
Contemporary Approaches to the Surgical Management of Pancreatic Neuroendocrine Tumors

Abstract
:Simple Summary
Abstract
1. Introduction
2. Histologic Classification
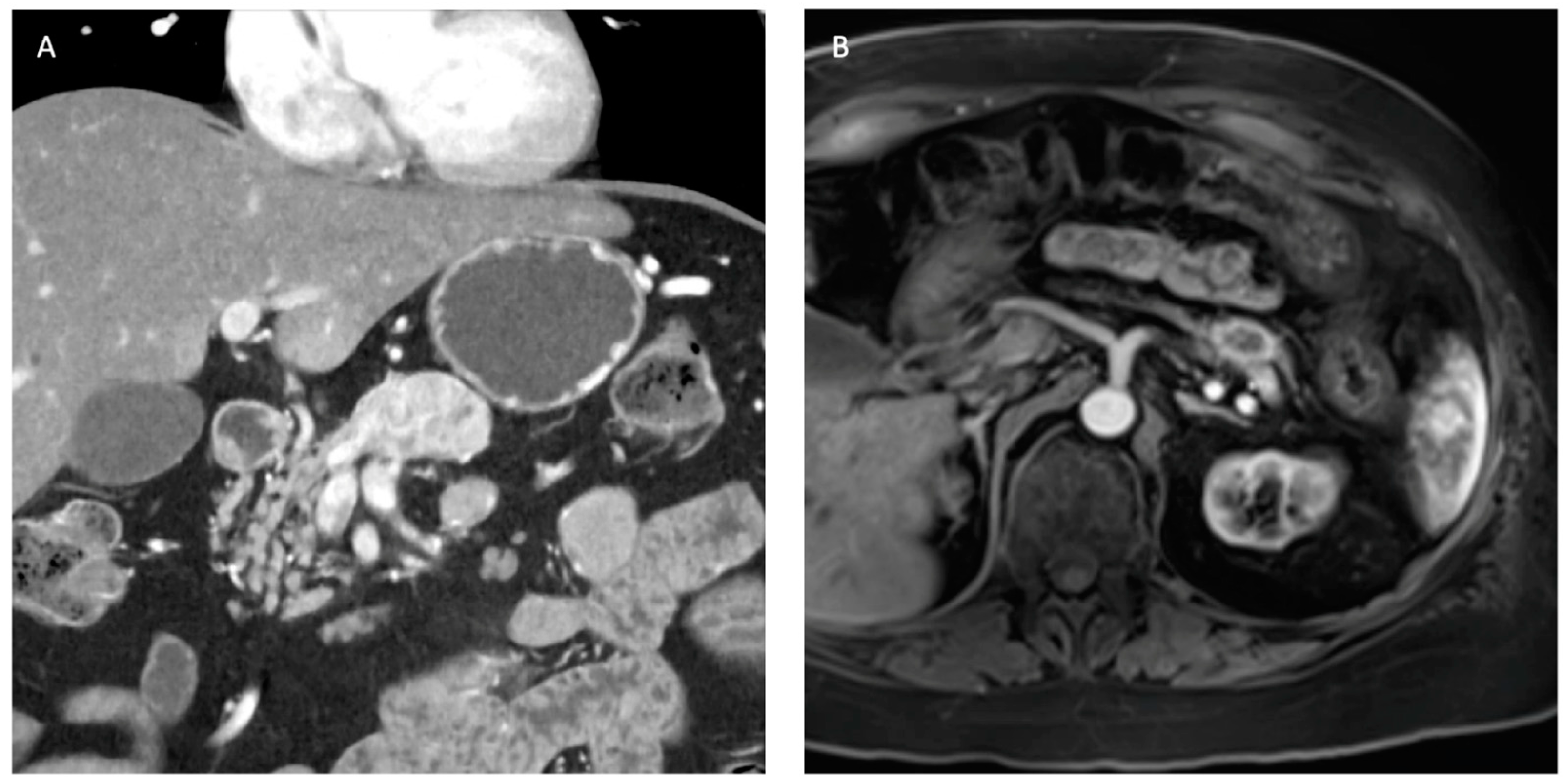
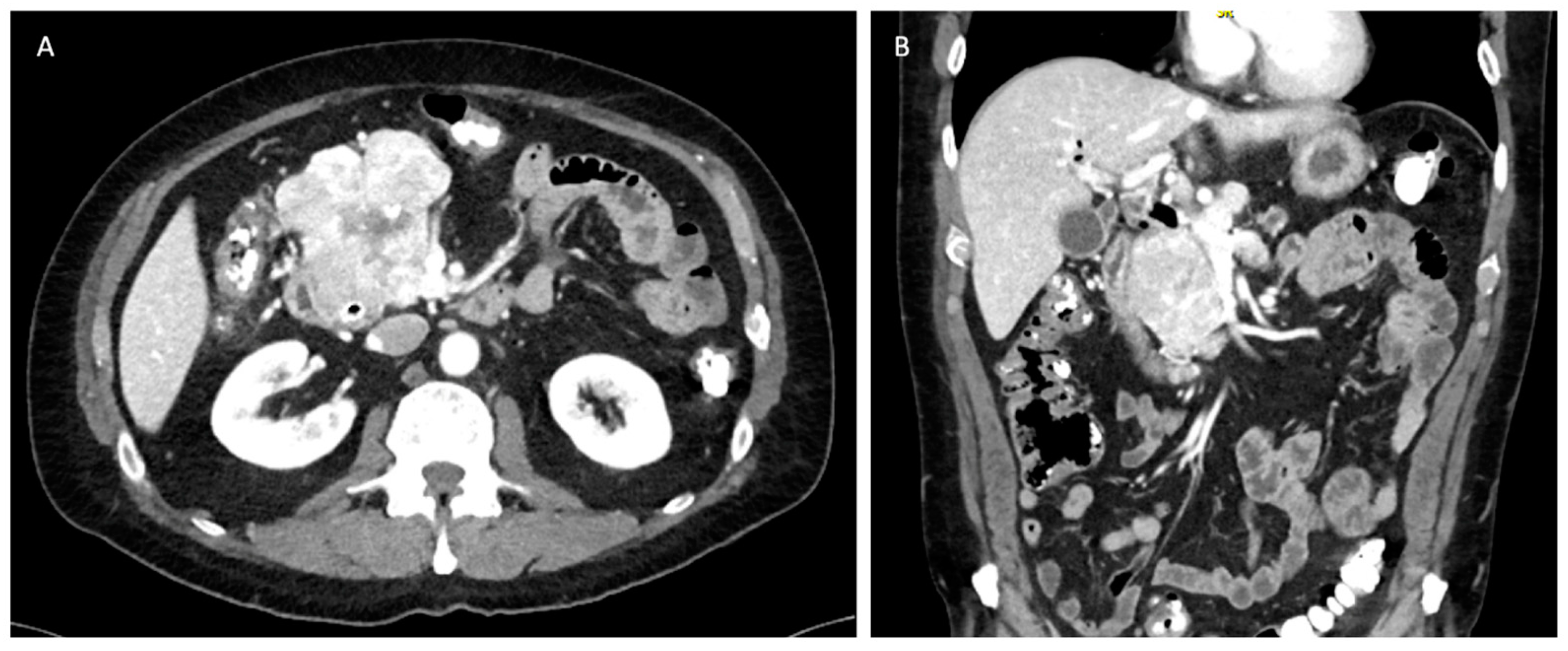
3. Radiographic Diagnostics
4. Clinical Staging
5. Non-Functional PNETs
5.1. Diagnosis of Non-Functional PNETs
5.2. Role of Tumor Markers
5.3. Surgical Management of NF-PNETs
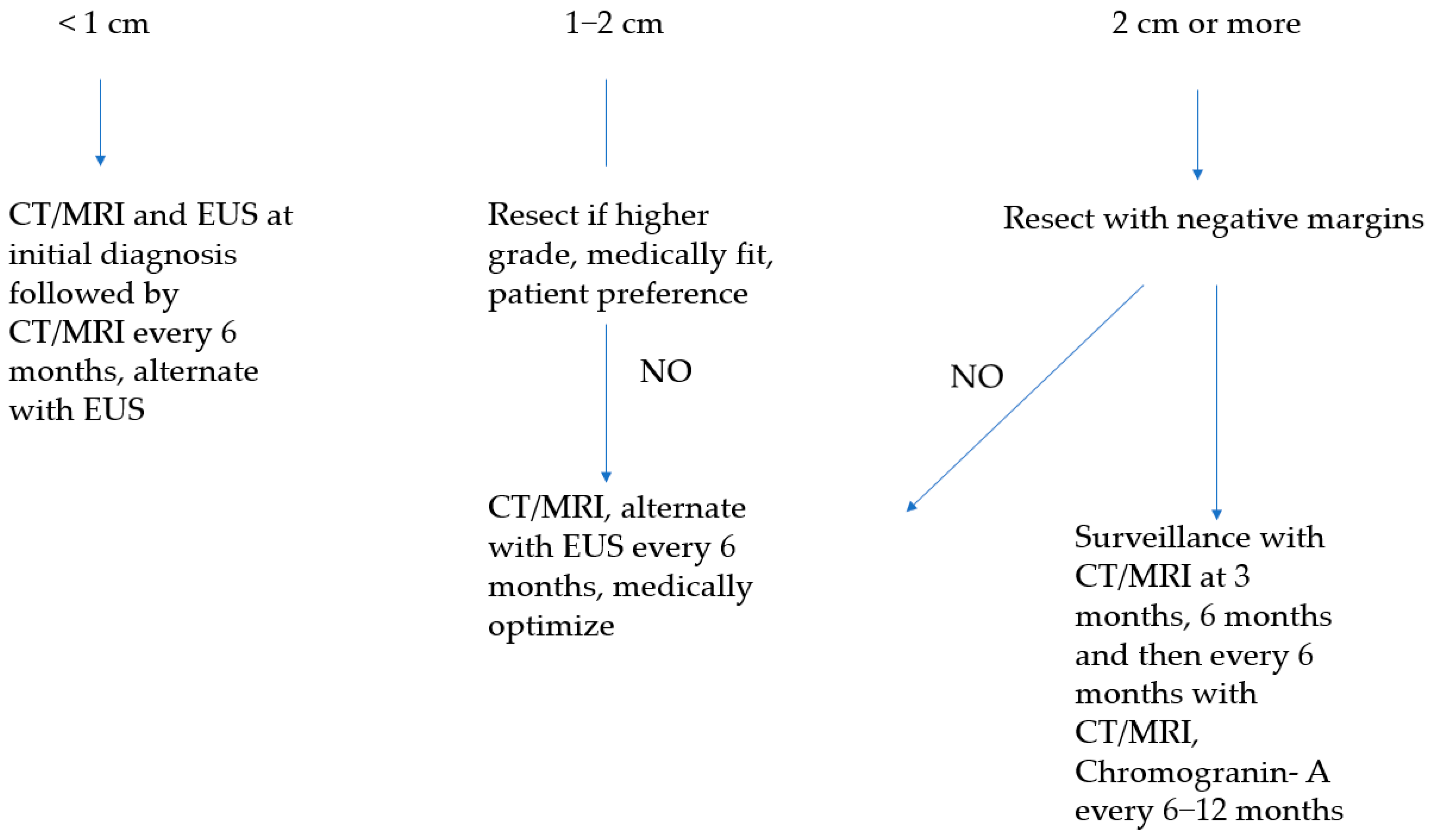
5.3.1. Sporadic Asymptomatic Tumors of Less Than 1 cm
5.3.2. Sporadic Asymptomatic Tumors of 1–2 cm
5.3.3. Sporadic Asymptomatic Tumors of 2 cm or Larger
5.3.4. Tumors with Evidence of Nodal Metastasis
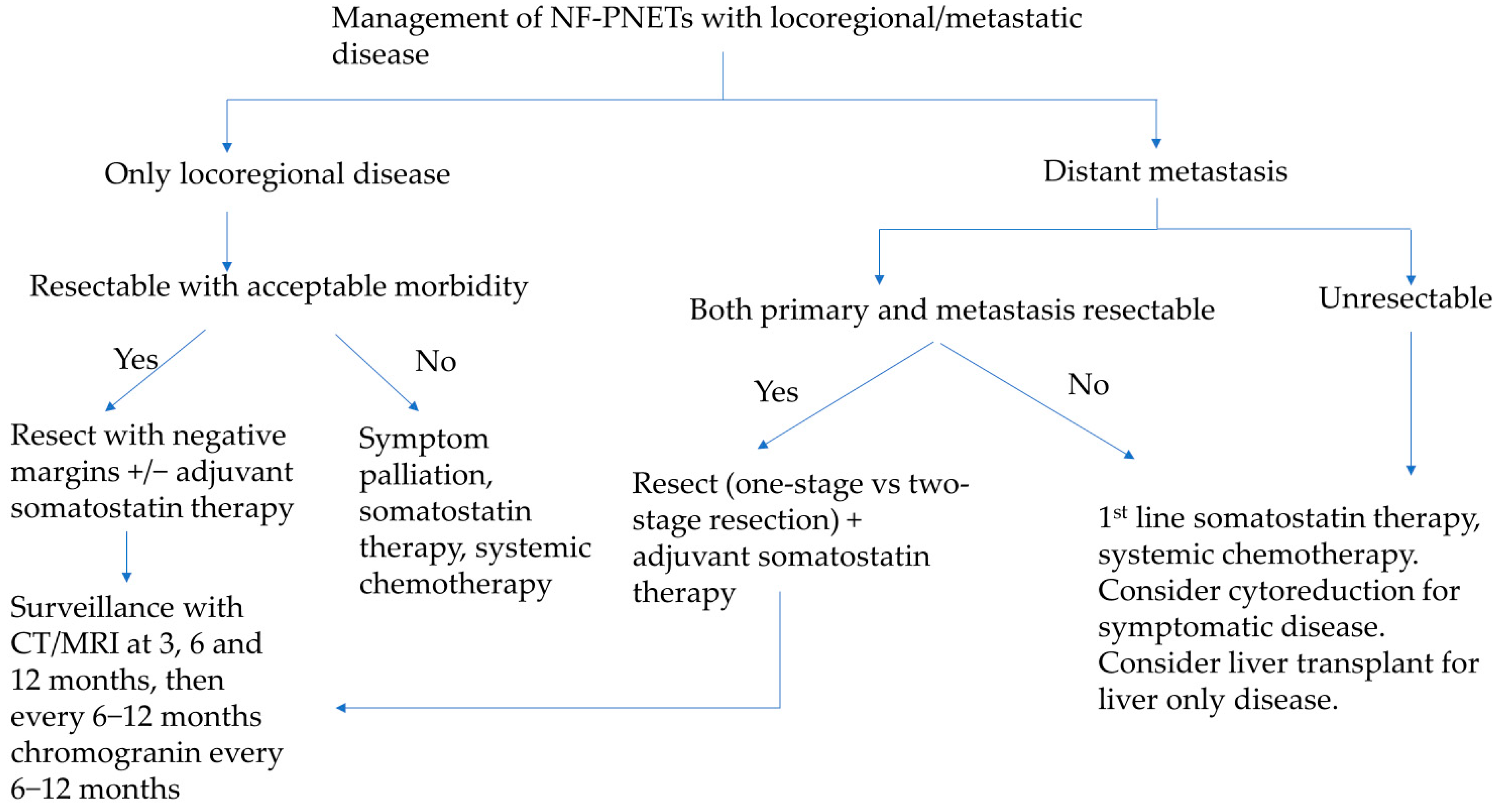
5.3.5. Tumors with Liver Metastasis
5.3.6. Role of Liver-Directed Therapies for Liver Metastasis

5.3.7. Open Versus Minimally Invasive Resection
5.3.8. Role of Neoadjuvant and Adjuvant Therapies for High Grade PNETs/NECs
6. Functional PNETs
6.1. Insulinomas
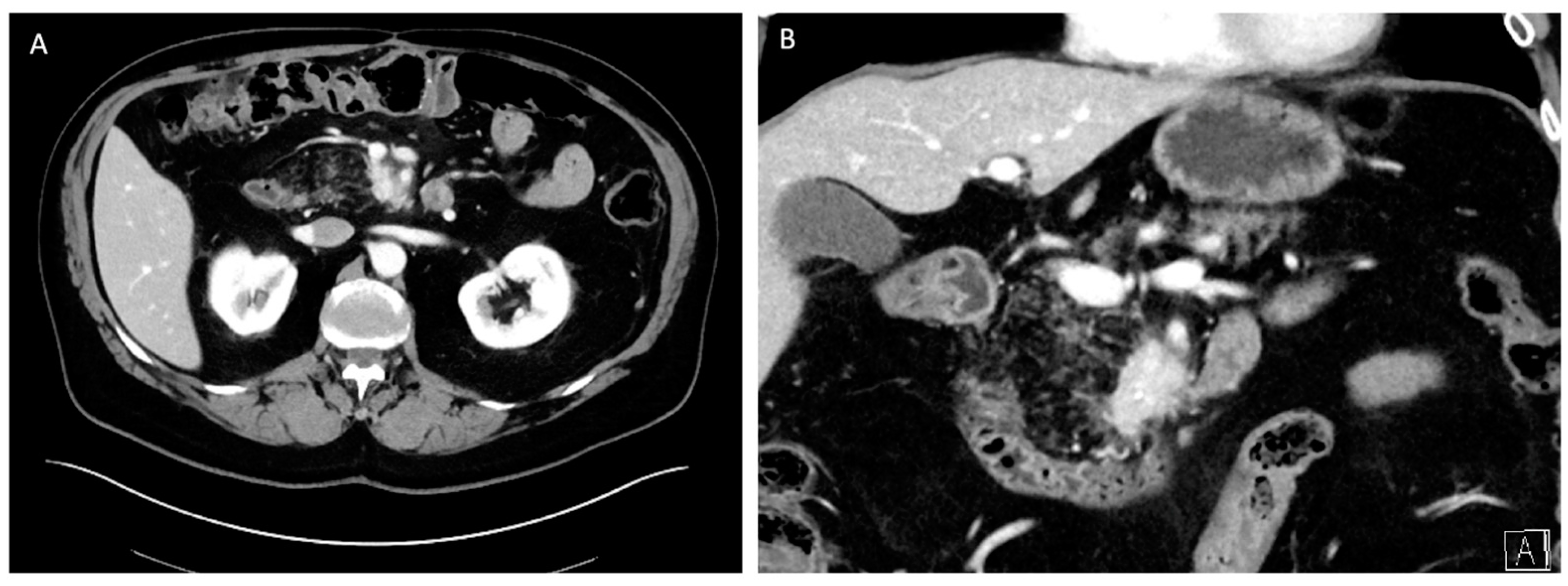
6.1.1. Diagnosis
6.1.2. Management
6.2. Gastrinomas
6.2.1. Diagnosis
6.2.2. Management
6.3. Glucagonoma
6.3.1. Diagnosis
6.3.2. Management
6.4. VIPoma
6.4.1. Diagnosis
6.4.2. Management
6.5. Somatostatinoma
6.5.1. Diagnosis
6.5.2. Management
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic Neuroendocrine Tumors (PNETs): Incidence, Prognosis and Recent Trend toward Improved Survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef]
- Franko, J.; Feng, W.; Yip, L.; Genovese, E.; Moser, A.J. Non-Functional Neuroendocrine Carcinoma of the Pancreas: Incidence, Tumor Biology, and Outcomes in 2,158 Patients. J. Gastrointest. Surg. 2010, 14, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Cheema, A.; Weber, J.; Strosberg, J.R. Incidental Detection of Pancreatic Neuroendocrine Tumors: An Analysis of Incidence and Outcomes. Ann. Surg. Oncol. 2012, 19, 2932–2936. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Partelli, S.; Maire, F.; D’Onofrio, M.; Larroque, B.; Tamburrino, D.; Sauvanet, A.; Falconi, M.; Ruszniewski, P. Observational Study of Natural History of Small Sporadic Nonfunctioning Pancreatic Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2013, 98, 4784–4789. [Google Scholar] [CrossRef] [PubMed]
- Vagefi, P.A.; Razo, O.; Deshpande, V.; McGrath, D.J.; Lauwers, G.Y.; Thayer, S.P.; Warshaw, A.L.; Castillo, C.F. Evolving Patterns in the Detection and Outcomes of Pancreatic Neuroendocrine Neoplasms. Arch. Surg. 2007, 142, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.; Gustafsson, B.I.; Chan, A.; Svejda, B.; Kidd, M.; Modlin, I.M. The Epidemiology of Gastroenteropancreatic Neuroendocrine Tumors. Endocrinol. Metab. Clin. N. Am. 2011, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.J.; Lee, K.-O.; Kaltsas, G. Epidemiology and Classification of Neuroendocrine Tumors of the Pancreas. In The Pancreas; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 919–929. ISBN 978-1-119-18842-1. [Google Scholar]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; for the WHO Classification of Tumours Editorial Board. The 2019 WHO Classification of Tumours of the Digestive System. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Kos-Kudła, B.; Blicharz-Dorniak, J.; Strzelczyk, J.; Bałdys-Waligórska, A.; Bednarczuk, T.; Bolanowski, M.; Boratyn-Nowicka, A.; Borowska, M.; Cichocki, A.; Ćwikła, J.B.; et al. Diagnostic and Therapeutic Guidelines for Gastro-Entero-Pancreatic Neuroendocrine Neoplasms (Recommended by the Polish Network of Neuroendocrine Tumours). Endokrynol. Pol. 2017, 68, 79–110. [Google Scholar] [CrossRef] [PubMed]
- Khanna, L.; Prasad, S.R.; Sunnapwar, A.; Kondapaneni, S.; Dasyam, A.; Tammisetti, V.S.; Salman, U.; Nazarullah, A.; Katabathina, V.S. Pancreatic Neuroendocrine Neoplasms: 2020 Update on Pathologic and Imaging Findings and Classification. RadioGraphics 2020, 40, 1240–1262. [Google Scholar] [CrossRef]
- Howe, J.R.; Merchant, N.B.; Conrad, C.; Keutgen, X.M.; Hallet, J.; Drebin, J.A.; Minter, R.M.; Lairmore, T.C.; Tseng, J.F.; Zeh, H.J.; et al. The North American Neuroendocrine Tumor Society Consensus Paper on the Surgical Management of Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, 1–33. [Google Scholar] [CrossRef]
- Deppen, S.A.; Blume, J.; Bobbey, A.J.; Shah, C.; Graham, M.M.; Lee, P.; Delbeke, D.; Walker, R.C. 68Ga-DOTATATE Compared with 111In-DTPA-Octreotide and Conventional Imaging for Pulmonary and Gastroenteropancreatic Neuroendocrine Tumors: A Systematic Review and Meta-Analysis. J. Nucl. Med. 2016, 57, 872–878. [Google Scholar] [CrossRef]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef]
- Panagiotidis, E.; Alshammari, A.; Michopoulou, S.; Skoura, E.; Naik, K.; Maragkoudakis, E.; Mohmaduvesh, M.; Al-Harbi, M.; Belda, M.; Caplin, M.E.; et al. Comparison of the Impact of 68Ga-DOTATATE and 18F-FDG PET/CT on Clinical Management in Patients with Neuroendocrine Tumors. J. Nucl. Med. 2017, 58, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Hindié, E. The NETPET Score: Combining FDG and Somatostatin Receptor Imaging for Optimal Management of Patients with Metastatic Well-Differentiated Neuroendocrine Tumors. Theranostics 2017, 7, 1159–1163. [Google Scholar] [CrossRef]
- You, Y.; Jang, J.-Y.; Kim, S.C.; Yoon, Y.-S.; Park, J.S.; Cho, C.K.; Park, S.-J.; Yang, J.D.; Lee, W.J.; Hong, T.H.; et al. Validation of the 8th AJCC Cancer Staging System for Pancreas Neuroendocrine Tumors Using Korean Nationwide Surgery Database. Cancer Res. Treat. 2019, 51, 1639–1652. [Google Scholar] [CrossRef]
- Foltyn, W.; Zajęcki, W.; Marek, B.; Kajdaniuk, D.; Siemińska, L.; Zemczak, A.; Kos-Kudła, B. The Value of the Ki-67 Proliferation Marker as a Prognostic Factor in Gastroenteropancreatic Neuroendocrine Tumours. Endokrynol. Pol. 2012, 63, 362–366. [Google Scholar]
- La Rosa, S. Diagnostic, Prognostic, and Predictive Role of Ki67 Proliferative Index in Neuroendocrine and Endocrine Neoplasms: Past, Present, and Future. Endocr. Pathol. 2023, 34, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.A.; Liu, T.-C.; Cavatiao, A.; Mawad, K.; Chen, L.; Strasberg, S.S.; Linehan, D.C.; Cao, D. Ki-67 Predicts Disease Recurrence and Poor Prognosis in Pancreatic Neuroendocrine Tumors. Surgery 2012, 152, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Aguiar, A.G.; Ethun, C.G.; Postlewait, L.M.; Zhelnin, K.; Krasinskas, A.; El-Rayes, B.F.; Russell, M.C.; Sarmiento, J.M.; Kooby, D.A.; Staley, C.A.; et al. Redefining the Ki-67 Index Stratification for Low-Grade Pancreatic Neuroendocrine Tumors: Improving Its Prognostic Value for Recurrence of Disease. Ann. Surg. Oncol. 2018, 25, 290–298. [Google Scholar] [CrossRef]
- Shyr, B.-S.; Shyr, B.-U.; Chen, S.-C.; Shyr, Y.-M.; Wang, S.-E. Impact of Tumor Grade on Pancreatic Neuroendocrine Tumors. Asian J. Surg. 2022, 45, 2659–2663. [Google Scholar] [CrossRef]
- Bilimoria, K.Y.; Tomlinson, J.S.; Merkow, R.P.; Stewart, A.K.; Ko, C.Y.; Talamonti, M.S.; Bentrem, D.J. Clinicopathologic Features and Treatment Trends of Pancreatic Neuroendocrine Tumors: Analysis of 9,821 Patients. J. Gastrointest. Surg. 2007, 11, 1460–1467; discussion 1467–1469. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, X.; Lu, X.; Zhao, Y.; Guan, W. Molecular Biology of Pancreatic Neuroendocrine Tumors: From Mechanism to Translation. Front. Oncol. 2022, 12, 967071. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.; Beyens, M.; Boons, G.; Schepers, A.; Kamp, K.; Biermann, K.; Pauwels, P.; De Herder, W.W.; Hofland, L.J.; Peeters, M.; et al. Hotspot DAXX, PTCH2 and CYFIP2 Mutations in Pancreatic Neuroendocrine Neoplasms. Endocr. Relat. Cancer 2019, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Totoki, Y.; Noë, M.; Nakatani, Y.; Horie, M.; Kawasaki, K.; Nakamura, H.; Saito-Adachi, M.; Suzuki, M.; Takai, E.; et al. Comprehensive Genomic Profiling of Neuroendocrine Carcinomas of the Gastrointestinal System. Cancer Discov. 2022, 12, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Choi, Y.H.; Kang, J.; Paik, W.H.; Lee, S.H.; Ryu, J.K.; Kim, Y.-T. Natural History of Small Pancreatic Lesions Suspected to Be Nonfunctioning Pancreatic Neuroendocrine Tumors. Pancreas 2018, 47, 1357. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, G.; Fu, D.; Jin, C.; Hao, S.; Yang, F.; Wang, X.; Yao, L.; Ni, Q. Preoperative Diagnosis of Nonfunctioning Pancreatic Neuroendocrine Tumors. Med. Oncol. 2011, 28, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Fujii, T.; Kanazumi, N.; Takeda, S.; Nomoto, S.; Kasuya, H.; Sugimoto, H.; Yamada, S.; Nakao, A. Nonfunctioning Neuroendocrine Pancreatic Tumors: Our Experience and Management. J. Hepatobiliary Pancreat. Surg. 2009, 16, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Molasy, B.; Zemła, P.; Mrowiec, S.; Grudzińska, E.; Kuśnierz, K. Evaluation of Risk Factors for Distant and Lymph Node Metastasis of Pancreatic Neuroendocrine Tumors. Ther. Clin. Risk Manag. 2022, 18, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Zerbi, A.; Falconi, M.; Rindi, G.; Delle Fave, G.; Tomassetti, P.; Pasquali, C.; Capitanio, V.; Boninsegna, L.; Di Carlo, V.; AISP-Network Study Group. Clinicopathological Features of Pancreatic Endocrine Tumors: A Prospective Multicenter Study in Italy of 297 Sporadic Cases. Am. J. Gastroenterol. 2010, 105, 1421–1429. [Google Scholar] [CrossRef]
- Jensen, R.T.; Cadiot, G.; Brandi, M.L.; de Herder, W.W.; Kaltsas, G.; Komminoth, P.; Scoazec, J.-Y.; Salazar, R.; Sauvanet, A.; Kianmanesh, R.; et al. ENETS Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Neoplasms: Functional Pancreatic Endocrine Tumor Syndromes. Neuroendocrinology 2011, 95, 98–119. [Google Scholar] [CrossRef]
- Sorbye, H.; Grande, E.; Pavel, M.; Tesselaar, M.; Fazio, N.; Reed, N.S.; Knigge, U.; Christ, E.; Ambrosini, V.; Couvelard, A.; et al. European Neuroendocrine Tumor Society (ENETS) 2023 Guidance Paper for Digestive Neuroendocrine Carcinoma. J. Neuroendocrinol. 2023, 35, e13249. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yan, H.; Xu, L.; Li, M.; Gao, W.; Jiang, K.; Wu, J.; Miao, Y. Correlation between Radiologic Features on Contrast-Enhanced CT and Pathological Tumor Grades in Pancreatic Neuroendocrine Neoplasms. J. Biomed. Res. 2021, 35, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Sallinen, V.; Le Large, T.Y.S.; Galeev, S.; Kovalenko, Z.; Tieftrunk, E.; Araujo, R.; Ceyhan, G.O.; Gaujoux, S. Surveillance Strategy for Small Asymptomatic Non-Functional Pancreatic Neuroendocrine Tumors—A Systematic Review and Meta-Analysis. HPB 2017, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Assi, H.A.; Mukherjee, S.; Kunz, P.L.; Machiorlatti, M.; Vesely, S.; Pareek, V.; Hatoum, H. Surgery Versus Surveillance for Well-Differentiated, Nonfunctional Pancreatic Neuroendocrine Tumors: An 11-Year Analysis of the National Cancer Database. Oncologist 2020, 25, e276–e283. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Rodriguez Franco, S.; Kirsch, M.J.; Colborn, K.L.; Ishida, J.; Grandi, S.; Al-Musawi, M.H.; Gleisner, A.; Del Chiaro, M.; Schulick, R.D. Evaluation of Survival Following Surgical Resection for Small Nonfunctional Pancreatic Neuroendocrine Tumors. JAMA Netw. Open 2023, 6, e234096. [Google Scholar] [CrossRef]
- Heidsma, C.M.; Engelsman, A.F.; van Dieren, S.; Stommel, M.W.J.; de Hingh, I.; Vriens, M.; Hol, L.; Festen, S.; Mekenkamp, L.; Hoogwater, F.J.H.; et al. Watchful Waiting for Small Non-Functional Pancreatic Neuroendocrine Tumours: Nationwide Prospective Cohort Study (PANDORA). Br. J. Surg. 2021, 108, 888–891. [Google Scholar] [CrossRef]
- Shah, M.H.; Goldner, W.S.; Halfdanarson, T.R.; Bergsland, E.; Berlin, J.D.; Halperin, D.; Chan, J.; Kulke, M.H.; Benson, A.B.; Blaszkowsky, L.S.; et al. NCCN Guidelines Insights: Neuroendocrine and Adrenal Tumors, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dey, C.; Kennecke, H.; Kocha, W.; Maroun, J.; Metrakos, P.; Mukhtar, T.; Pasieka, J.; Rayson, D.; Rowsell, C.; et al. Consensus Recommendations for the Diagnosis and Management of Pancreatic Neuroendocrine Tumors: Guidelines from a Canadian National Expert Group. Ann. Surg. Oncol. 2015, 22, 2685–2699. [Google Scholar] [CrossRef]
- Perinel, J.; Nappo, G.; Zerbi, A.; Heidsma, C.M.; Nieveen van Dijkum, E.J.M.; Han, H.S.; Yoon, Y.-S.; Satoi, S.; Demir, I.E.; Friess, H.; et al. Sporadic Nonfunctional Pancreatic Neuroendocrine Tumors: Risk of Lymph Node Metastases and Aggressiveness According to Tumor Size: A Multicenter International Study. Surgery 2022, 172, 975–981. [Google Scholar] [CrossRef]
- Heeger, K.; Falconi, M.; Partelli, S.; Waldmann, J.; Crippa, S.; Fendrich, V.; Bartsch, D.K. Increased Rate of Clinically Relevant Pancreatic Fistula after Deep Enucleation of Small Pancreatic Tumors. Langenbecks Arch. Surg. 2014, 399, 315–321. [Google Scholar] [CrossRef]
- Bolm, L.; Nebbia, M.; Wei, A.C.; Zureikat, A.H.; Fernández-del Castillo, C.; Zheng, J.; Pulvirenti, A.; Javed, A.A.; Sekigami, Y.; Petruch, N.; et al. Long-Term Outcomes of Parenchyma-Sparing and Oncologic Resections in Patients With Nonfunctional Pancreatic Neuroendocrine Tumors <3 Cm in a Large Multicenter Cohort. Ann. Surg. 2022, 276, 522. [Google Scholar] [CrossRef] [PubMed]
- Sallinen, V.J.; Le Large, T.Y.S.; Tieftrunk, E.; Galeev, S.; Kovalenko, Z.; Haugvik, S.-P.; Antila, A.; Franklin, O.; Martinez-Moneo, E.; Robinson, S.M.; et al. Prognosis of Sporadic Resected Small (≤2 Cm) Nonfunctional Pancreatic Neuroendocrine Tumors—A Multi-Institutional Study. HPB 2018, 20, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hashim, Y.M.; Trinkaus, K.M.; Linehan, D.C.; Strasberg, S.S.; Fields, R.C.; Cao, D.; Hawkins, W.G. Regional Lymphadenectomy Is Indicated in the Surgical Treatment of Pancreatic Neuroendocrine Tumors (PNETs). Ann. Surg. 2014, 259, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.R. Lymphadenectomy for Pancreatic Neuroendocrine Tumors: Is That the Relevant Debate? Ann. Surg. 2014, 259, 213. [Google Scholar] [CrossRef]
- Mansour, J.C.; Chavin, K.; Morris-Stiff, G.; Warner, S.G.; Cardona, K.; Fong, Z.V.; Maker, A.; Libutti, S.K.; Warren, R.; St Hill, C.; et al. Management of Asymptomatic, Well-Differentiated PNETs: Results of the Delphi Consensus Process of the Americas Hepato-Pancreato-Biliary Association. HPB 2019, 21, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.J.; Turrini, O.; Vigano, L.; Russolillo, N.; Autret, A.; Moutardier, V.; Capussotti, L.; Le Treut, Y.-P.; Delpero, J.-R.; Hardwigsen, J. Surgical Management of Advanced Pancreatic Neuroendocrine Tumors: Short-Term and Long-Term Results from an International Multi-Institutional Study. Ann. Surg. Oncol. 2015, 22, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Lesurtel, M.; Nagorney, D.M.; Mazzaferro, V.; Jensen, R.T.; Poston, G.J. When Should a Liver Resection Be Performed in Patients with Liver Metastases from Neuroendocrine Tumours? A Systematic Review with Practice Recommendations. HPB 2015, 17, 17–22. [Google Scholar] [CrossRef]
- Gaujoux, S.; Gonen, M.; Tang, L.; Klimstra, D.; Brennan, M.F.; D’Angelica, M.; Dematteo, R.; Allen, P.J.; Jarnagin, W.; Fong, Y. Synchronous Resection of Primary and Liver Metastases for Neuroendocrine Tumors. Ann. Surg. Oncol. 2012, 19, 4270–4277. [Google Scholar] [CrossRef]
- Bento de Sousa, J.H.; Calil, I.L.; Tustumi, F.; da Cunha Khalil, D.; Felga, G.E.G.; de Arruda Pecora, R.A.; de Almeida, M.D. Comparison between Milan and UCSF Criteria for Liver Transplantation in Patients with Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis. Transl. Gastroenterol. Hepatol. 2021, 6, 11. [Google Scholar] [CrossRef]
- Frilling, A.; Modlin, I.M.; Kidd, M.; Russell, C.; Breitenstein, S.; Salem, R.; Kwekkeboom, D.; Lau, W.; Klersy, C.; Vilgrain, V.; et al. Recommendations for Management of Patients with Neuroendocrine Liver Metastases. Lancet Oncol. 2014, 15, e8–e21. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Pulvirenti, A.; Coppa, J. Neuroendocrine Tumors Metastatic to the Liver: How to Select Patients for Liver Transplantation? J. Hepatol. 2007, 47, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zimmerman, M.A.; Hong, J.C. Liver Transplantation in the Treatment of Unresectable Hepatic Metastasis from Neuroendocrine Tumors. J. Gastrointest. Oncol. 2020, 11, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; McCarthy, G.; Berthon, A.; Dinet, J. Patient-Reported Health State Utilities in Metastatic Gastroenteropancreatic Neuroendocrine Tumours—An Analysis Based on the CLARINET Study. Health Qual. Life Outcomes 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Naraev, B.G.; Mailman, J.; Halfdanarson, T.R.; Soares, H.P.; Mittra, E.S.; Hallet, J. Consideration of Quality of Life in the Treatment Decision-Making for Patients with Advanced Gastroenteropancreatic Neuroendocrine Tumors. Expert. Rev. Anticancer Ther. 2023, 23, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.; Bianchi, L.; Di Vece, F.; Tombesi, P. Liver-Directed Therapies for Liver Metastases from Neuroendocrine Neoplasms: Can Laser Ablation Play Any Role? World J. Gastroenterol. 2020, 26, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Cloyd, J.M.; Ejaz, A.; Konda, B.; Makary, M.S.; Pawlik, T.M. Neuroendocrine Liver Metastases: A Contemporary Review of Treatment Strategies. Hepatobiliary Surg. Nutr. 2020, 9, 44051–44451. [Google Scholar] [CrossRef]
- Gococo-Benore, D.A.; Kuhlman, J.; Parent, E.E.; Sharma, A.; Accurso, J.; Yang, M.; Kendi, A.T.; Johnson, G.; Sonbol, M.B.; Hobday, T.; et al. Evaluation of Hepatotoxicity from Peptide Receptor Radionuclide Therapy in Patients with Gastroenteropancreatic Neuroendocrine Tumors and a Very High Liver Tumor Burden. J. Nucl. Med. 2023, 64, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Cazzato, R.L.; Hubelé, F.; De Marini, P.; Ouvrard, E.; Salvadori, J.; Addeo, P.; Garnon, J.; Kurtz, J.-E.; Greget, M.; Mertz, L.; et al. Liver-Directed Therapy for Neuroendocrine Metastases: From Interventional Radiology to Nuclear Medicine Procedures. Cancers 2021, 13, 6368. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Pulvirenti, A.; Javed, A.A.; Michelakos, T.; Paniccia, A.; Lee, K.K.; Ferrone, C.R.; Wei, A.C.; He, J.; Zureikat, A.H.; et al. Minimally Invasive vs Open Pancreatectomy for Pancreatic Neuroendocrine Tumors: Multi-Institutional 10-Year Experience of 1,023 Patients. J. Am. Coll. Surg. 2022, 235, 315–330. [Google Scholar] [CrossRef]
- Kim, J.; Hwang, H.K.; Lee, W.J.; Kang, C.M. Minimally Invasive vs Open Pancreatectomy for Nonfunctioning Pancreatic Neuroendocrine Tumors. World J. Gastrointest. Oncol. 2020, 12, 1133–1145. [Google Scholar] [CrossRef]
- Sutton, T.L.; Pommier, R.F.; Mayo, S.C.; Gilbert, E.W.; Papavasiliou, P.; Babicky, M.; Gerry, J.; Sheppard, B.C.; Worth, P.J. Similar Outcomes in Minimally Invasive versus Open Management of Primary Pancreatic Neuroendocrine Tumors: A Regional, Multi-Institutional Collaborative Analysis. Cancers 2022, 14, 1387. [Google Scholar] [CrossRef] [PubMed]
- Lania, A.; Ferraù, F.; Rubino, M.; Modica, R.; Colao, A.; Faggiano, A. Neoadjuvant Therapy for Neuroendocrine Neoplasms: Recent Progresses and Future Approaches. Front. Endocrinol. 2021, 12, 651438. [Google Scholar] [CrossRef] [PubMed]
- Parghane, R.V.; Ostwal, V.; Ramaswamy, A.; Bhandare, M.; Chaudhari, V.; Talole, S.; Shrikhande, S.V.; Basu, S. Long-Term Outcome of “Sandwich” Chemo-PRRT: A Novel Treatment Strategy for Metastatic Neuroendocrine Tumors with Both FDG- and SSTR-Avid Aggressive Disease. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Staszczak, A.; Pach, D.; Chrzan, R.; Trofimiuk, M.; Stefańska, A.; Tomaszuk, M.; Kołodziej, M.; Mikołajczak, R.; Pawlak, D.; Hubalewska-Dydejczyk, A. Peptide Receptor Radionuclide Therapy as a Potential Tool for Neoadjuvant Therapy in Patients with Inoperable Neuroendocrine Tumours (NETs). Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1669–1674. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Fine, R.L.; Choi, J.; Nasir, A.; Coppola, D.; Chen, D.-T.; Helm, J.; Kvols, L. First-Line Chemotherapy with Capecitabine and Temozolomide in Patients with Metastatic Pancreatic Endocrine Carcinomas. Cancer 2011, 117, 268–275. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.I.; van Eijck, C.H.; de Krijger, R.R.; Nieveen van Dijkum, E.J.; Teunissen, J.J.; Kam, B.L.; de Herder, W.W.; Feelders, R.A.; Bonsing, B.A.; Brabander, T.; et al. Neoadjuvant Treatment of Nonfunctioning Pancreatic Neuroendocrine Tumors with [177Lu-DOTA0,Tyr3]Octreotate. J. Nucl. Med. 2015, 56, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Arrivi, G.; Verrico, M.; Roberto, M.; Barchiesi, G.; Faggiano, A.; Marchetti, P.; Mazzuca, F.; Tomao, S. Capecitabine and Temozolomide (CAPTEM) in Advanced Neuroendocrine Neoplasms (NENs): A Systematic Review and Pooled Analysis. Cancer Manag. Res. 2022, 14, 3507–3523. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Mayerson, E.; Chae, Y.K.; Strosberg, J.; Wang, J.; Konda, B.; Hayward, J.; McLeod, C.M.; Chen, H.X.; Sharon, E.; et al. A Phase II Basket Trial of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART) SWOG S1609: High-Grade Neuroendocrine Neoplasm Cohort. Cancer 2021, 127, 3194–3201. [Google Scholar] [CrossRef]
- Liu, J.B.; Baker, M.S. Surgical Management of Pancreatic Neuroendocrine Tumors. Surg. Clin. N. Am. 2016, 96, 1447–1468. [Google Scholar] [CrossRef]
- Grant, C.S. Insulinoma. Best. Pract. Res. Clin. Gastroenterol. 2005, 19, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Service, F.J.; McMAHON, M.M.; O’brien, P.C.; Ballard, D.J. Functioning Insulinoma—Incidence, Recurrence, and Long-Term Survival of Patients: A 60-Year Study. Mayo Clin. Proc. 1991, 66, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Shima, Y.; Sumiyoshi, T.; Kozuki, A.; Ito, S.; Ogawa, Y.; Kobayashi, M.; Hanazaki, K. Diagnosis and Management of Insulinoma. World J. Gastroenterol. 2013, 19, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Cigrovski Berkovic, M.; Ulamec, M.; Marinovic, S.; Balen, I.; Mrzljak, A. Malignant Insulinoma: Can We Predict the Long-Term Outcomes? World J. Clin. Cases 2022, 10, 5124–5132. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Ciccolallo, L.; De Angelis, R.; Bouvier, A.M.; Faivre, J.; Gatta, G.; EUROCARE working group. European Disparities in Malignant Digestive Endocrine Tumours Survival. Int. J. Cancer 2010, 126, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Whipple, A.O.; Frantz, V.K. Adenoma of islet cells with hyperinsulinism. Ann. Surg. 1935, 101, 1299–1335. [Google Scholar] [CrossRef] [PubMed]
- Bolanle Ademolu, A. Whipple Triad Its Limitations in Diagnosis and Management of Hypoglycemia as a Co-Morbidity in Covid-19 Diabetics and Diabetes Mellitus in General- A Review. Indones. J. Devot. Empower. 2020, 5, 23. [Google Scholar] [CrossRef]
- Cryer, P.E.; Axelrod, L.; Grossman, A.B.; Heller, S.R.; Montori, V.M.; Seaquist, E.R.; Service, F.J. Evaluation and Management of Adult Hypoglycemic Disorders: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2009, 94, 709–728. [Google Scholar] [CrossRef]
- Hirshberg, B.; Livi, A.; Bartlett, D.L.; Libutti, S.K.; Alexander, H.R.; Doppman, J.L.; Skarulis, M.C.; Gorden, P. Forty-Eight-Hour Fast: The Diagnostic Test for Insulinoma. J. Clin. Endocrinol. Metab. 2000, 85, 3222–3226. [Google Scholar] [CrossRef]
- Balci, N.C.; Semelka, R.C. Radiologic Features of Cystic, Endocrine and Other Pancreatic Neoplasms. Eur. J. Radiol. 2001, 38, 113–119. [Google Scholar] [CrossRef]
- Wang, H.; Ba, Y.; Xing, Q.; Cai, R.-C. Diagnostic Value of ASVS for Insulinoma Localization: A Systematic Review and Meta-Analysis. PLoS ONE 2019, 14, e0224928. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Machida, H.; Kuwatsuru, R.; Saito, N.; Suzuki, K.; Iihara, M.; Obara, T.; Mitsuhashi, N. Preoperative Localization of Pancreatic Insulinoma by Super Selective Arterial Stimulation with Venous Sampling. Abdom. Imaging 2007, 32, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.-M.; Chen, J.-Y.; Won, J.G.-S.; Tseng, H.-S.; Yang, A.-H.; Wang, S.-E.; Lee, C.-H. The Role of Intra-Arterial Calcium Stimulation Test with Hepatic Venous Sampling (IACS) in the Management of Occult Insulinomas. Ann. Surg. Oncol. 2007, 14, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Zerbi, A.; Boninsegna, L.; Capitanio, V.; Partelli, S.; Balzano, G.; Pederzoli, P.; Di Carlo, V.; Falconi, M. Surgical Management of Insulinomas: Short- and Long-Term Outcomes After Enucleations and Pancreatic Resections. Arch. Surg. 2012, 147, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Wiese, D.; Humburg, F.G.; Kann, P.H.; Rinke, A.; Luster, M.; Mahnken, A.; Bartsch, D.K. Changes in Diagnosis and Operative Treatment of Insulinoma over Two Decades. Langenbecks Arch. Surg. 2023, 408, 255. [Google Scholar] [CrossRef] [PubMed]
- de Nucci, G.; Imperatore, N.; Mandelli, E.D.; di Nuovo, F.; d’Urbano, C.; Manes, G. Endoscopic Ultrasound-Guided Radiofrequency Ablation of Pancreatic Neuroendocrine Tumors: A Case Series. Endosc. Int. Open 2020, 8, E1754–E1758. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, N.; de Nucci, G.; Mandelli, E.D.; de Leone, A.; Zito, F.P.; Lombardi, G.; Manes, G. Endoscopic Ultrasound-Guided Radiofrequency Ablation of Pancreatic Neuroendocrine Tumors: A Systematic Review of the Literature. Endosc. Int. Open 2020, 8, E1759–E1764. [Google Scholar] [CrossRef] [PubMed]
- Crinò, S.F.; Napoleon, B.; Facciorusso, A.; Lakhtakia, S.; Borbath, I.; Caillol, F.; Do-Cong Pham, K.; Rizzatti, G.; Forti, E.; Palazzo, L.; et al. Endoscopic Ultrasound-Guided Radiofrequency Ablation Versus Surgical Resection for Treatment of Pancreatic Insulinoma. Clin. Gastroenterol. Hepatol. 2023, 21, 2834–2843.e2. [Google Scholar] [CrossRef] [PubMed]
- Crinò, S.F.; Partelli, S.; Napoleon, B.; Conti Bellocchi, M.C.; Facciorusso, A.; Salvia, R.; Forti, E.; Cintolo, M.; Mazzola, M.; Ferrari, G.; et al. Study Protocol for a Multicenter Randomized Controlled Trial to Compare Radiofrequency Ablation with Surgical Resection for Treatment of Pancreatic Insulinoma. Dig. Liver Dis. 2023, 55, 1187–1193. [Google Scholar] [CrossRef]
- van Beek, D.J.; Nell, S.; Verkooijen, H.M.; Borel Rinkes, I.H.M.; Valk, G.D.; Vriens, M.R. Surgery for Multiple Endocrine Neoplasia Type 1-related Insulinoma: Long-term Outcomes in a Large International Cohort. Br. J. Surg. 2020, 107, 1489–1499. [Google Scholar] [CrossRef]
- Kulke, M.H.; Anthony, L.B.; Bushnell, D.L.; de Herder, W.W.; Goldsmith, S.J.; Klimstra, D.S.; Marx, S.J.; Pasieka, J.L.; Pommier, R.F.; Yao, J.C.; et al. NANETS Treatment Guidelines: Well-Differentiated Neuroendocrine Tumors of the Stomach and Pancreas. Pancreas 2010, 39, 735–752. [Google Scholar] [CrossRef] [PubMed]
- van Beek, D.; Nell, S.; Pieterman, C.R.C.; de Herder, W.W.; van de Ven, A.C.; Dekkers, O.M.; van der Horst-Schrivers, A.N.; Drent, M.L.; Bisschop, P.H.; Havekes, B.; et al. Prognostic Factors and Survival in MEN1 Patients with Gastrinomas: Results from the DutchMEN Study Group (DMSG). J. Surg. Oncol. 2019, 120, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Chatzipanagiotou, O.; Schizas, D.; Vailas, M.; Tsoli, M.; Sakarellos, P.; Sotiropoulou, M.; Papalambros, A.; Felekouras, E. All You Need to Know about Gastrinoma Today | Gastrinoma and Zollinger-Ellison Syndrome: A Thorough Update. J. Neuroendocrinol. 2023, 35, e13267. [Google Scholar] [CrossRef] [PubMed]
- Berna, M.J.; Hoffmann, K.M.; Serrano, J.; Gibril, F.; Jensen, R.T. Serum Gastrin in Zollinger-Ellison Syndrome: I. Prospective Study of Fasting Serum Gastrin in 309 Patients from the National Institutes of Health and Comparison with 2229 Cases from the Literature. Medicine 2006, 85, 295–330. [Google Scholar] [CrossRef] [PubMed]
- Metz, D.C.; Cadiot, G.; Poitras, P.; Ito, T.; Jensen, R.T. Diagnosis of Zollinger-Ellison Syndrome in the Era of PPIs, Faulty Gastrin Assays, Sensitive Imaging and Limited Access to Acid Secretory Testing. Int. J. Endocr. Oncol. 2017, 4, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Gibril, F.; Jensen, R.T. Zollinger-Ellison Syndrome Revisited: Diagnosis, Biologic Markers, Associated Inherited Disorders, and Acid Hypersecretion. Curr. Gastroenterol. Rep. 2004, 6, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Elvevi, A.; Citterio, D.; Coppa, J.; Invernizzi, P.; Mazzaferro, V.; Massironi, S. Gastrinoma and Zollinger Ellison Syndrome: A Roadmap for the Management between New and Old Therapies. World J. Gastroenterol. 2021, 27, 5890–5907. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Takahashi, K.; Adachi, H.; Minematsu, S.; Shimada, Y.; Naito, M.; Suzuki, T.; Tobe, T.; Azuma, T. Usefulness of Selective Arterial Secretin Injection Test for Localization of Gastrinoma in the Zollinger-Ellison Syndrome. Ann. Surg. 1987, 205, 230–239. [Google Scholar] [CrossRef]
- Norton, J.A.; Fraker, D.L.; Alexander, H.R.; Jensen, R.T. Value of Surgery In Patients With Negative Imaging And Sporadic Zollinger-Ellison Syndrome (ZES). Ann. Surg. 2012, 256, 509–517. [Google Scholar] [CrossRef]
- Norton, J.A.; Harris, E.J.; Chen, Y.; Visser, B.C.; Poultsides, G.A.; Kunz, P.C.; Fisher, G.A.; Jensen, R.T. Pancreatic Endocrine Tumors with Major Vascular Abutment, Involvement, or Encasement and Indication for Resection. Arch. Surg. 2011, 146, 724–732. [Google Scholar] [CrossRef]
- Ellison, E.C.; Sparks, J.; Verducci, J.S.; Johnson, J.A.; Muscarella, P.; Bloomston, M.; Melvin, W.S. 50-Year Appraisal of Gastrinoma: Recommendations for Staging and Treatment. J. Am. Coll. Surg. 2006, 202, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Langer, P.; Waldmann, J.; Bartsch, D.K.; Rothmund, M. Management of Sporadic and Multiple Endocrine Neoplasia Type 1 Gastrinomas. Br. J. Surg. 2007, 94, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.J.; Pasieka, J.L.; Dixon, E.; Rorstad, O. The Palliative Benefit of Aggressive Surgical Intervention for Both Hepatic and Mesenteric Metastases from Neuroendocrine Tumors. Surgery 2008, 144, 645–653. [Google Scholar] [CrossRef]
- Guarnotta, V.; Martini, C.; Davì, M.V.; Pizza, G.; Colao, A.; Faggiano, A. NIKE group The Zollinger-Ellison Syndrome: Is There a Role for Somatostatin Analogues in the Treatment of the Gastrinoma? Endocrine 2018, 60, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.-H.; Schade-Brittinger, C.; Klose, K.-J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.-F.; Bläker, M.; et al. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors: A Report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Kindmark, H.; Sundin, A.; Granberg, D.; Dunder, K.; Skogseid, B.; Janson, E.T.; Welin, S.; Oberg, K.; Eriksson, B. Endocrine Pancreatic Tumors with Glucagon Hypersecretion: A Retrospective Study of 23 Cases during 20 Years. Med. Oncol. 2007, 24, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic Neuroendocrine Tumors: Clinical Features, Diagnosis and Medical Treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef]
- Wermers, R.A.; Fatourechi, V.; Kvols, L.K. Clinical Spectrum of Hyperglucagonemia Associated with Malignant Neuroendocrine Tumors. Mayo Clin. Proc. 1996, 71, 1030–1038. [Google Scholar] [CrossRef]
- Eldor, R.; Glaser, B.; Fraenkel, M.; Doviner, V.; Salmon, A.; Gross, D.J. Glucagonoma and the Glucagonoma Syndrome—Cumulative Experience with an Elusive Endocrine Tumour. Clin. Endocrinol. 2011, 74, 593–598. [Google Scholar] [CrossRef]
- Wermers, R.A.; Fatourechi, V.; Wynne, A.G.; Kvols, L.K.; Lloyd, R.V. The Glucagonoma Syndrome. Clinical and Pathologic Features in 21 Patients. Medicine 1996, 75, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1 and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Su, Y.; Cheng, Y.; Jiang, X.; Peng, Y.; Li, Y.; Lu, J.; Gu, Y.; Zhang, C.; Cao, Y.; et al. Conditional Deletion of Men1 in the Pancreatic β-Cell Leads to Glucagon-Expressing Tumor Development. Endocrinology 2015, 156, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lévy-Bohbot, N.; Merle, C.; Goudet, P.; Delemer, B.; Calender, A.; Jolly, D.; Thiéfin, G.; Cadiot, G. Prevalence, Characteristics and Prognosis of MEN 1-Associated Glucagonomas, VIPomas, and Somatostatinomas: Study from the GTE (Groupe des Tumeurs Endocrines) Registry. Gastroenterol. Clin. Biol. 2004, 28, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- John, A.M.; Schwartz, R.A. Glucagonoma Syndrome: A Review and Update on Treatment. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 2016–2022. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, Y.; Zhang, T.; Liao, Q.; Cong, L. Clinical experience in diagnosis and treatment of glucagonoma. Zhonghua Wai Ke Za Zhi 2009, 47, 333–336. [Google Scholar]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Verner, J.V.; Morrison, A.B. Islet Cell Tumor and a Syndrome of Refractory Watery Diarrhea and Hypokalemia. Am. J. Med. 1958, 25, 374–380. [Google Scholar] [CrossRef]
- Yao, J.C.; Eisner, M.P.; Leary, C.; Dagohoy, C.; Phan, A.; Rashid, A.; Hassan, M.; Evans, D.B. Population-Based Study of Islet Cell Carcinoma. Ann. Surg. Oncol. 2007, 14, 3492–3500. [Google Scholar] [CrossRef]
- Barbezat, G.O.; Grossman, M.I. Intestinal Secretion: Stimulation by Peptides. Science 1971, 174, 422–424. [Google Scholar] [CrossRef]
- de Herder, W.W.; Hofland, J. Vasoactive Intestinal Peptide-Secreting Tumor (VIPoma). In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Soga, J.; Yakuwa, Y. Vipoma/Diarrheogenic Syndrome: A Statistical Evaluation of 241 Reported Cases. J. Exp. Clin. Cancer Res. 1998, 17, 389–400. [Google Scholar]
- Schizas, D.; Mastoraki, A.; Bagias, G.; Patras, R.; Moris, D.; Lazaridis, I.I.; Arkadopoulos, N.; Felekouras, E. Clinicopathological Data and Treatment Modalities for Pancreatic Vipomas: A Systematic Review. J. BUON 2019, 24, 415–423. [Google Scholar] [PubMed]
- Angelousi, A.; Koffas, A.; Grozinsky-Glasberg, S.; Gertner, J.; Kassi, E.; Alexandraki, K.; Caplin, M.E.; Kaltsas, G.; Toumpanakis, C. Diagnostic and Management Challenges in Vasoactive Intestinal Peptide Secreting Tumors: A Series of 15 Patients. Pancreas 2019, 48, 934. [Google Scholar] [CrossRef] [PubMed]
- Abdullayeva, L. VIPoma: Mechanisms, Clinical Presentation, Diagnosis and Treatment (Review). World Acad. Sci. J. 2019, 1, 229–235. [Google Scholar] [CrossRef]
- Azizian, A.; König, A.; Ghadimi, M. Treatment Options of Metastatic and Nonmetastatic VIPoma: A Review. Langenbecks Arch. Surg. 2022, 407, 2629–2636. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.Y.; Li, J.T.; Liu, Y.B.; Fang, H.Q.; Wu, Y.L.; Peng, C.H.; Wang, X.B.; Qian, H.R. Diagnosis and Treatment of VIPoma in China: (Case Report and 31 Cases Review) Diagnosis and Treatment of VIPoma. Pancreas 2004, 28, 93. [Google Scholar] [CrossRef] [PubMed]
- O’Dorisio, T.M.; Gaginella, T.S.; Mekhjian, H.S.; Rao, B.; O’Dorisio, M.S. Somatostatin and Analogues in the Treatment of VIPoma. Ann. N. Y. Acad. Sci. 1988, 527, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Ruskoné, A.; René, E.; Chayvialle, J.A.; Bonin, N.; Pignal, F.; Kremer, M.; Bonfils, S.; Rambaud, J.C. Effect of Somatostatin on Diarrhea and on Small Intestinal Water and Electrolyte Transport in a Patient with Pancreatic Cholera. Dig. Dis. Sci. 1982, 27, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Kaltsas, G.; Androulakis, I.I.; de Herder, W.W.; Grossman, A.B. Paraneoplastic Syndromes Secondary to Neuroendocrine Tumours. Endocr. Relat. Cancer 2010, 17, R173–R193. [Google Scholar] [CrossRef]
- Oberg, K.; Couvelard, A.; Delle Fave, G.; Gross, D.; Grossman, A.; Jensen, R.T.; Pape, U.-F.; Perren, A.; Rindi, G.; Ruszniewski, P.; et al. ENETS Consensus Guidelines for Standard of Care in Neuroendocrine Tumours: Biochemical Markers. Neuroendocrinology 2017, 105, 201–211. [Google Scholar] [CrossRef]
- Arrojo e Drigo, R.; Jacob, S.; García-Prieto, C.F.; Zheng, X.; Fukuda, M.; Nhu, H.T.T.; Stelmashenko, O.; Peçanha, F.L.M.; Rodriguez-Diaz, R.; Bushong, E.; et al. Structural Basis for Delta Cell Paracrine Regulation in Pancreatic Islets. Nat. Commun. 2019, 10, 3700. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Thorn, C.; Spalding, D.; Williamson, R. Pancreatic and Peripancreatic Somatostatinomas. Ann. R. Coll. Surg. Engl. 2011, 93, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Garbrecht, N.; Anlauf, M.; Schmitt, A.; Henopp, T.; Sipos, B.; Raffel, A.; Eisenberger, C.F.; Knoefel, W.T.; Pavel, M.; Fottner, C.; et al. Somatostatin-Producing Neuroendocrine Tumors of the Duodenum and Pancreas: Incidence, Types, Biological Behavior, Association with Inherited Syndromes, and Functional Activity. Endocr.-Relat. Cancer 2008, 15, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Sandru, F.; Carsote, M.; Valea, A.; Albu, S.E.; Petca, R.-C.; Dumitrascu, M.C. Somatostatinoma: Beyond Neurofibromatosis Type 1 (Review). Exp. Ther. Med. 2020, 20, 3383–3388. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.; Pappo, A.; Reiss, U.; Shulkin, B.L.; Zhuang, Z.; Pacak, K.; Bahrami, A. Clinical Manifestations of Pacak-Zhuang Syndrome in a Male Pediatric Patient. Pediatr. Blood Cancer 2020, 67, e28096. [Google Scholar] [CrossRef]
- Vianna, P.M.; Ferreira, C.R.; Campos, F.P.F.D. Somatostatinoma Syndrome: A Challenging Differential Diagnosis among Pancreatic Tumors. ACR 2013, 3, 29–37. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terminology | Differentiation | Grade | Mitotic Rate * (Mitoses/2 mm2) | Ki-67 Index * |
|---|---|---|---|---|
| NET, G1 | Well differentiated | Low | <2 | <3% |
| NET, G2 | Intermediate | 2–20 | 3–20% | |
| NET, G3 | High | >20 | >20% | |
| NEC, small-cell type (SCNEC) | Poorly differentiated | High † | >20 | >20% |
| NEC, large-cell type (LCNEC) | >20 | >20% | ||
| MiNEN | Well or poorly differentiated ‡ | Variable ‡ | Variable ‡ | Variable ‡ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartik, A.; Armstrong, V.L.; Stucky, C.-C.; Wasif, N.; Fong, Z.V. Contemporary Approaches to the Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers 2024, 16, 1501. https://doi.org/10.3390/cancers16081501
Kartik A, Armstrong VL, Stucky C-C, Wasif N, Fong ZV. Contemporary Approaches to the Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers. 2024; 16(8):1501. https://doi.org/10.3390/cancers16081501
Chicago/Turabian StyleKartik, Akash, Valerie L. Armstrong, Chee-Chee Stucky, Nabil Wasif, and Zhi Ven Fong. 2024. "Contemporary Approaches to the Surgical Management of Pancreatic Neuroendocrine Tumors" Cancers 16, no. 8: 1501. https://doi.org/10.3390/cancers16081501
APA StyleKartik, A., Armstrong, V. L., Stucky, C. -C., Wasif, N., & Fong, Z. V. (2024). Contemporary Approaches to the Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers, 16(8), 1501. https://doi.org/10.3390/cancers16081501