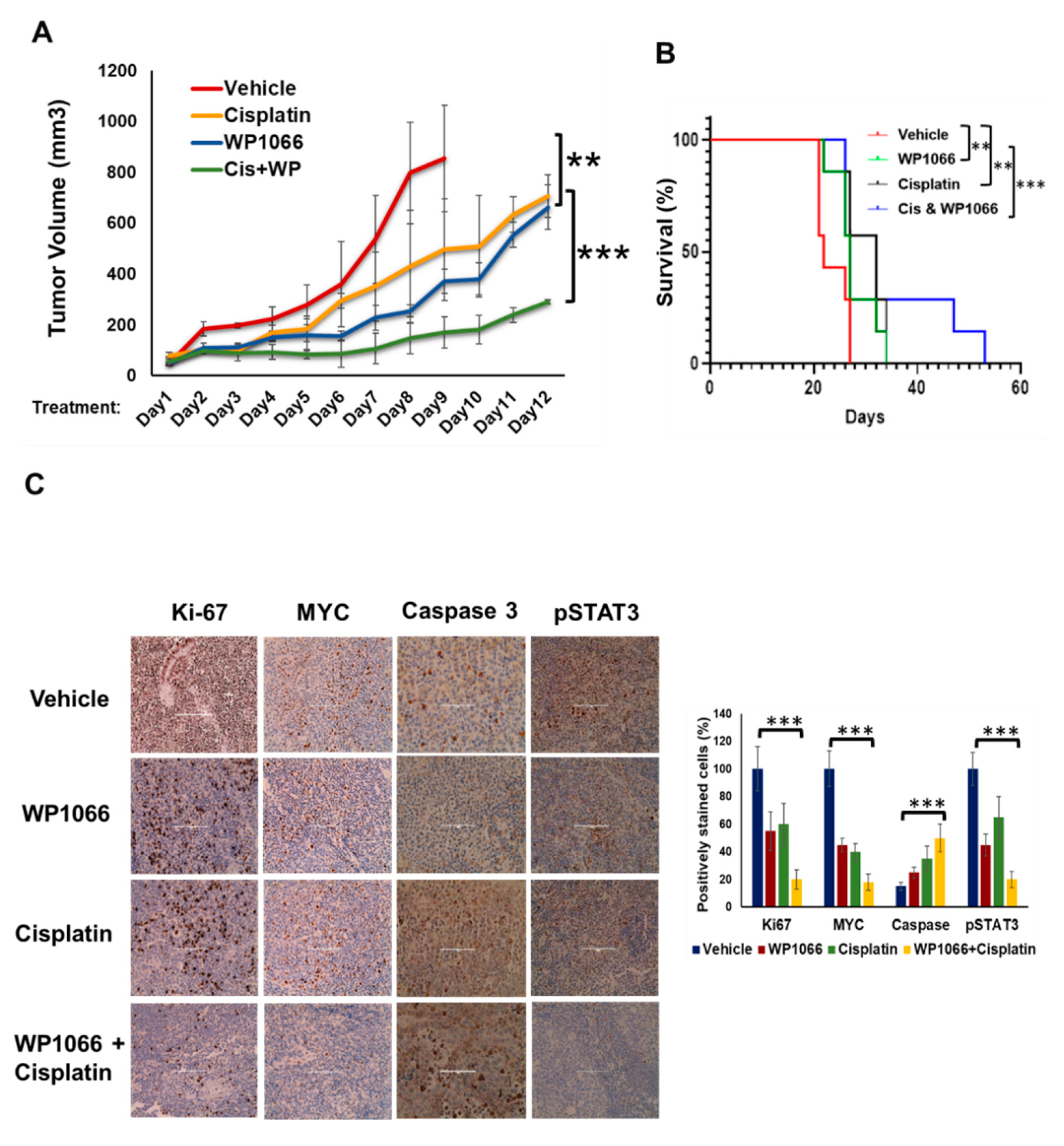
3.7. WP1066 Decreases MB Tumor Burden in Subcutaneous and Intracranial Orthotopic Xenograft Models and Augments MB Chemosensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newton, H.B. Review of the molecular genetics and chemotherapeutic treatment of adult and paediatric medulloblastoma. Expert Opin. Investig. Drugs 2001, 10, 2089–2104. [Google Scholar] [CrossRef]
- Ray, S.; Chaturvedi, N.K.; Bhakat, K.K.; Rizzino, A.; Mahapatra, S. Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment. Diagnostics 2021, 12, 61. [Google Scholar] [CrossRef]
- Stavrou, T.; Bromley, C.M.; Nicholson, H.S.; Byrne, J.; Packer, R.J.; Goldstein, A.M.; Reaman, G.H. Prognostic factors and secondary malignancies in childhood medulloblastoma. J. Pediatr. Hematol. Oncol. 2001, 23, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Ellison, D.W. The origins of medulloblastoma subtypes. Annu. Rev. Pathol. 2008, 3, 341–365. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saran, A. Medulloblastoma: Role of developmental pathways, DNA repair signaling, and other players. Curr. Mol. Med. 2009, 9, 1046–1057. [Google Scholar] [CrossRef]
- Bromberg, J.F. Activation of STAT proteins and growth control. Bioessays 2001, 23, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Levy, C.; Lee, Y.N.; Nechushtan, H.; Schueler-Furman, O.; Sonnenblick, A.; Hacohen, S.; Razin, E. Identifying a common molecular mechanism for inhibition of MITF and STAT3 by PIAS3. Blood 2006, 107, 2839–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Coulter, D.W.; Gray, S.D.; Sughroue, J.A.; Roychoudhury, S.; McIntyre, E.M.; Chaturvedi, N.K.; Bhakat, K.K.; Joshi, S.S.; McGuire, T.R.; et al. Suppression of STAT3 NH2-terminal domain chemosensitizes medulloblastoma cells by activation of protein inhibitor of activated STAT3 via de-repression by microRNA-21. Mol. Carcinog. 2018, 57, 536–548. [Google Scholar] [CrossRef]
- Markou, A.; Zavridou, M.; Lianidou, E.S. miRNA-21 as a novel therapeutic target in lung cancer. Lung. Cancer (Auckl.) 2016, 7, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Jeong, A.J.; Ye, S.K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref, D.; Croul, S. Medulloblastoma: Recurrence and metastasis. CNS Oncol. 2013, 2, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Labak, C.M.; Jain, N.; Purvis, I.J.; Guda, M.R.; Bach, S.E.; Tsung, A.J.; Asuthkar, S.; Velpula, K.K. OTX2 expression contributes to proliferation and progression in Myc-amplified medulloblastoma. Am. J. Cancer Res. 2017, 7, 647–656. [Google Scholar]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Kiuchi, N.; Nakajima, K.; Ichiba, M.; Fukada, T.; Narimatsu, M.; Mizuno, K.; Hibi, M.; Hirano, T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med. 1999, 189, 63–73. [Google Scholar] [CrossRef]
- Kidder, B.L.; Yang, J.; Palmer, S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS ONE 2008, 3, e3932. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasan, L.; Wang, H.; Yap, S.Q.; Leclair, P.; Tam, A.; Lim, C.J. Autocrine IL-6/STAT3 signaling aids development of acquired drug resistance in Group 3 medulloblastoma. Cell Death Dis. 2020, 11, 1035. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressel, S.; Schwalb, B.; Decker, T.M.; Qin, W.; Leonhardt, H.; Eick, D.; Cramer, P. CDK9-dependent RNA polymerase II pausing controls transcription initiation. eLife 2017, 6, e29736. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Boldogh, I.; Brasier, A.R. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 2005, 129, 1616–1632. [Google Scholar] [CrossRef]
- Ray, S.; Sherman, C.T.; Lu, M.; Brasier, A.R. Angiotensinogen gene expression is dependent on signal transducer and activator of transcription 3-mediated p300/cAMP response element binding protein-binding protein coactivator recruitment and histone acetyltransferase activity. Mol. Endocrinol. 2002, 16, 824–836. [Google Scholar] [CrossRef]
- Ray, S.; Zhao, Y.; Jamaluddin, M.; Edeh, C.B.; Lee, C.; Brasier, A.R. Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell. Signal. 2014, 26, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, N.K.; Kling, M.J.; Griggs, C.N.; Kesherwani, V.; Shukla, M.; McIntyre, E.M.; Ray, S.; Liu, Y.; McGuire, T.R.; Sharp, J.G.; et al. A Novel Combination Approach Targeting an Enhanced Protein Synthesis Pathway in MYC-driven (Group 3) Medulloblastoma. Mol. Cancer Ther. 2020, 19, 1351–1362. [Google Scholar] [CrossRef]
- Ray, S.; Ju, X.; Sun, H.; Finnerty, C.C.; Herndon, D.N.; Brasier, A.R. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. J. Investig. Dermatol. 2013, 133, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Lee, C.; Hou, T.; Bhakat, K.K.; Brasier, A.R. Regulation of signal transducer and activator of transcription 3 enhanceosome formation by apurinic/apyrimidinic endonuclease 1 in hepatic acute phase response. Mol. Endocrinol. 2010, 24, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Bhakat, R.; Kling, M.J.; Coulter, D.W.; Chaturvedi, N.K.; Ray, S.; Joshi, S.S. Targeting cyclin-dependent kinase 9 sensitizes medulloblastoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 2019, 520, 250–256. [Google Scholar] [CrossRef]
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619–16633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramanik, S.; Chen, Y.; Song, H.; Khutsishvili, I.; Marky, L.A.; Ray, S.; Natarajan, A.; Singh, P.K.; Bhakat, K.K. The human AP-endonuclease 1 (APE1) is a DNA G-quadruplex structure binding protein and regulates KRAS expression in pancreatic ductal adenocarcinoma cells. Nucleic Acids Res. 2022, 50, 3394–3412. [Google Scholar] [CrossRef]
- Song, H.; Xi, S.; Chen, Y.; Pramanik, S.; Zeng, J.; Roychoudhury, S.; Harris, H.; Akbar, A.; Elhag, S.S.; Coulter, D.W.; et al. Histone chaperone FACT complex inhibitor CBL0137 interferes with DNA damage repair and enhances sensitivity of medulloblastoma to chemotherapy and radiation. Cancer Lett. 2021, 520, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, Identification, and Characterization of Cancer Stem Cells: A Review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Zanini, C.; Ercole, E.; Mandili, G.; Salaroli, R.; Poli, A.; Renna, C.; Papa, V.; Cenacchi, G.; Forni, M. Medullospheres from DAOY, UW228 and ONS-76 cells: Increased stem cell population and proteomic modifications. PLoS ONE 2013, 8, e63748. [Google Scholar] [CrossRef] [Green Version]
- Mehrazma, M.; Madjd, Z.; Kalantari, E.; Panahi, M.; Hendi, A.; Shariftabrizi, A. Expression of stem cell markers, CD133 and CD44, in pediatric solid tumors: A study using tissue microarray. Fetal Pediatr. Pathol. 2013, 32, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Ray, S.; Lee, C.; Brasier, A.R. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J. Biol. Chem. 2008, 283, 30725–30734. [Google Scholar] [CrossRef] [Green Version]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Sweha, S.R.; Chung, C.; Natarajan, S.K.; Panwalkar, P.; Pun, M.; Ghali, A.; Bayliss, J.; Pratt, D.; Shankar, A.; Ravikumar, V.; et al. Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Sci. Transl. Med. 2021, 13, eabf7860. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Zaiter, A.; Audi, Z.F.; Shawraba, F.; Saker, Z.; Bahmad, H.F.; Nabha, R.H.; Harati, H.; Nabha, S.M. STAT3 in medulloblastoma: A key transcriptional regulator and potential therapeutic target. Mol. Biol. Rep. 2022, 49, 10635–10652. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Bakhshinyan, D.; Venugopal, C.; Mahendram, S.; Rosa, D.A.; Vijayakumar, T.; Manoranjan, B.; Hallett, R.; McFarlane, N.; Delaney, K.H.; et al. CD133+ brain tumor-initiating cells are dependent on STAT3 signaling to drive medulloblastoma recurrence. Oncogene 2017, 36, 606–617. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Pan, L.; Wei, J.; Zhang, R.; Yang, X.; Song, J.; Bai, R.Y.; Fu, S.; Pierson, C.R.; Finlay, J.L.; et al. LLL12B, a small molecule STAT3 inhibitor, induces growth arrest, apoptosis, and enhances cisplatin-mediated cytotoxicity in medulloblastoma cells. Sci. Rep. 2021, 11, 6517. [Google Scholar] [CrossRef]
- Wei, J.; Ma, L.; Li, C.; Pierson, C.R.; Finlay, J.L.; Lin, J. Targeting Upstream Kinases of STAT3 in Human Medulloblastoma Cells. Curr. Cancer Drug Targets 2019, 19, 571–582. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Ray, S.; Brasier, A.R. The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human gamma-fibrinogen gene expression. J. Biol. Chem. 2007, 282, 37091–37102. [Google Scholar] [CrossRef] [Green Version]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [Green Version]
- Bres, V.; Yoh, S.M.; Jones, K.A. The multi-tasking P-TEFb complex. Curr. Opin. Cell Biol. 2008, 20, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, S.; Bertolini, F. Characterization of Cancer Stem Cells. Methods Mol. Biol. 2016, 1464, 49–62. [Google Scholar] [CrossRef]
- Konrad, C.V.; Murali, R.; Varghese, B.A.; Nair, R. The role of cancer stem cells in tumor heterogeneity and resistance to therapy. Can. J. Physiol. Pharmacol. 2017, 95, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.C.; Liao, P.N.; Ho, A.S.; Lim, K.H.; Chang, J.; Su, Y.W.; Chen, C.G.; Chiang, Y.W.; Yang, B.L.; Lin, H.C.; et al. STAT3 exacerbates survival of cancer stem-like tumorspheres in EGFR-positive colorectal cancers: RNAseq analysis and therapeutic screening. J. Biomed. Sci. 2018, 25, 60. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, L.; Lin, K.; Zhang, Y.; Deng, A.; Liang, Y.; Li, C.; Wen, T. The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells. Clin. Cancer Res. 2017, 23, 6673–6685. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohrer, K.A.; Song, H.; Akbar, A.; Chen, Y.; Pramanik, S.; Wilder, P.J.; McIntyre, E.M.; Chaturvedi, N.K.; Bhakat, K.K.; Rizzino, A.; et al. STAT3 Inhibition Attenuates MYC Expression by Modulating Co-Activator Recruitment and Suppresses Medulloblastoma Tumor Growth by Augmenting Cisplatin Efficacy In Vivo. Cancers 2023, 15, 2239. https://doi.org/10.3390/cancers15082239
Rohrer KA, Song H, Akbar A, Chen Y, Pramanik S, Wilder PJ, McIntyre EM, Chaturvedi NK, Bhakat KK, Rizzino A, et al. STAT3 Inhibition Attenuates MYC Expression by Modulating Co-Activator Recruitment and Suppresses Medulloblastoma Tumor Growth by Augmenting Cisplatin Efficacy In Vivo. Cancers. 2023; 15(8):2239. https://doi.org/10.3390/cancers15082239
Chicago/Turabian StyleRohrer, Kyle A., Heyu Song, Anum Akbar, Yingling Chen, Suravi Pramanik, Phillip J. Wilder, Erin M. McIntyre, Nagendra K. Chaturvedi, Kishor K. Bhakat, Angie Rizzino, and et al. 2023. "STAT3 Inhibition Attenuates MYC Expression by Modulating Co-Activator Recruitment and Suppresses Medulloblastoma Tumor Growth by Augmenting Cisplatin Efficacy In Vivo" Cancers 15, no. 8: 2239. https://doi.org/10.3390/cancers15082239
APA StyleRohrer, K. A., Song, H., Akbar, A., Chen, Y., Pramanik, S., Wilder, P. J., McIntyre, E. M., Chaturvedi, N. K., Bhakat, K. K., Rizzino, A., Coulter, D. W., & Ray, S. (2023). STAT3 Inhibition Attenuates MYC Expression by Modulating Co-Activator Recruitment and Suppresses Medulloblastoma Tumor Growth by Augmenting Cisplatin Efficacy In Vivo. Cancers, 15(8), 2239. https://doi.org/10.3390/cancers15082239