
A Phase Ib Expansion Cohort Evaluating Aurora A Kinase Inhibitor Alisertib and Dual TORC1/2 Inhibitor Sapanisertib in Patients with Advanced Solid Tumors

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection
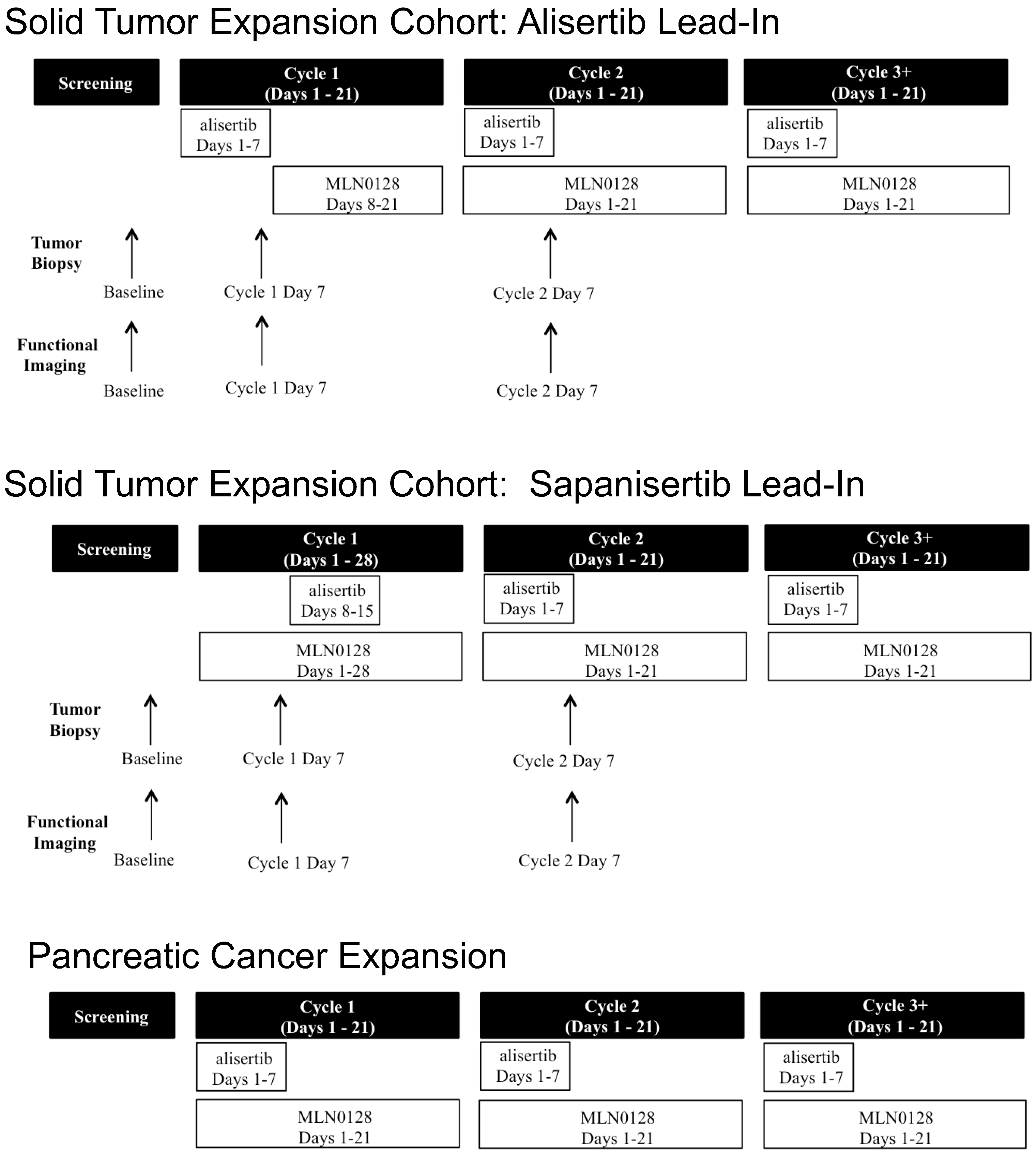
2.2. Study Design
2.3. Correlative Studies
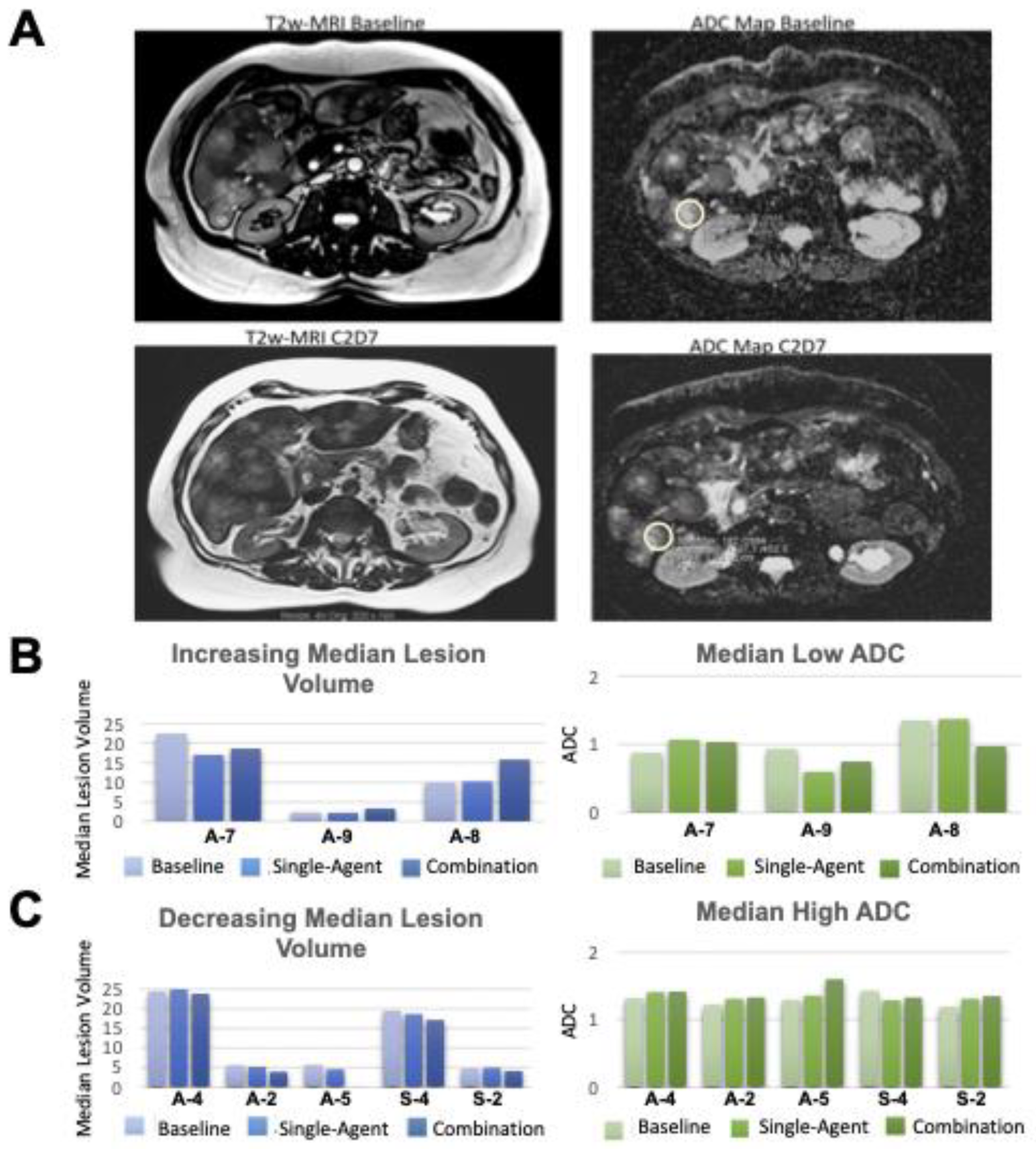
2.3.1. Functional Imaging
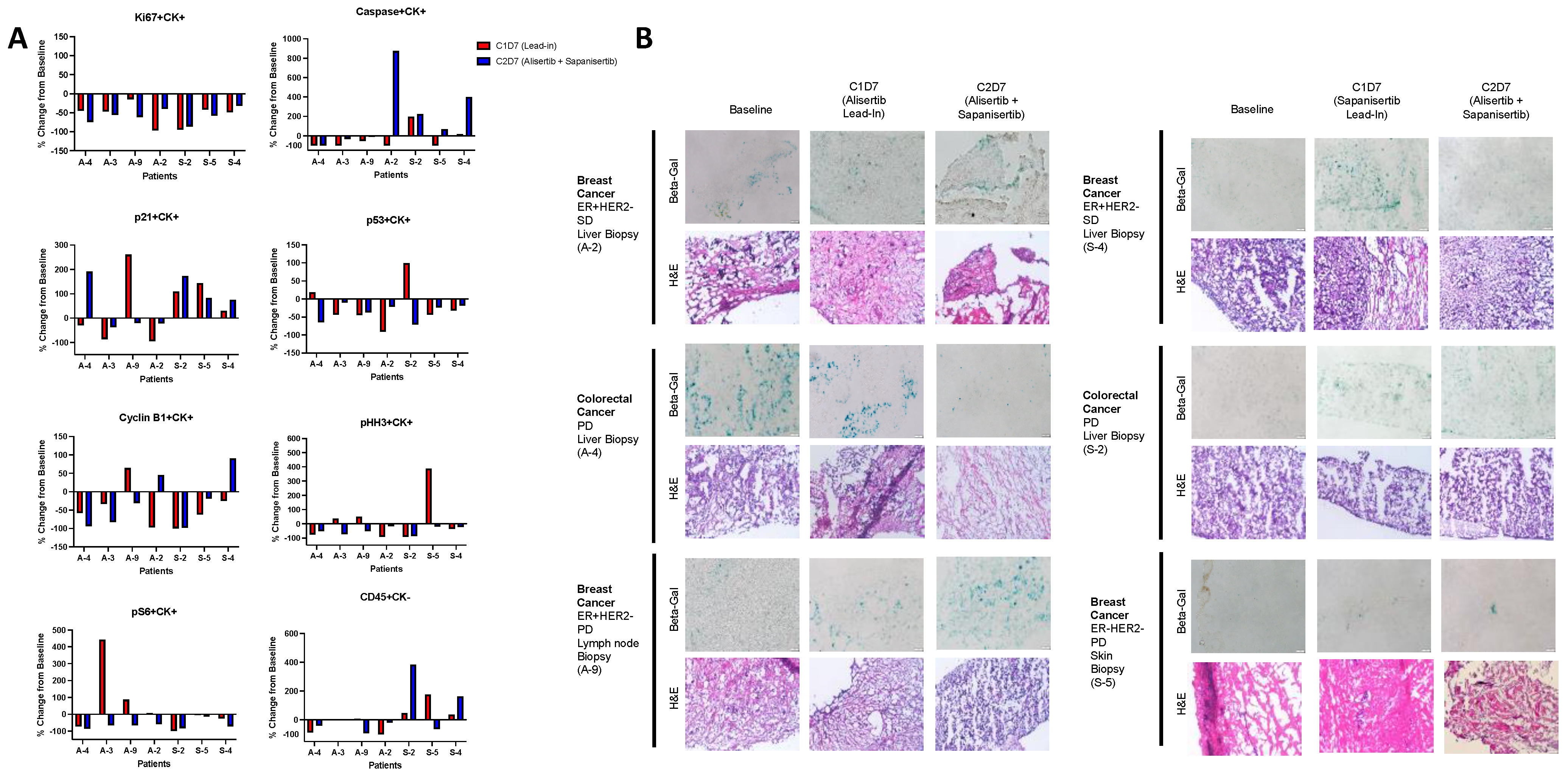
2.3.2. Tumor Tissue Evaluation
Fluorescence Microscopy
SA-β-Gal and H and E
2.4. Statistical Methods
3. Results
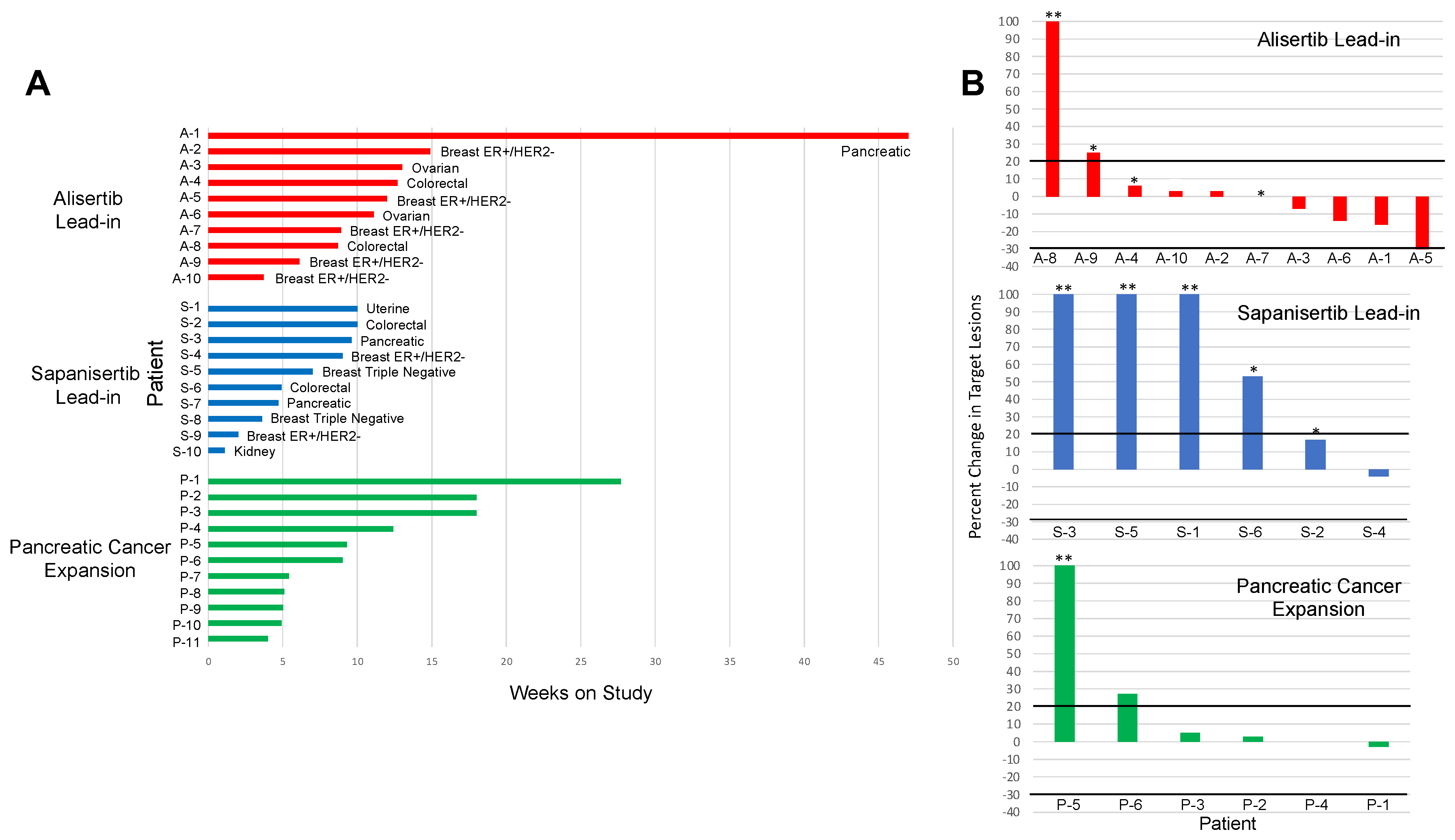
3.1. Clinical Outcomes
3.1.1. Solid Tumor Expansion Cohort
3.1.2. Pancreatic Cancer Expansion Cohort
3.2. Toxicities
3.3. Correlative Studies
3.3.1. Functional Imaging
3.3.2. Tumor Tissue Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef] [PubMed]
- Karthigeyan, D.; Prasad, S.B.B.; Shandilya, J.; Agrawal, S.; Kundu, T.K. Biology of Aurora A kinase: Implications in cancer manifestation and therapy. Med. Res. Rev. 2010, 31, 757–793. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Li, J.; Yan, H.; Zhang, G.; Li, W.; Chu, E.; Wei, N. Emerging roles of Aurora-A kinase in cancer therapy resistance. Acta Pharm. Sin. B 2023, 13, 2826–2843. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Puma Biotechnology Receives FDA Orphan Drug Designation for Alisertib for the Treatment of Small Cell Lung Cancer. Available online: https://investor.pumabiotechnology.com/news-releases/news-details/2023/Puma-Biotechnology-Receives-FDA-Orphan-Drug-Designation-for-Alisertib-for-the-Treatment-of-Small-Cell-Lung-Cancer/ (accessed on 21 September 2023).
- Tayyar, Y.; Jubair, L.; Fallaha, S.; McMillan, N.A. Critical risk-benefit assessment of the novel anti-cancer aurora a kinase inhibitor alisertib (MLN8237): A comprehensive review of the clinical data. Crit. Rev. Oncol. 2017, 119, 59–65. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd. Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target. Oncol. 2011, 6, 17–27. [Google Scholar] [CrossRef]
- Kirubakaran, S.; Shaik, A. Evolution of PIKK family kinase inhibitors A new age cancer therapeutics. Front. Biosci. 2020, 25, 1510–1537. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-competitive inhibitors of mTOR: An update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Ionkina, A.A.; Tentler, J.J.; Kim, J.; Capasso, A.; Pitts, T.M.; Ryall, K.A.; Howison, R.R.; Kabos, P.; Sartorius, C.A.; Tan, A.C.; et al. Efficacy and Molecular Mechanisms of Differentiated Response to the Aurora and Angiogenic Kinase Inhibitor ENMD-2076 in Preclinical Models of p53-Mutated Triple-Negative Breast Cancer. Front. Oncol. 2017, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xia, D.; Li, Z.; Zhou, T.; Chen, T.; Wu, Z.; Zhou, W.; Li, Z.; Li, L.; Xu, J. Aurora-A/ERK1/2/mTOR axis promotes tumor progression in triple-negative breast cancer and dual-targeting Aurora-A/mTOR shows synthetic lethality. Cell Death Dis. 2019, 10, 606. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-Z.; Long, Z.-J.; Peng, F.; Liu, Y.; Xu, J.; Wang, C.; Jiang, L.; Guo, T.; Kamran, M.; Li, S.-S.; et al. Aurora kinase A suppresses metabolic stress-induced autophagic cell death by activating mTOR signaling in breast cancer cells. Oncotarget 2014, 5, 7498–7511. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.L.; Ionkina, A.A.; Bagby, S.M.; Orth, J.D.; Gittleman, B.; Marcus, J.M.; Lam, E.T.; Corr, B.R.; O’Bryant, C.L.; Glode, A.E.; et al. Preclinical and Dose-Finding Phase I Trial Results of Combined Treatment with a TORC1/2 Inhibitor (TAK-228) and Aurora A Kinase Inhibitor (Alisertib) in Solid Tumors. Clin. Cancer Res. 2020, 26, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Taouli, B.; Koh, D.-M. Diffusion-weighted MR Imaging of the Liver. Radiology 2010, 254, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Padhani, A.R.; Liu, G.; Mu-Koh, D.; Chenevert, T.L.; Thoeny, H.C.; Takahara, T.; Dzik-Jurasz, A.; Ross, B.D.; Van Cauteren, M.; Collins, D.; et al. Diffusion-Weighted Magnetic Resonance Imaging as a Cancer Biomarker: Consensus and Recommendations. Neoplasia 2009, 11, 102–125. [Google Scholar] [CrossRef]
- Jordan, K.R.; Sikora, M.J.; Slansky, J.E.; Minic, A.; Richer, J.K.; Moroney, M.R.; Hu, J.; Wolsky, R.J.; Watson, Z.L.; Yamamoto, T.M.; et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin. Cancer Res. 2020, 26, 6362–6373. [Google Scholar] [CrossRef]
- Johnson, K. phenoptrReports: Create Reports Using Phenoptics Data. 2022. Available online: https://akoyabio.github.io/phenoptrReports/ (accessed on 7 February 2024).
- syngo.MR General Engine. Available online: https://www.siemens-healthineers.com/en-us/magnetic-resonance-imaging/options-and-upgrades/clinical-applications/syngo-mr-general-engine (accessed on 7 February 2024).
- Bilreiro, C.; Andrade, L.; Marques, R.M.; Matos, C. Diffusion-weighted imaging for determining response to neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis. Eur. Radiol. 2023, 1–11. [Google Scholar] [CrossRef]
- Schreuder, S.M.; Lensing, R.; Stoker, J.; Bipat, S. Monitoring treatment response in patients undergoing chemoradiotherapy for locally advanced uterine cervical cancer by additional diffusion-weighted imaging: A systematic review. J. Magn. Reson. Imaging 2015, 42, 572–594. [Google Scholar] [CrossRef]
- Tentler, J.J.; Ionkina, A.A.; Tan, A.C.; Newton, T.P.; Pitts, T.M.; Glogowska, M.J.; Kabos, P.; Sartorius, C.A.; Sullivan, K.D.; Espinosa, J.M.; et al. p53 Family Members Regulate Phenotypic Response to Aurora Kinase A Inhibition in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2015, 14, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Haddad, T.C.; Suman, V.J.; D’Assoro, A.B.; Carter, J.M.; Giridhar, K.V.; McMenomy, B.P.; Santo, K.; Mayer, E.L.; Karuturi, M.S.; Morikawa, A.; et al. Evaluation of Alisertib Alone or Combined with Fulvestrant in Patients With Endocrine-Resistant Advanced Breast Cancer: The Phase 2 TBCRC041 Randomized Clinical Trial. JAMA Oncol. 2023, 9, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.-C.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M.; et al. Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. JNCI J. Natl. Cancer Inst. 2015, 108, djv406. [Google Scholar] [CrossRef]
- Yin, T.; Zhao, Z.-B.; Guo, J.; Wang, T.; Yang, J.-B.; Wang, C.; Long, J.; Ma, S.; Huang, Q.; Zhang, K.; et al. Aurora A Inhibition Eliminates Myeloid Cell–Mediated Immunosuppression and Enhances the Efficacy of Anti–PD-L1 Therapy in Breast Cancer. Cancer Res 2019, 79, 3431–3444. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Jiang, Z.; Wang, C.; Chen, X.; Li, R.; Sun, N.; Liu, X.; Wang, H.; Hong, L.; Zheng, K.; et al. Inhibition of Aurora-A Promotes CD8+ T-Cell Infiltration by Mediating IL10 Production in Cancer Cells. Mol. Cancer Res. 2020, 18, 1589–1602. [Google Scholar] [CrossRef]
- Yu, J.; Green, M.D.; Li, S.; Sun, Y.; Journey, S.N.; Choi, J.E.; Rizvi, S.M.; Qin, A.; Waninger, J.J.; Lang, X.; et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 2021, 27, 152–164. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients (%) | |||
|---|---|---|---|
| Characteristic | Alisertib Lead-In (n = 10) Patients A-1 through A-10 | Sapanisertib Lead-In (n = 10) Patients S-1 through S-10 | Pancreatic Cancer Expansion (n = 11) Patients P-1 through P-11 |
| Age | |||
| Median (Range) | 59 (51–71) | 63 (39–74) | 56 (38–74) |
| Sex | |||
| Male | 2 (20%) | 4 (40%) | 9 (82%) |
| Female | 8 (80%) | 6 (60%) | 2 (18%) |
| Race/Ethnicity | |||
| Caucasian | 9 (90%) | 7 (70%) | 7 (64%) |
| Hispanic | 1 (10%) | 1 (10%) | 3 (27%) |
| African American | 0 (0%) | 1 (10%) | 1 (9%) |
| Asian | 0 (0%) | 1 (10%) | 0 (0%) |
| Tumor type | |||
| Breast adenocarcinoma | 5 (50%) | 4 (40%) | 0 (0%) |
| Colorectal adenocarcinoma | 2 (20%) | 2 (20%) | 0 (0%) |
| Ovarian serous carcinoma | 2 (20%) | 1 (0%) | 0 (0%) |
| Pancreatic adenocarcinoma | 1 (10%) | 2 (20%) | 11 (100%) |
| Uterine serous carcinoma | 0 (0%) | 1 (10%) | 0 (0%) |
| Renal cell carcinoma | 0 (0%) | 1 (10%) | 0 (0%) |
| Baseline ECOG Performance Status | |||
| 0 | 2 (20%) | 3 (30%) | 4 (36%) |
| 1 | 8 (80%) | 7 (70%) | 7 (64%) |
| Prior Lines of Therapy for Metastatic Disease | |||
| Median (Range) | 4 (3–14) | 3 (2–9) | 2 (1–3) |
| 1 | 0 (0%) | 0 (0%) | 1 (9%) |
| 2 | 0 (0%) | 3 (30%) | 7 (64%) |
| 3 | 2 (20%) | 3 (30%) | 3 (27%) |
| 4 | 4 (40%) | 0 (0%) | 0 (0%) |
| 5 or more | 4 (40%) | 4 (40%) | 0 (0%) |
| Alisertib Lead-In N = 10 | Sapanisertib Lead-In N = 10 | Pancreatic Cancer Expansion N = 11 | All Treatment Groups N = 31 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Number (percent) | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Total |
| Fatigue | 3 | 1 | 7 | 0 | 7 | 0 | 17 (55%) | 1 (3%) | 18 (58%) |
| Diarrhea | 3 | 1 | 2 | 0 | 6 | 1 | 11 (35%) | 2 (6%) | 13 (42%) |
| Nausea | 2 | 0 | 4 | 0 | 7 | 0 | 13 (42%) | 0 | 13 (42%) |
| Abdominal pain | 2 | 1 | 2 | 0 | 3 | 2 | 7 (23%) | 3 (10%) | 10 (32%) |
| Mucositis | 3 | 2 | 2 | 0 | 1 | 1 | 6 (19%) | 3 (10%) | 9 (29%) |
| Hyperglycemia | 2 | 1 | 2 | 0 | 3 | 0 | 7 (23%) | 1 (3%) | 8 (26%) |
| Anorexia | 2 | 0 | 2 | 0 | 4 | 0 | 8 (26%) | 0 | 8 (26%) |
| Hypokalemia | 2 | 0 | 0 | 0 | 4 | 1 | 6 (19%) | 1 (3%) | 7 (23%) |
| Neutropenia | 0 | 1 | 2 | 0 | 0 | 3 | 2 (6%) | 4 (13%) | 6 (20%) |
| Cognitive disturbance | 0 | 0 | 2 | 1 | 3 | 0 | 5 (16%) | 1 (3%) | 6 (20%) |
| Dyspnea | 3 | 0 | 0 | 1 | 2 | 0 | 5 (16%) | 1 (3%) | 6 (20%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, S.L.; Messersmith, W.A.; Purcell, W.T.; Lam, E.T.; Corr, B.R.; Leal, A.D.; Lieu, C.H.; O’Bryant, C.L.; Smoots, S.G.; Dus, E.D.; et al. A Phase Ib Expansion Cohort Evaluating Aurora A Kinase Inhibitor Alisertib and Dual TORC1/2 Inhibitor Sapanisertib in Patients with Advanced Solid Tumors. Cancers 2024, 16, 1456. https://doi.org/10.3390/cancers16081456
Davis SL, Messersmith WA, Purcell WT, Lam ET, Corr BR, Leal AD, Lieu CH, O’Bryant CL, Smoots SG, Dus ED, et al. A Phase Ib Expansion Cohort Evaluating Aurora A Kinase Inhibitor Alisertib and Dual TORC1/2 Inhibitor Sapanisertib in Patients with Advanced Solid Tumors. Cancers. 2024; 16(8):1456. https://doi.org/10.3390/cancers16081456
Chicago/Turabian StyleDavis, S. Lindsey, Wells A. Messersmith, W. Thomas Purcell, Elaine T. Lam, Bradley R. Corr, Alexis D. Leal, Christopher H. Lieu, Cindy L. O’Bryant, Stephen G. Smoots, Evan D. Dus, and et al. 2024. "A Phase Ib Expansion Cohort Evaluating Aurora A Kinase Inhibitor Alisertib and Dual TORC1/2 Inhibitor Sapanisertib in Patients with Advanced Solid Tumors" Cancers 16, no. 8: 1456. https://doi.org/10.3390/cancers16081456
APA StyleDavis, S. L., Messersmith, W. A., Purcell, W. T., Lam, E. T., Corr, B. R., Leal, A. D., Lieu, C. H., O’Bryant, C. L., Smoots, S. G., Dus, E. D., Jordan, K. R., Serkova, N. J., Pitts, T. M., & Diamond, J. R. (2024). A Phase Ib Expansion Cohort Evaluating Aurora A Kinase Inhibitor Alisertib and Dual TORC1/2 Inhibitor Sapanisertib in Patients with Advanced Solid Tumors. Cancers, 16(8), 1456. https://doi.org/10.3390/cancers16081456