

Detection of Genomic Structural Variations Associated with Drug Sensitivity and Resistance in Acute Leukemia
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Primary Leukemia Cell Samples
2.2. Ultra-High Molecular Weight DNA Isolation and Optical Genome Mapping
2.3. Bionano Access/Rare Variant Analysis Pipeline
2.4. SV Filtering and Identification of Disrupted Gene Loci
2.5. Curve-Fitting of Drug Sensitivity Data
tryCatch (nplr(x = x, y = y/100),
error = function(e){ −1; })
}
drug.auc <- unlist (getAUC(nplr.res)[1]) * 0.5
2.6. Gene–Drug Interaction Significance Test
2.7. Drug Collections
2.8. Cell Viability Assay and Drug Screening
2.9. Data Sharing Statement
3. Results and Discussion
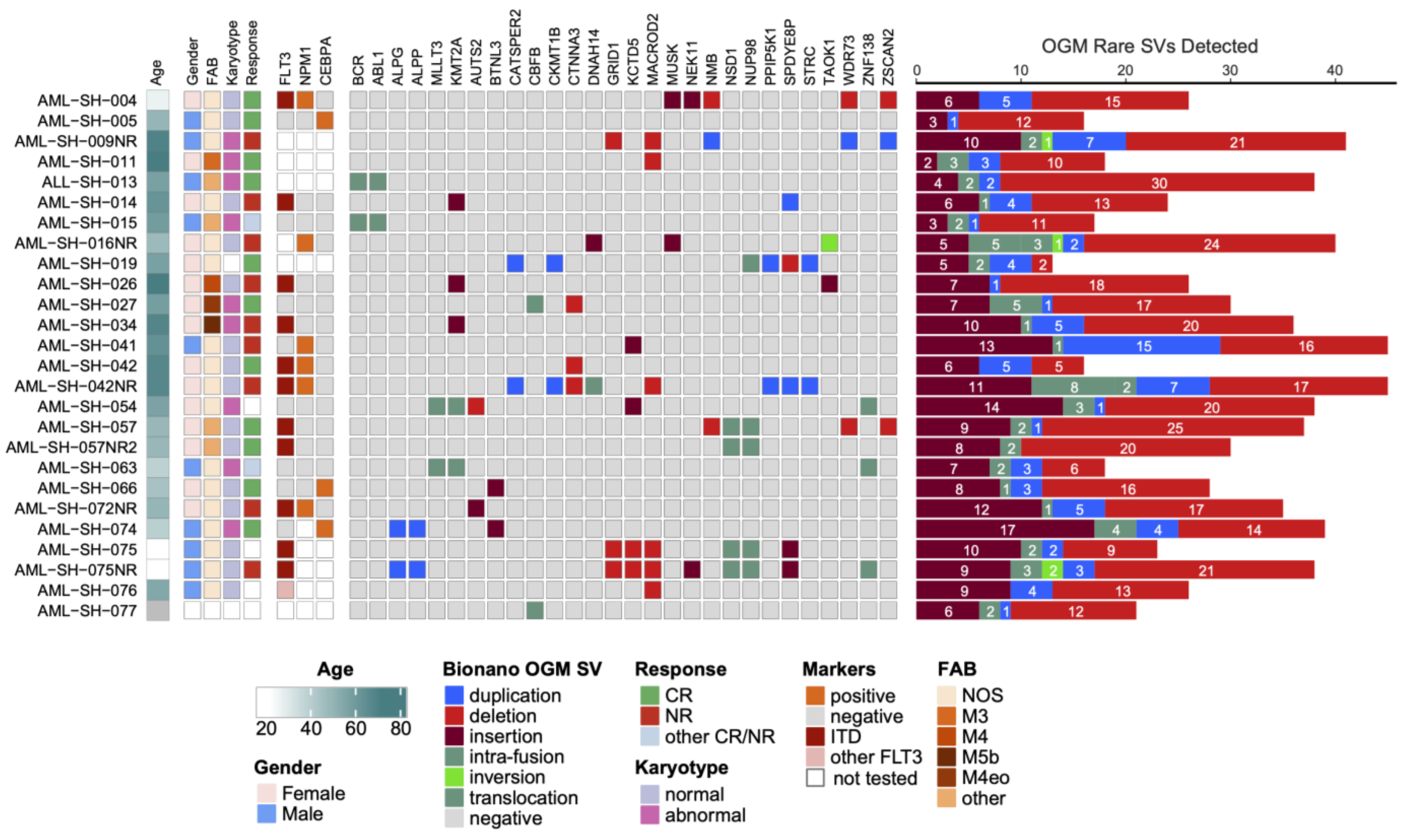
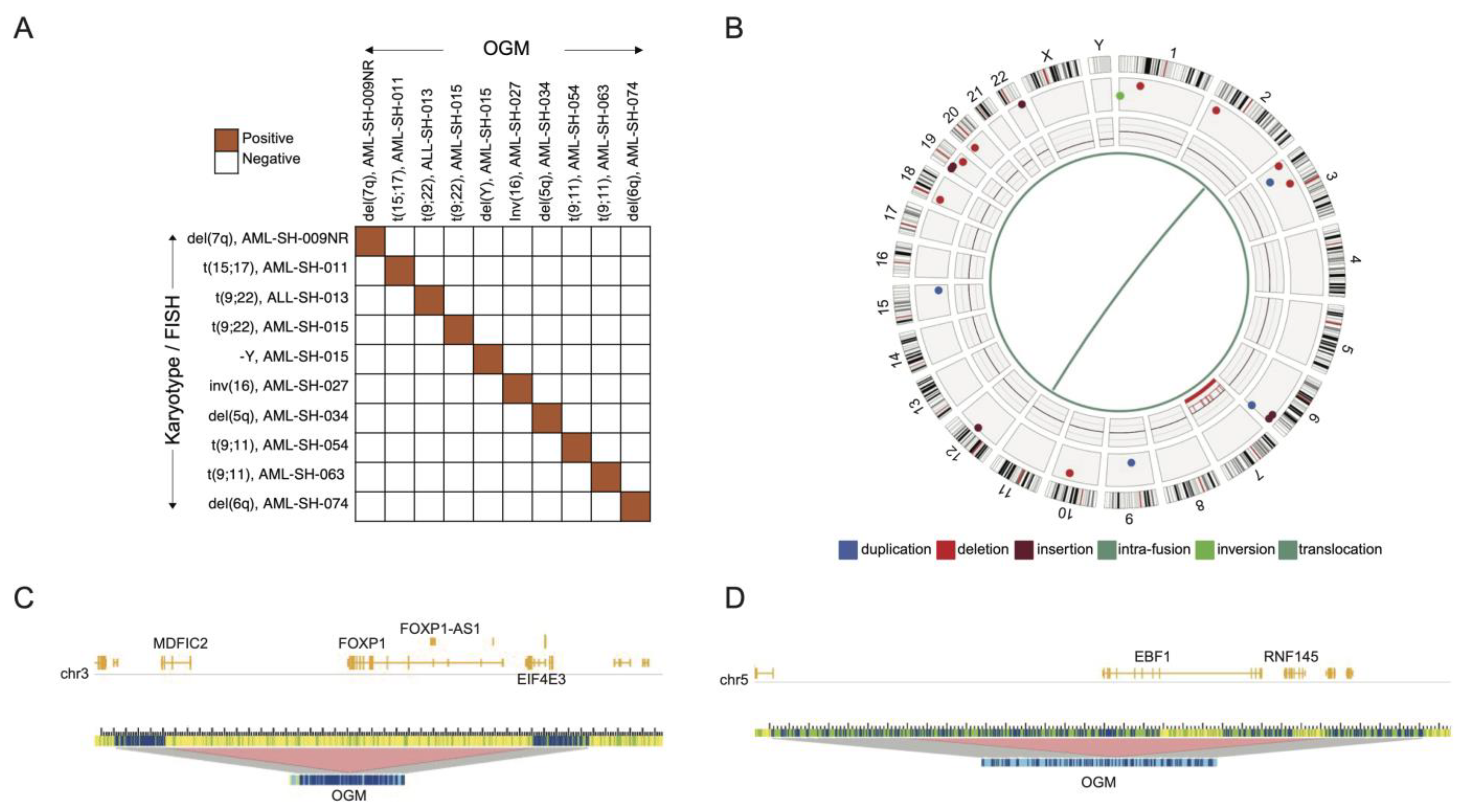
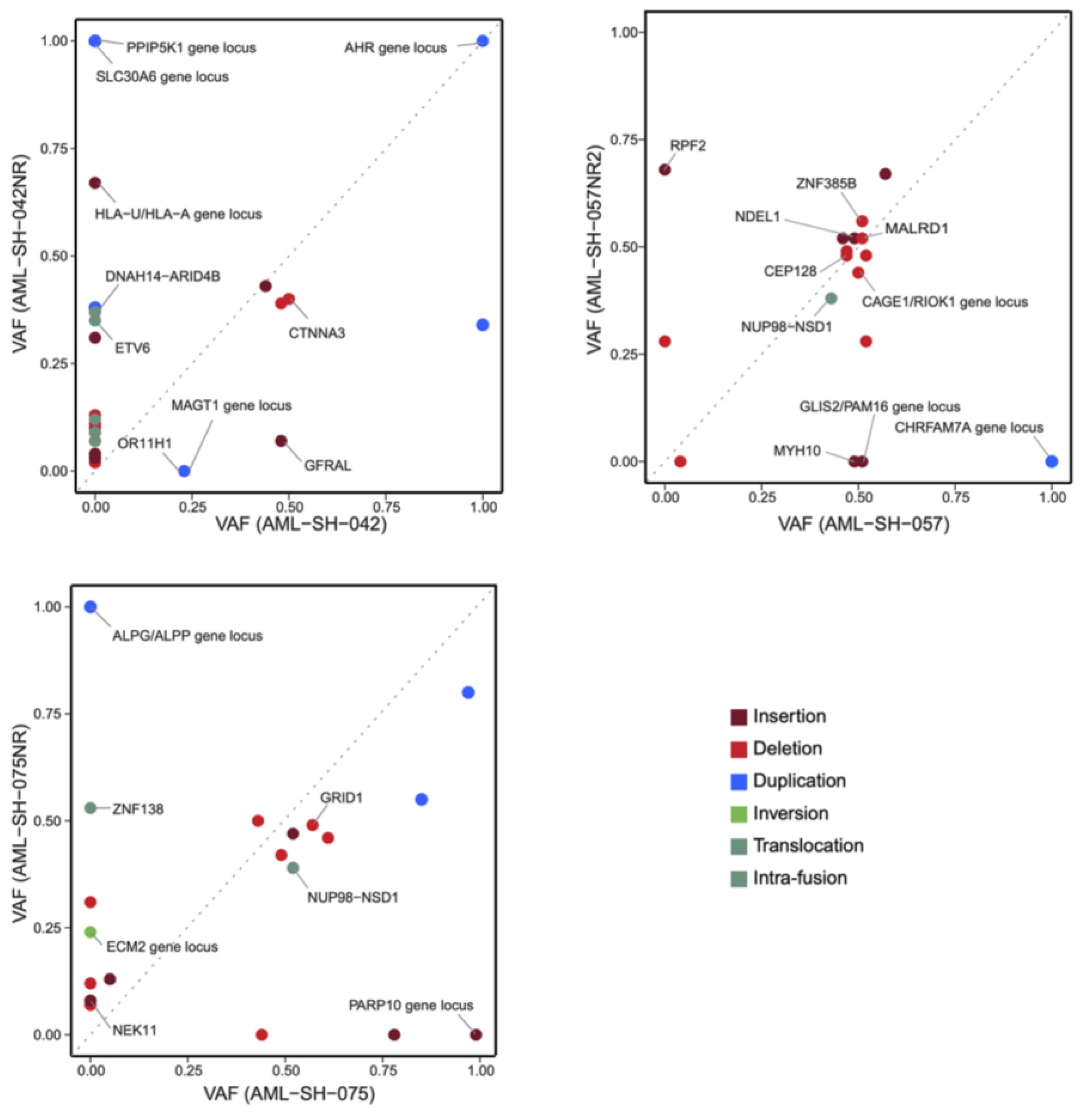
3.1. Optical Genome Mapping Detects All Structural Variants That Had Been Observed via the Standard of Care and Multiple Additional Variants Not Previously Reported
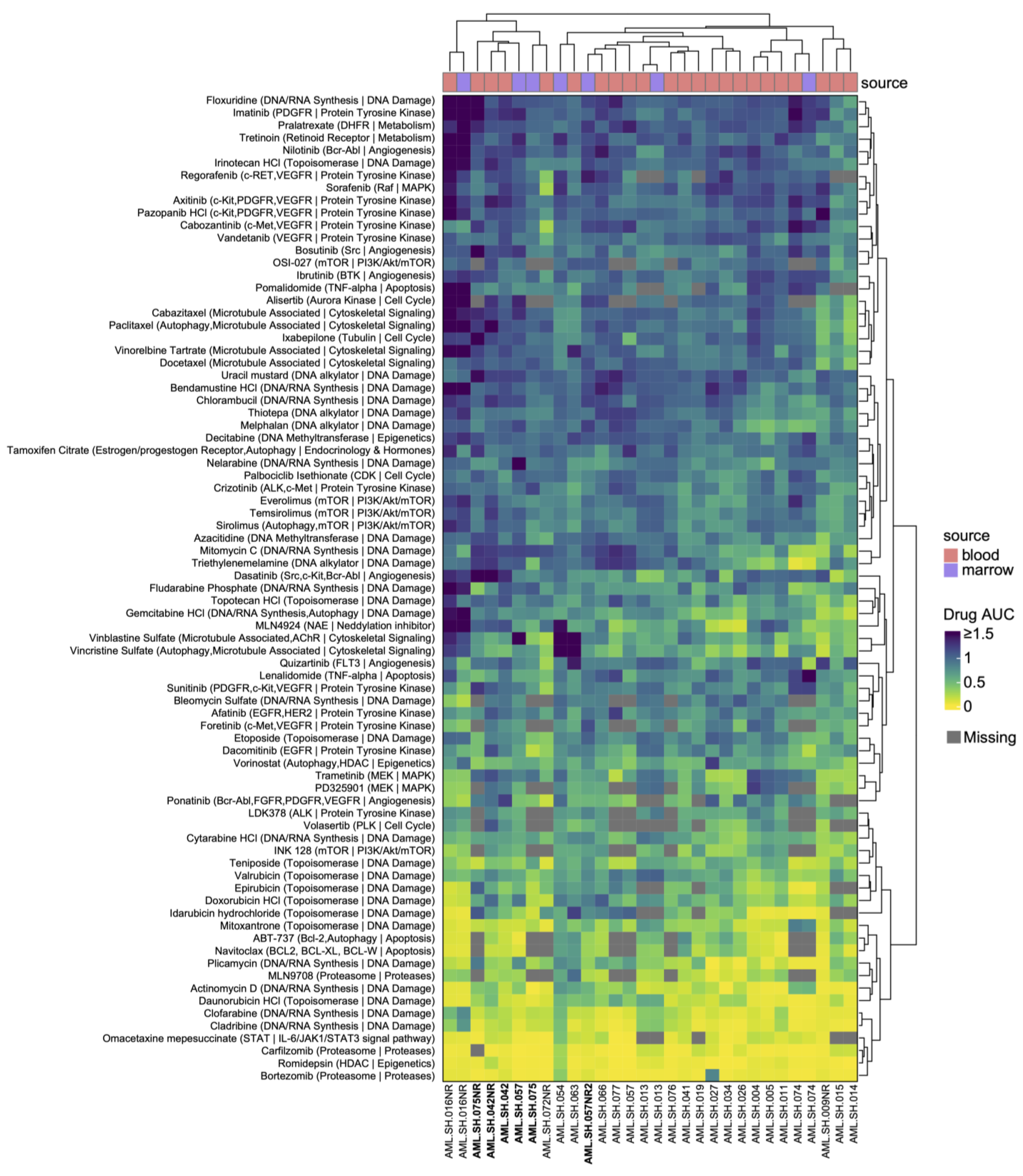
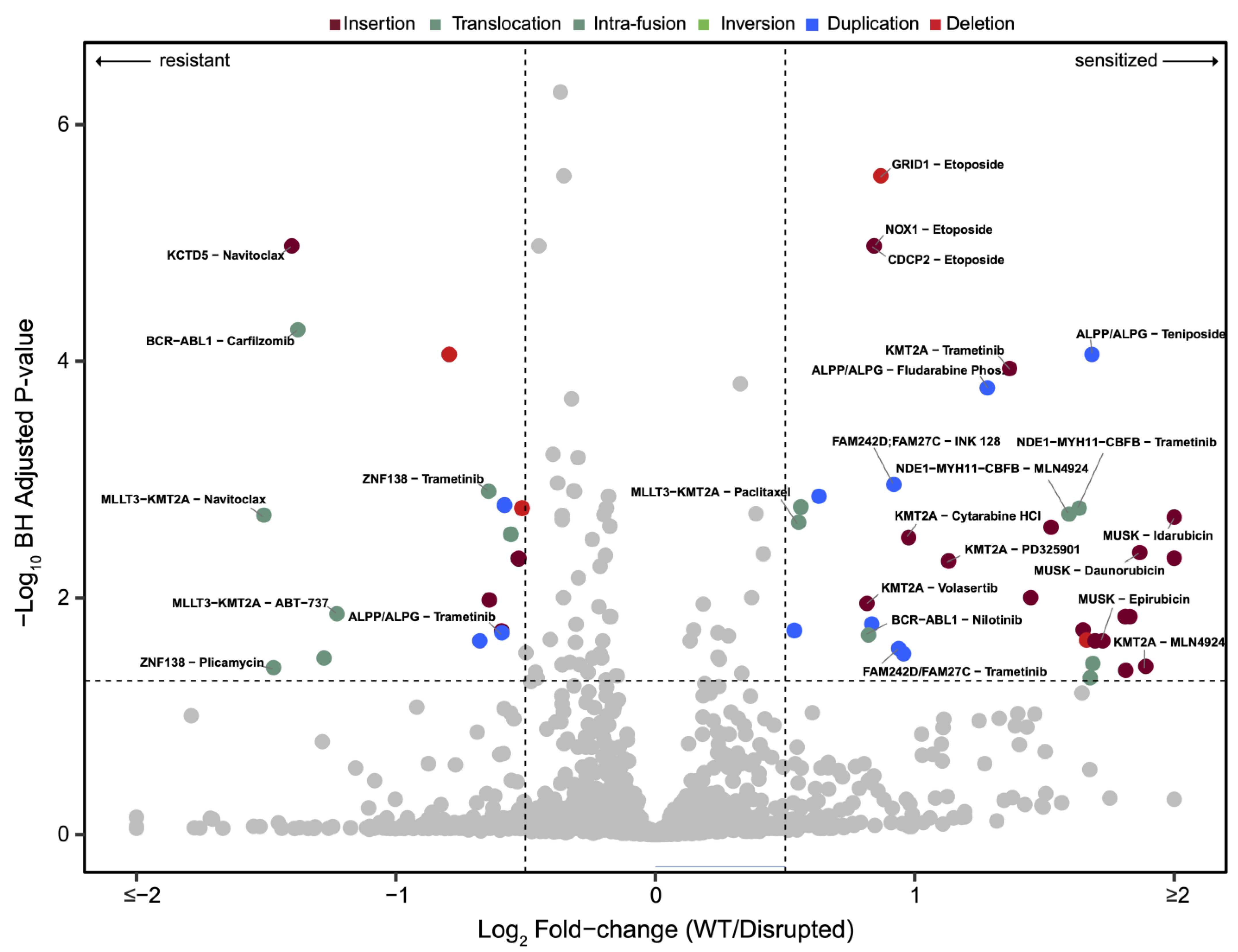
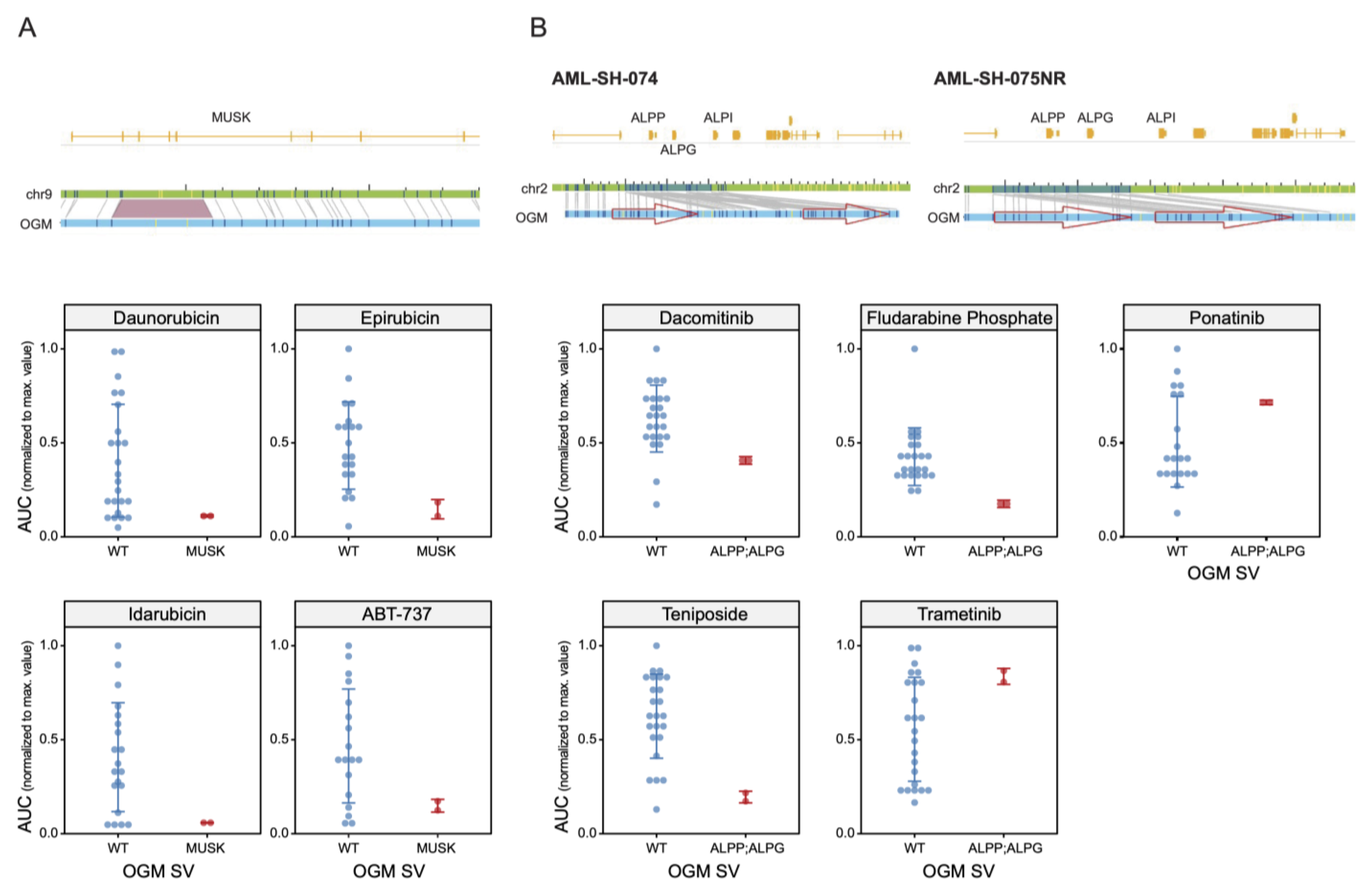
3.2. Drug Sensitivities Are Associated with Novel Genomic Rearrangements
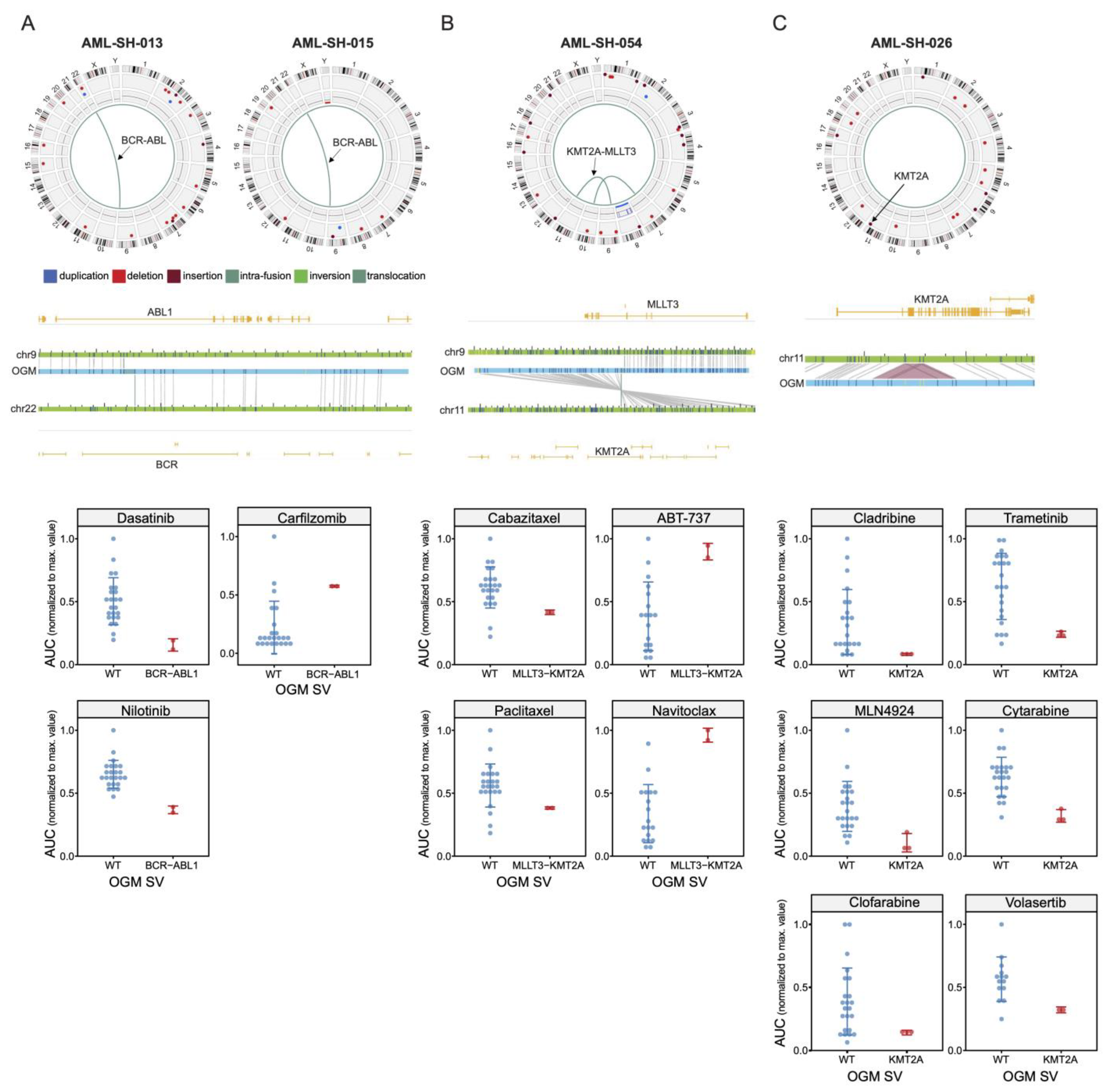
3.3. BCR-ABL1 Translocated Leukemia Samples Are Sensitive to Nilotinib but Exhibit Resistance to Proteasome Inhibitors
3.4. Drug Sensitivities and Resistance of Leukemia Samples with KMT2A Variants
3.5. Unbiased Chemosensitivity Associations with SVs Unveil Potential New Treatment Strategies for Leukemia
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef]
- Seo, J.S.; Rhie, A.; Kim, J.; Lee, S.; Sohn, M.H.; Kim, C.U.; Hastie, A.; Cao, H.; Yun, J.Y.; Kim, J.; et al. De novo assembly and phasing of a Korean human genome. Nature 2016, 538, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Li, P.; Wang, Z.; Liang, F.; Yang, F.; Fang, L.; Huang, Y.; Huang, S.; Zhou, J.; Wang, D.; et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). J. Med. Genet. 2020, 57, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Stence, A.A.; Thomason, J.G.; Pruessner, J.A.; Sompallae, R.R.; Snow, A.N.; Ma, D.; Moore, S.A.; Bossler, A.D. Validation of Optical Genome Mapping for the Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy. J. Mol. Diagn. 2021, 23, 1506–1514. [Google Scholar] [CrossRef]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef]
- Soto, D.C.; Shew, C.; Mastoras, M.; Schmidt, J.M.; Sahasrabudhe, R.; Kaya, G.; Andrés, A.M.; Dennis, M.Y. Identification of Structural Variation in Chimpanzees Using Optical Mapping and Nanopore Sequencing. Genes 2020, 11, 276. [Google Scholar] [CrossRef]
- Kronenberg, Z.N.; Fiddes, I.T.; Gordon, D.; Murali, S.; Cantsilieris, S.; Meyerson, O.S.; Underwood, J.G.; Nelson, B.J.; Chaisson, M.J.P.; Dougherty, M.L.; et al. High-resolution comparative analysis of great ape genomes. Science 2018, 360, eaar6343. [Google Scholar] [CrossRef]
- Dixon, J.R.; Xu, J.; Dileep, V.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; Clark, R.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Jaratlerdsiri, W.; Chan, E.K.F.; Petersen, D.C.; Yang, C.; Croucher, P.I.; Bornman, M.S.R.; Sheth, P.; Hayes, V.M. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 2017, 8, 23588–23602. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.K.F.; Cameron, D.L.; Petersen, D.C.; Lyons, R.J.; Baldi, B.F.; Papenfuss, A.T.; Thomas, D.M.; Hayes, V.M. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res. 2018, 28, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.; Xu, J.; Schleicher, E.; Song, F.; Pool, C.; Hennessy, M.; Sheldon, K.; Annageldiyev, C.; Sharma, A.; Chang, Y.; et al. An Integrated Framework for Genome Analysis Reveals Numerous Previously Unrecognizable Structural Variants in Leukemia Patient Samples. FASEB J. 2019, 33, 474.1. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Suttorp, J.; Lühmann, J.L.; Behrens, Y.L.; Göhring, G.; Steinemann, D.; Reinhardt, D.; Neuhoff, N.V.; Schneider, M. Optical Genome Mapping as a Diagnostic Tool in Pediatric Acute Myeloid Leukemia. Cancers 2022, 14, 2058. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Ramos-Campoy, S.; Kamaso, J.; de la Rosa, M.; Salido, M.; Melero, C.; Rodríguez-Rivera, M.; Bougeon, S.; Collado, R.; Gimeno, E.; et al. Optical Genome Mapping: A Promising New Tool to Assess Genomic Complexity in Chronic Lymphocytic Leukemia (CLL). Cancers 2022, 14, 3376. [Google Scholar] [CrossRef]
- Valkama, A.; Vorimo, S.; Kumpula, T.; Räsänen, H.; Savolainen, E.R.; Pylkäs, K.; Mantere, T. Optical Genome Mapping as an Alternative to FISH-Based Cytogenetic Assessment in Chronic Lymphocytic Leukemia. Cancers 2023, 15, 1294. [Google Scholar] [CrossRef]
- Soler, G.; Ouedraogo, Z.; Goumy, C.; Lebecque, B.; Aspas Requena, G.; Ravinet, A.; Kanold, J.; Véronèse, L.; Tchirkov, A. Optical Genome Mapping in Routine Cytogenetic Diagnosis of Acute Leukemia. Cancers 2023, 15, 2131. [Google Scholar] [CrossRef]
- Tembrink, M.; Gerding, W.; Wieczorek, S.; Mika, T.; Schroers, R.; Nguyen, H.P.; Vangala, D.B.; Nilius-Eliliwi, V. Novel NUP98::ASH1L Gene Fusion in Acute Myeloid Leukemia Detected by Optical Genome Mapping. Cancers 2023, 15, 2942. [Google Scholar] [CrossRef]
- Sahajpal, N.; Mondal, A.; Singh, H.; Vashisht, A.; Ananth, S.; Saul, D.; Hastie, A.R.; Hilton, B.; DuPont, B.R.; Savage, N.M.; et al. Clinical Utility of Optical Genome Mapping and 523-Gene Next Generation Sequencing Panel for Comprehensive Evaluation of Myeloid Cancers. Cancers 2023, 15, 3214. [Google Scholar] [CrossRef] [PubMed]
- Giguère, A.; Raymond-Bouchard, I.; Collin, V.; Claveau, J.S.; Hébert, J.; LeBlanc, R. Optical Genome Mapping Reveals the Complex Genetic Landscape of Myeloma. Cancers 2023, 15, 4687. [Google Scholar] [CrossRef] [PubMed]
- Rusert, J.M.; Juarez, E.F.; Brabetz, S.; Jensen, J.; Garancher, A.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Wahab, S.; Udaka, Y.T.; Finlay, D.; et al. Functional precision medicine identifies new therapeutic candidates for medulloblastoma. Cancer Res. 2020, 80, 5393–5407. [Google Scholar] [CrossRef] [PubMed]
- Khateb, A.; Deshpande, A.; Feng, Y.; Finlay, D.; Lee, J.S.; Lazar, I.; Fabre, B.; Li, Y.; Fujita, Y.; Zhang, T.; et al. The ubiquitin ligase RNF5 determines acute myeloid leukemia growth and susceptibility to histone deacetylase inhibitor. Nat. Commun. 2021, 12, 5397. [Google Scholar] [CrossRef]
- Benitez, J.A.; Finlay, D.; Castanza, A.; Parisian, A.; Ma, J.; Longobardi, C.; Campos, A.; Vadla, R.; Izurieta, A.; Scerra, G.; et al. PTEN deficiency leads to proteasome addiction, a novel vulnerability in Glioblastoma. Neuro Oncol. 2021, 23, 1072–1086. [Google Scholar] [CrossRef]
- Lestringant, V.; Duployez, N.; Penther, D.; Luquet, I.; Derrieux, C.; Lutun, A.; Preudhomme, C.; West, M.; Ouled-Haddou, H.; Devoldere, C.; et al. Optical genome mapping, a promising alternative to gold standard cytogenetic approaches in a series of acute lymphoblastic leukemias. Genes Chromosomes Cancer 2021, 60, 657–667. [Google Scholar] [CrossRef]
- Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Göhring, G.; Möricke, A.; Cario, G.; et al. The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia. Cancers 2021, 13, 4388. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Rack, K.; De Bie, J.; Ameye, G.; Gielen, O.; Demeyer, S.; Cools, J.; De Keersmaecker, K.; Vermeesch, J.R.; Maertens, J.; Segers, H.; et al. Optimizing the diagnostic workflow for acute lymphoblastic leukemia by optical genome mapping. Am. J. Hematol. 2022, 97, 548–561. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Tvrdik, T.; Hauenstein, J.; Shi, H.; Deeb, K.K.; Saxe, D.; Hastie, A.R.; Chaubey, A.; Savage, N.M.; et al. Clinical Validation and Diagnostic Utility of Optical Genome Mapping for Enhanced Cytogenomic Analysis of Hematological Neoplasms. J. Mol. Diagn. 2022, 24, 1279–1291. [Google Scholar] [CrossRef]
- Vangala, D.B.; Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P. Optical Genome Mapping in MDS and AML as tool for structural variant profiling-comment and data update on Yang et al.: “High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance”. Leukemia 2023, 37, 248–249. [Google Scholar] [CrossRef]
- Vieler, L.M.; Nilius-Eliliwi, V.; Schroers, R.; Vangala, D.B.; Nguyen, H.P.; Gerding, W.M. Optical Genome Mapping Reveals and Characterizes Recurrent Aberrations and New Fusion Genes in Adult ALL. Genes 2023, 14, 686. [Google Scholar] [CrossRef] [PubMed]
- Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P.; Vangala, D.B. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers 2023, 15, 1684. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical genome mapping in acute myeloid leukemia: A multicenter evaluation. Blood Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Wapner, J. The Philadelphia Chromosome: A Genetic Mystery, a Lethal Cancer, and the Improbable Invention of a Lifesaving Treatment; The Experiment, LLC: New York, NY, USA, 2014; ISBN 9781615191970. [Google Scholar]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Roberts, A.W.; Huang, D. Targeting BCL2 with BH3 Mimetics: Basic Science and Clinical Application of Venetoclax in Chronic Lymphocytic Leukemia and Related B Cell Malignancies. Clin. Pharmacol. Ther. 2017, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Vanhuele, S.; Kirova, Y.; Hamy-Petit, A.-S.; Rapinat, A.; Gentien, D.; Reyal, F.; Vincent-Salomon, A.; Eeckhoutte, A.; Rodrigues, M.; Popova, T.; et al. Optical Genome Mapping for detecting Homologous Recombination Deficiency (HRD) in human breast cancers. bioRxiv 2022. [Google Scholar] [CrossRef]
- Bose, P.; Grant, S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leuk. Res. 2014, 38, 862–865. [Google Scholar] [CrossRef]
- Pigneux, A.; Perreau, V.; Jourdan, E.; Vey, N.; Dastugue, N.; Huguet, F.; Sotto, J.J.; Salmi, L.R.; Ifrah, N.; Reiffers, J. Adding lomustine to idarubicin and cytarabine for induction chemotherapy in older patients with acute myeloid leukemia: The BGMT 95 trial results. Haematologica 2007, 92, 1327–1334. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finlay, D.; Murad, R.; Hong, K.; Lee, J.; Pang, A.W.C.; Lai, C.-Y.; Clifford, B.; Burian, C.; Mason, J.; Hastie, A.R.; et al. Detection of Genomic Structural Variations Associated with Drug Sensitivity and Resistance in Acute Leukemia. Cancers 2024, 16, 418. https://doi.org/10.3390/cancers16020418
Finlay D, Murad R, Hong K, Lee J, Pang AWC, Lai C-Y, Clifford B, Burian C, Mason J, Hastie AR, et al. Detection of Genomic Structural Variations Associated with Drug Sensitivity and Resistance in Acute Leukemia. Cancers. 2024; 16(2):418. https://doi.org/10.3390/cancers16020418
Chicago/Turabian StyleFinlay, Darren, Rabi Murad, Karl Hong, Joyce Lee, Andy Wing Chun Pang, Chi-Yu Lai, Benjamin Clifford, Carol Burian, James Mason, Alex R. Hastie, and et al. 2024. "Detection of Genomic Structural Variations Associated with Drug Sensitivity and Resistance in Acute Leukemia" Cancers 16, no. 2: 418. https://doi.org/10.3390/cancers16020418
APA StyleFinlay, D., Murad, R., Hong, K., Lee, J., Pang, A. W. C., Lai, C. -Y., Clifford, B., Burian, C., Mason, J., Hastie, A. R., Yin, J., & Vuori, K. (2024). Detection of Genomic Structural Variations Associated with Drug Sensitivity and Resistance in Acute Leukemia. Cancers, 16(2), 418. https://doi.org/10.3390/cancers16020418