
Lysine Methyltransferase 9 (KMT9) Is an Actionable Target in Muscle-Invasive Bladder Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Transfection with siRNA
2.4. Cell Proliferation Assay
2.5. Cell Migration and Invasion Assay
2.6. Organoid Isolation
2.7. Organoid Culture and Size Assessment
2.8. Preparation of Cell Extracts
2.9. Western Blot
2.10. RNA Preparation and RNAseq
2.11. RNA Preparation for Quantitative RT-PCR and Analysis
2.12. MTT Assay
2.13. Growth of Xenograft Tumors in NOD/SCID Mice
2.14. Statistical Analysis
3. Results
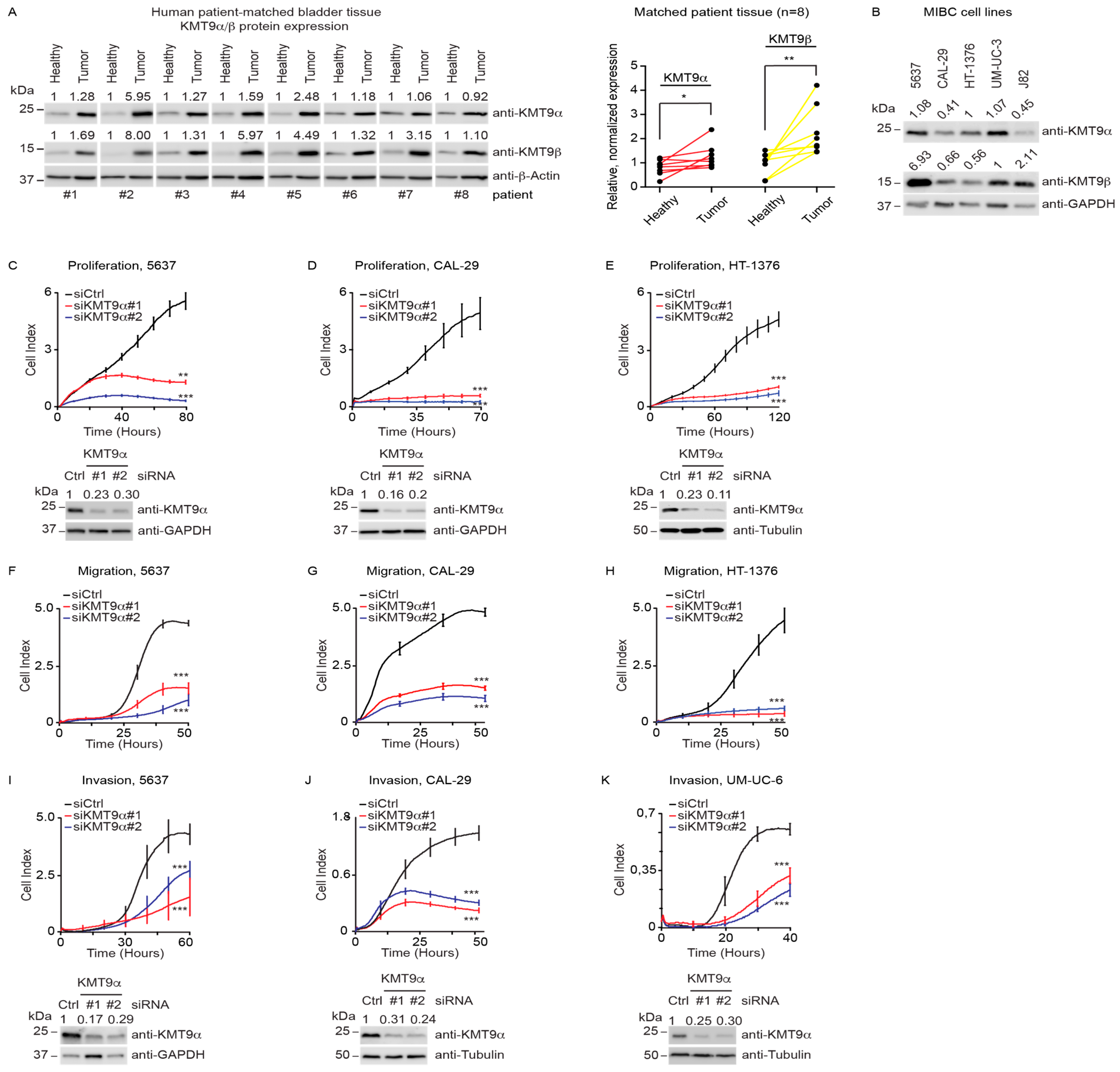
3.1. KMT9α Depletion Blocks Proliferation and Migration of MIBC Cell Lines
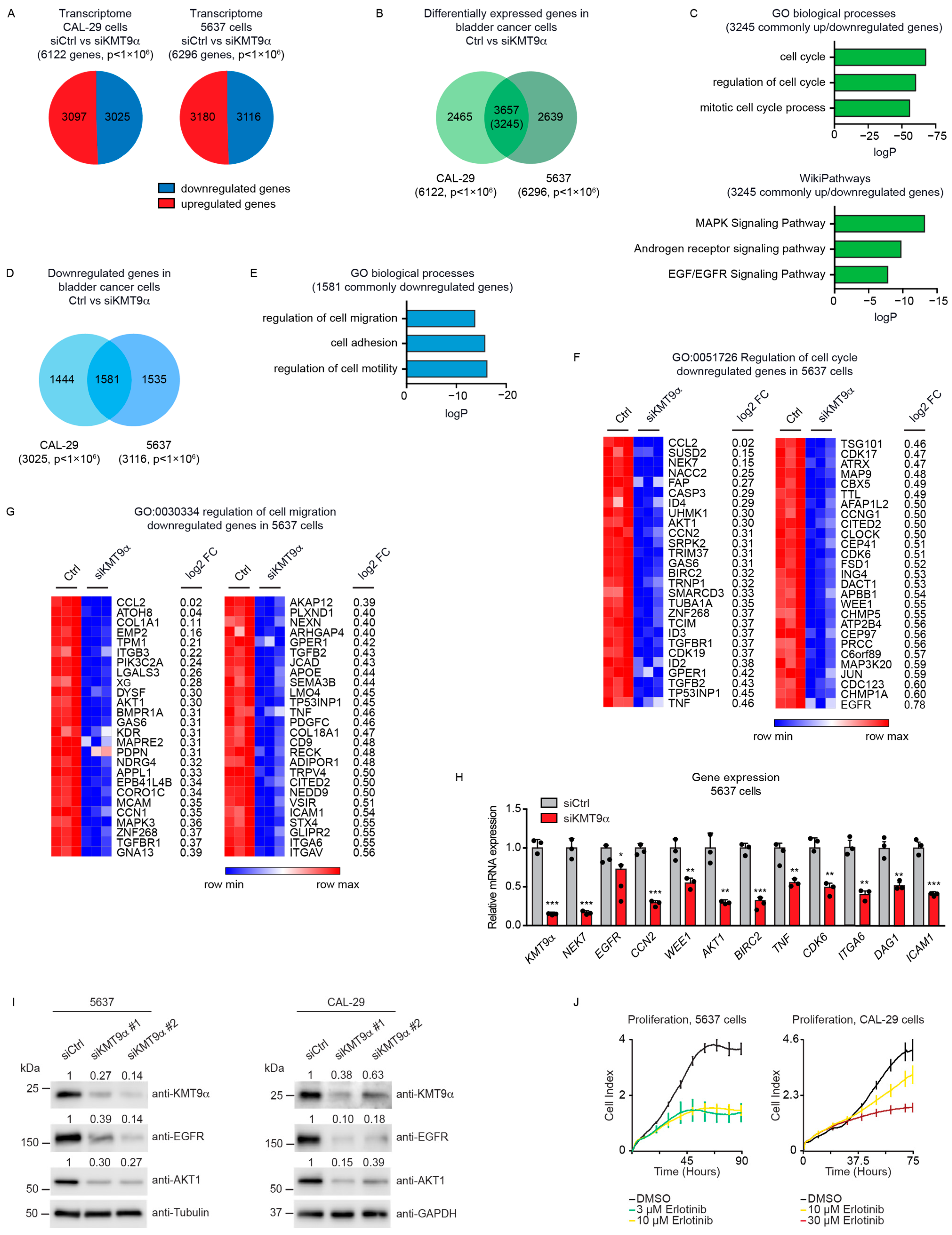
3.2. Knockdown of KMT9α Affects Expression of Proliferation- and Migration-Related Genes
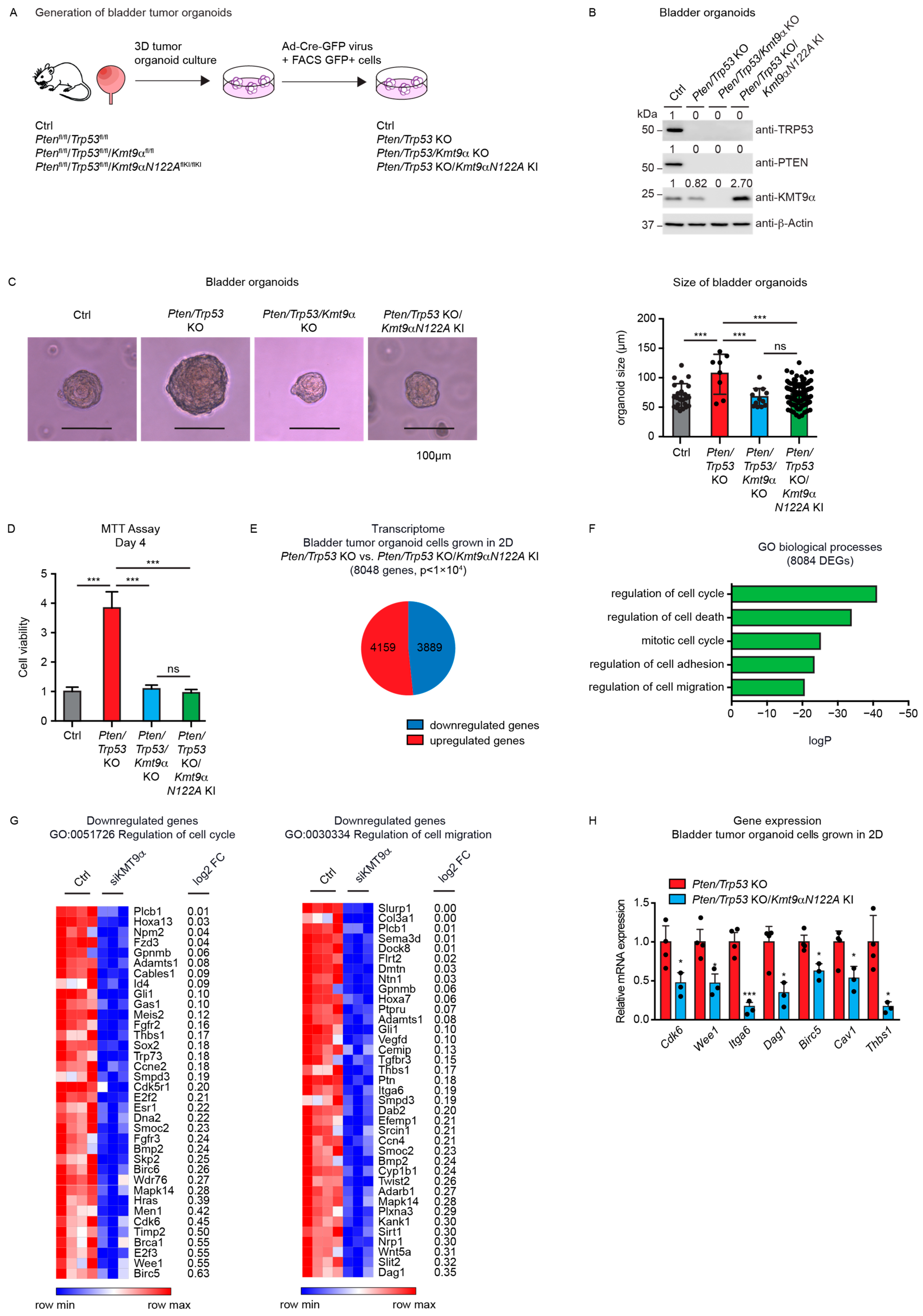
3.3. Cell Proliferation in Mouse Bladder Tumor Organoids Depends on the Catalytic Activity of KMT9
3.4. KMT9α Depletion Impairs BC Growth in Immunodeficient NOD SCID Mice
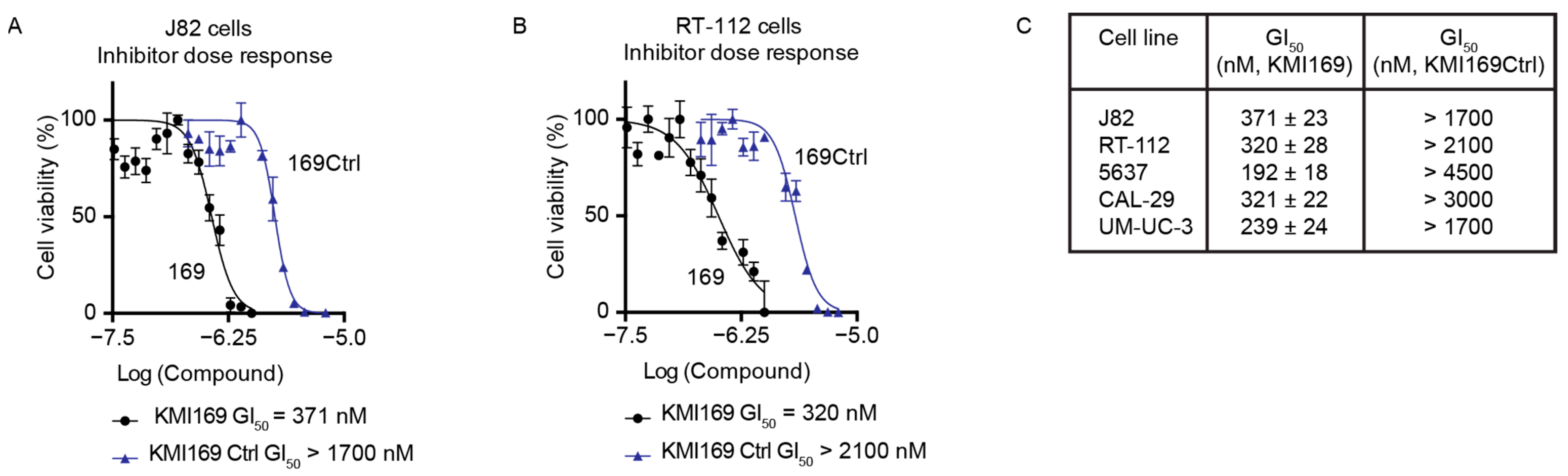
3.5. Inhibition of KMT9 Activity Blocks Proliferation of MIBC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sanli, O.; Dobruch, J.; Knowles, M.A.; Burger, M.; Alemozaffar, M.; Nielsen, M.E.; Lotan, Y. Bladder Cancer. Nat. Rev. Dis. Primers 2017, 3, 17022. [Google Scholar] [CrossRef]
- Schwarzova, L.; Varchulova Novakova, Z.; Danisovic, L.; Ziaran, S. Molecular Classification of Urothelial Bladder Carcinoma. Mol. Biol. Rep. 2023, 50, 7867–7877. [Google Scholar] [CrossRef]
- Chang, S.S.; Boorjian, S.A.; Chou, R.; Clark, P.E.; Daneshmand, S.; Konety, B.R.; Pruthi, R.; Quale, D.Z.; Ritch, C.R.; Seigne, J.D.; et al. Diagnosis and Treatment of Non-Muscle Invasive Bladder Cancer: Aua/Suo Guideline. J. Urol. 2016, 196, 1021–1029. [Google Scholar] [CrossRef]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Chang, S.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; et al. Bladder Cancer, Version 3. 2020, Nccn Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 329–354. [Google Scholar] [CrossRef]
- Kamoun, A.; De Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-Invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Shoemaker, R.; Kim, J. Urobiome: An Outlook on the Metagenome of Urological Diseases. Investig. Clin. Urol. 2021, 62, 611. [Google Scholar] [CrossRef]
- Cumberbatch, M.G.K.; Jubber, I.; Black, P.C.; Esperto, F.; Figueroa, J.D.; Kamat, A.M.; Kiemeney, L.; Lotan, Y.; Pang, K.; Silverman, D.T.; et al. Epidemiology of Bladder Cancer: A Systematic Review and Contemporary Update of Risk Factors in 2018. Eur. Urol. 2018, 74, 784–795. [Google Scholar] [CrossRef]
- Pezzuto, A.; Citarella, F.; Croghan, I.; Tonini, G. The Effects of Cigarette Smoking Extracts on Cell Cycle and Tumor Spread: Novel Evidence. Future Sci. OA 2019, 5, FSO394. [Google Scholar] [CrossRef]
- Friedrich, V.; Choi, H.W. The Urinary Microbiome: Role in Bladder Cancer and Treatment. Diagnostics 2022, 12, 2068. [Google Scholar] [CrossRef]
- Tabayoyong, W.; Kamat, A.M. Current Use and Promise of Urinary Markers for Urothelial Cancer. Curr. Urol. Rep. 2018, 19, 96. [Google Scholar] [CrossRef]
- Ng, K.; Stenzl, A.; Sharma, A.; Vasdev, N. Urinary Biomarkers in Bladder Cancer: A Review of the Current Landscape and Future Directions. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 41–51. [Google Scholar] [CrossRef]
- Nardelli, C.; Aveta, A.; Pandolfo, S.D.; Tripodi, L.; Russo, F.; Imbimbo, C.; Castaldo, G.; Pastore, L. Microbiome Profiling in Bladder Cancer Patients Using the First-Morning Urine Sample. Eur. Urol. Open Sci. 2024, 59, 18–26. [Google Scholar] [CrossRef]
- Hussein, A.A.; Elsayed, A.S.; Durrani, M.; Jing, Z.; Iqbal, U.; Gomez, E.C.; Singh, P.K.; Liu, S.; Smith, G.; Tang, L.; et al. Investigating the Association between the Urinary Microbiome and Bladder Cancer: An Exploratory Study. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 370.e9–370.e19. [Google Scholar] [CrossRef]
- Oresta, B.; Braga, D.; Lazzeri, M.; Frego, N.; Saita, A.; Faccani, C.; Fasulo, V.; Colombo, P.; Guazzoni, G.; Hurle, R.; et al. The Microbiome of Catheter Collected Urine in Males with Bladder Cancer According to Disease Stage. J. Urol. 2021, 205, 86–93. [Google Scholar] [CrossRef]
- Babjuk, M.; Burger, M.; Capoun, O.; Cohen, D.; Compérat, E.M.; Dominguez Escrig, J.L.; Gontero, P.; Liedberg, F.; Masson-Lecomte, A.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Non–Muscle-Invasive Bladder Cancer (Ta, T1, and Carcinoma in Situ). Eur. Urol. 2022, 81, 75–94. [Google Scholar] [CrossRef]
- Eau Guidelines Office—Uroweb. Available online: https://uroweb.org/eau-guidelines (accessed on 9 April 2024).
- Kustrimovic, N.; Bilato, G.; Mortara, L.; Baci, D. The Urinary Microbiome in Health and Disease: Relevance for Bladder Cancer. Int. J. Mol. Sci. 2024, 25, 1732. [Google Scholar] [CrossRef]
- Russo, A.E.; Memon, A.; Ahmed, S. Bladder Cancer and the Urinary Microbiome—New Insights and Future Directions: A Review. Clin. Genitourin. Cancer 2024, 22, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Bochner, B.H.; Chou, R.; Dreicer, R.; Kamat, A.M.; Lerner, S.P.; Lotan, Y.; Meeks, J.J.; Michalski, J.M.; Morgan, T.M.; et al. Treatment of Non-Metastatic Muscle-Invasive Bladder Cancer: Aua/Asco/Astro/Suo Guideline. J. Urol. 2017, 198, 552–559. [Google Scholar] [CrossRef]
- Guercio, B.J.; Iyer, G.; Rosenberg, J.E. Developing Precision Medicine for Bladder Cancer. Hematol. Oncol. Clin. N. Am. 2021, 35, 633–653. [Google Scholar] [CrossRef]
- Sjödahl, G.; Abrahamsson, J.; Holmsten, K.; Bernardo, C.; Chebil, G.; Eriksson, P.; Johansson, I.; Kollberg, P.; Lindh, C.; Lövgren, K.; et al. Different Responses to Neoadjuvant Chemotherapy in Urothelial Carcinoma Molecular Subtypes. Eur. Urol. 2022, 81, 523–532. [Google Scholar] [CrossRef]
- Sjödahl, G.; Eriksson, P.; Patschan, O.; Marzouka, N.; Jakobsson, L.; Bernardo, C.; Lövgren, K.; Chebil, G.; Zwarthoff, E.; Liedberg, F.; et al. Molecular Changes during Progression from Nonmuscle Invasive to Advanced Urothelial Carcinoma. Int. J. Cancer 2020, 146, 2636–2647. [Google Scholar] [CrossRef]
- Al Hussein Al Awamlh, B.; Chang, S.S. Novel Therapies for High-Risk Non-Muscle Invasive Bladder Cancer. Curr. Oncol. Rep. 2023, 25, 83–91. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cookson, M.S.; Guercio, B.J.; Cheng, L. Advances in Diagnosis and Treatment of Bladder Cancer. BMJ 2024, 384, e076743. [Google Scholar] [CrossRef]
- Bukhari, N.; Al-Shamsi, H.O.; Azam, F. Update on the Treatment of Metastatic Urothelial Carcinoma. Sci. World J. 2018, 2018, 5682078. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of Muscle-invasive and Advanced Bladder Cancer in 2020. CA A Cancer J Clin. 2020, 70, 404–423. [Google Scholar] [CrossRef]
- Faltas, B.; Goldenberg, D.M.; Ocean, A.J.; Govindan, S.V.; Wilhelm, F.; Sharkey, R.M.; Hajdenberg, J.; Hodes, G.; Nanus, D.M.; Tagawa, S.T. Sacituzumab Govitecan, a Novel Antibody–Drug Conjugate, in Patients with Metastatic Platinum-Resistant Urothelial Carcinoma. Clin. Genitourin. Cancer 2016, 14, e75–e79. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Fléchon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A Phase II Open-Label Study of Sacituzumab Govitecan in Patients with Metastatic Urothelial Carcinoma Progressing after Platinum-Based Chemotherapy and Checkpoint Inhibitors. J. Clin. Oncol. 2021, 39, 2474–2485. [Google Scholar] [CrossRef]
- Bellmunt, J.; De Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Powles, T.; Csőszi, T.; Özgüroğlu, M.; Matsubara, N.; Géczi, L.; Cheng, S.Y.-S.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Morales Barrera, R.; et al. Pembrolizumab Alone or Combined with Chemotherapy versus Chemotherapy as First-Line Therapy for Advanced Urothelial Carcinoma (KEYNOTE-361): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 931–945. [Google Scholar] [CrossRef]
- Al-Obaidy, K.I.; Cheng, L. Fibroblast Growth Factor Receptor (Fgfr) Gene: Pathogenesis and Treatment Implications in Urothelial Carcinoma of the Bladder. J. Clin. Pathol. 2021, 74, 491–495. [Google Scholar] [CrossRef]
- Knowles, M.A.; Hurst, C.D. Molecular Biology of Bladder Cancer: New Insights into Pathogenesis and Clinical Diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Cordon-Cardo, C. Molecular Alterations Associated with Bladder Cancer Initiation and Progression. Scand. J. Urol. Nephrol. 2008, 42, 154–165. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M.; Castillo-Martin, M.; Kinkade, C.W.; Wang, X.; Shen, T.H.; Matos, T.; Shen, M.M.; Cordon-Cardo, C.; Abate-Shen, C. Inactivation of P53 and Pten Promotes Invasive Bladder Cancer. Genes Dev. 2009, 23, 675–680. [Google Scholar] [CrossRef]
- Park, S.; Rong, L.; Owczarek, T.B.; Bernardo, M.D.; Shoulson, R.L.; Chua, C.-W.; Kim, J.Y.; Lankarani, A.; Chakrapani, P.; Syed, T.; et al. Novel Mouse Models of Bladder Cancer Identify a Prognostic Signature Associated with Risk of Disease Progression. Cancer Res. 2021, 81, 5161–5175. [Google Scholar] [CrossRef]
- He, F.; Zhang, F.; Liao, Y.; Tang, M.; Wu, X.-R. Structural or Functional Defects of PTEN in Urothelial Cells Lacking P53 Drive Basal/Squamous-Subtype Muscle-Invasive Bladder Cancer. Cancer Lett. 2022, 550, 215924. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef]
- Mu, P.; Zhou, S.; Lv, T.; Xia, F.; Shen, L.; Wan, J.; Wang, Y.; Zhang, H.; Cai, S.; Peng, J.; et al. Newly Developed 3D in Vitro Models to Study Tumor–Immune Interaction. J. Exp. Clin. Cancer Res. 2023, 42, 81. [Google Scholar] [CrossRef]
- Mullenders, J.; De Jongh, E.; Brousali, A.; Roosen, M.; Blom, J.P.A.; Begthel, H.; Korving, J.; Jonges, T.; Kranenburg, O.; Meijer, R.; et al. Mouse and Human Urothelial Cancer Organoids: A Tool for Bladder Cancer Research. Proc. Natl. Acad. Sci. USA 2019, 116, 4567–4574. [Google Scholar] [CrossRef]
- Van Batavia, J.; Yamany, T.; Molotkov, A.; Dan, H.; Mansukhani, M.; Batourina, E.; Schneider, K.; Oyon, D.; Dunlop, M.; Wu, X.-R.; et al. Bladder Cancers Arise from Distinct Urothelial Sub-Populations. Nat. Cell Biol. 2014, 16, 982–991. [Google Scholar] [CrossRef]
- Metzger, E.; Wang, S.; Urban, S.; Willmann, D.; Schmidt, A.; Offermann, A.; Allen, A.; Sum, M.; Obier, N.; Cottard, F.; et al. KMT9 Monomethylates Histone H4 Lysine 12 and Controls Proliferation of Prostate Cancer Cells. Nat. Struct. Mol. Biol. 2019, 26, 361–371. [Google Scholar] [CrossRef]
- Liu, P.; Nie, S.; Li, B.; Yang, Z.-Q.; Xu, Z.-M.; Fei, J.; Lin, C.; Zeng, R.; Xu, G.-L. Deficiency in a Glutamine-Specific Methyltransferase for Release Factor Causes Mouse Embryonic Lethality. Mol. Cell. Biol. 2010, 30, 4245–4253. [Google Scholar] [CrossRef]
- Heurgué-Hamard, V.; Graille, M.; Scrima, N.; Ulryck, N.; Champ, S.; Van Tilbeurgh, H.; Buckingham, R.H. The Zinc Finger Protein Ynr046w Is Plurifunctional and a Component of the Erf1 Methyltransferase in Yeast. J. Biol. Chem. 2006, 281, 36140–36148. [Google Scholar] [CrossRef]
- Figaro, S.; Scrima, N.; Buckingham, R.H.; Heurgué-Hamard, V. HemK2 Protein, Encoded on Human Chromosome 21, Methylates Translation Termination Factor eRF1. FEBS Lett. 2008, 582, 2352–2356. [Google Scholar] [CrossRef]
- Baumert, H.M.; Metzger, E.; Fahrner, M.; George, J.; Thomas, R.K.; Schilling, O.; Schüle, R. Depletion of Histone Methyltransferase KMT9 Inhibits Lung Cancer Cell Proliferation by Inducing Non-Apoptotic Cell Death. Cancer Cell Int. 2020, 20, 52. [Google Scholar] [CrossRef]
- Berlin, C.; Cottard, F.; Willmann, D.; Urban, S.; Tirier, S.M.; Marx, L.; Rippe, K.; Schmitt, M.; Petrocelli, V.; Greten, F.R.; et al. Kmt9 Controls Stemness and Growth of Colorectal Cancer. Cancer Res. 2022, 82, 210–220. [Google Scholar] [CrossRef]
- Wang, S.; Klein, S.O.; Urban, S.; Staudt, M.; Barthes, N.P.F.; Willmann, D.; Bacher, J.; Sum, M.; Bauer, H.; Peng, L.; et al. Structure-Guided Design of a Selective Inhibitor of the Methyltransferase KMT9 with Cellular Activity. Nat. Commun. 2024, 15, 43. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 Demethylates Repressive Histone Marks to Promote Androgen-Receptor-Dependent Transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Roshan Moniri, M.; Young, A.; Reinheimer, K.; Rayat, J.; Dai, L.-J.; Warnock, G.L. Dynamic Assessment of Cell Viability, Proliferation and Migration Using Real Time Cell Analyzer System (Rtca). Cytotechnology 2015, 67, 379–386. [Google Scholar] [CrossRef]
- Zuiverloon, T.C.M.; De Jong, F.C.; Costello, J.C.; Theodorescu, D. Systematic Review: Characteristics and Preclinical Uses of Bladder Cancer Cell Lines. Bladder Cancer 2018, 4, 169–183. [Google Scholar] [CrossRef]
- Seront, E.; Pinto, A.; Bouzin, C.; Bertrand, L.; Machiels, J.-P.; Feron, O. PTEN Deficiency Is Associated with Reduced Sensitivity to mTOR Inhibitor in Human Bladder Cancer through the Unhampered Feedback Loop Driving PI3K/Akt Activation. Br. J. Cancer 2013, 109, 1586–1592. [Google Scholar] [CrossRef]
- Chaux, A.; Cohen, J.S.; Schultz, L.; Albadine, R.; Jadallah, S.; Murphy, K.M.; Sharma, R.; Schoenberg, M.P.; Netto, G.J. High Epidermal Growth Factor Receptor Immunohistochemical Expression in Urothelial Carcinoma of the Bladder Is Not Associated with EGFR Mutations in Exons 19 and 21: A Study Using Formalin-Fixed, Paraffin-Embedded Archival Tissues. Hum. Pathol. 2012, 43, 1590–1595. [Google Scholar] [CrossRef]
- Chow, N.-H.; Liu, H.-S.; Yang, H.-B.; Chan, S.-H.; Su, I.-J. Expression Patterns of erbB Receptor Family in Normal Urothelium and Transitional Cell Carcinoma. Virchows Arch. 1997, 430, 461–466. [Google Scholar] [CrossRef]
- Rebouissou, S.; Bernard-Pierrot, I.; De Reyniès, A.; Lepage, M.-L.; Krucker, C.; Chapeaublanc, E.; Hérault, A.; Kamoun, A.; Caillault, A.; Letouzé, E.; et al. EGFR as a Potential Therapeutic Target for a Subset of Muscle-Invasive Bladder Cancers Presenting a Basal-like Phenotype. Sci. Transl. Med. 2014, 6, 244ra91. [Google Scholar] [CrossRef]
- Koll, F.J.; Metzger, E.; Hamann, J.; Ramos-Triguero, A.; Bankov, K.; Köllermann, J.; Döring, C.; Chun, F.K.H.; Schüle, R.; Wild, P.J.; et al. Overexpression of Kmt9α Is Associated with Aggressive Basal-like Muscle-Invasive Bladder Cancer. Cells 2023, 12, 589. [Google Scholar] [CrossRef]
- Mellon, K.; Wright, C.; Kelly, P.; Horne, C.H.W.; Neal, D.E. Original Articles: Bladder Cancer: Long-Term Outcome Related to Epidermal Growth Factor Receptor Status in Bladder Cancer. J. Urol. 1995, 153, 919–925. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and Cancer Prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Chen, M.; Wu, C.; Fu, Z.; Liu, S. ICAM1 Promotes Bone Metastasis via Integrin-mediated TGF-β/EMT Signaling in Triple-negative Breast Cancer. Cancer Sci. 2022, 113, 3751–3765. [Google Scholar] [CrossRef]
- Lai, H.; Cheng, X.; Liu, Q.; Luo, W.; Liu, M.; Zhang, M.; Miao, J.; Ji, Z.; Lin, G.N.; Song, W.; et al. Single-cell RNA Sequencing Reveals the Epithelial Cell Heterogeneity and Invasive Subpopulation in Human Bladder Cancer. Int. J. Cancer 2021, 149, 2099–2115. [Google Scholar] [CrossRef]
- Jin, H.; Ying, X.; Que, B.; Wang, X.; Chao, Y.; Zhang, H.; Yuan, Z.; Qi, D.; Lin, S.; Min, W.; et al. N6-Methyladenosine Modification of ITGA6 mRNA Promotes the Development and Progression of Bladder Cancer. EBioMedicine 2019, 47, 195–207. [Google Scholar] [CrossRef]
- Khademi, R.; Malekzadeh, H.; Bahrami, S.; Saki, N.; Khademi, R.; Villa-Diaz, L.G. Regulation and Functions of A6-Integrin (Cd49f) in Cancer Biology. Cancers 2023, 15, 3466. [Google Scholar] [CrossRef]
- Wu, Y.; Tan, X.; Liu, P.; Yang, Y.; Huang, Y.; Liu, X.; Meng, X.; Yu, B.; Wu, M.; Jin, H. ITGA6 and RPSA Synergistically Promote Pancreatic Cancer Invasion and Metastasis via PI3K and MAPK Signaling Pathways. Exp. Cell Res. 2019, 379, 30–47. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Role of Integrins in Cell Invasion and Migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. Egfr in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Baart, V.M.; Van Duijn, C.; Van Egmond, S.L.; Dijckmeester, W.A.; Jansen, J.C.; Vahrmeijer, A.L.; Sier, C.F.M.; Cohen, D. Egfr and Avβ6 as Promising Targets for Molecular Imaging of Cutaneous and Mucosal Squamous Cell Carcinoma of the Head and Neck Region. Cancers 2020, 12, 1474. [Google Scholar] [CrossRef]
- Rao, T.C.; Pui-Yan Ma, V.; Blanchard, A.; Urner, T.M.; Grandhi, S.; Salaita, K.; Mattheyses, A.L. EGFR Activation Attenuates the Mechanical Threshold for Integrin Tension and Focal Adhesion Formation. J. Cell Sci. 2020, 133, jcs.238840. [Google Scholar] [CrossRef]
- Gan, Y.; Shi, C.; Inge, L.; Hibner, M.; Balducci, J.; Huang, Y. Differential Roles of ERK and Akt Pathways in Regulation of EGFR-Mediated Signaling and Motility in Prostate Cancer Cells. Oncogene 2010, 29, 4947–4958. [Google Scholar] [CrossRef]
- Pruthi, R.S.; Nielsen, M.; Heathcote, S.; Wallen, E.M.; Rathmell, W.K.; Godley, P.; Whang, Y.; Fielding, J.; Schultz, H.; Grigson, G.; et al. A Phase II Trial of Neoadjuvant Erlotinib in Patients with Muscle-invasive Bladder Cancer Undergoing Radical Cystectomy: Clinical and Pathological Results. BJU Int. 2010, 106, 349–354. [Google Scholar] [CrossRef]
- Wong, Y.-N.; Litwin, S.; Vaughn, D.; Cohen, S.; Plimack, E.R.; Lee, J.; Song, W.; Dabrow, M.; Brody, M.; Tuttle, H.; et al. Phase Ii Trial of Cetuximab with or without Paclitaxel in Patients with Advanced Urothelial Tract Carcinoma. J. Clin. Oncol. 2012, 30, 3545–3551. [Google Scholar] [CrossRef]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of Deaths from Cancer Caused by Metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef]
- Steeg, P.S. Targeting Metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Abufaraj, M.; Gust, K.; Moschini, M.; Foerster, B.; Soria, F.; Mathieu, R.; Shariat, S.F. Management of Muscle Invasive, Locally Advanced and Metastatic Urothelial Carcinoma of the Bladder: A Literature Review with Emphasis on the Role of Surgery. Transl. Androl. Urol. 2016, 5, 735–744. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Totonji, S.; Ramos-Triguero, A.; Willmann, D.; Sum, M.; Urban, S.; Bauer, H.; Rieder, A.; Wang, S.; Greschik, H.; Metzger, E.; et al. Lysine Methyltransferase 9 (KMT9) Is an Actionable Target in Muscle-Invasive Bladder Cancer. Cancers 2024, 16, 1532. https://doi.org/10.3390/cancers16081532
Totonji S, Ramos-Triguero A, Willmann D, Sum M, Urban S, Bauer H, Rieder A, Wang S, Greschik H, Metzger E, et al. Lysine Methyltransferase 9 (KMT9) Is an Actionable Target in Muscle-Invasive Bladder Cancer. Cancers. 2024; 16(8):1532. https://doi.org/10.3390/cancers16081532
Chicago/Turabian StyleTotonji, Sainab, Anna Ramos-Triguero, Dominica Willmann, Manuela Sum, Sylvia Urban, Helena Bauer, Astrid Rieder, Sheng Wang, Holger Greschik, Eric Metzger, and et al. 2024. "Lysine Methyltransferase 9 (KMT9) Is an Actionable Target in Muscle-Invasive Bladder Cancer" Cancers 16, no. 8: 1532. https://doi.org/10.3390/cancers16081532
APA StyleTotonji, S., Ramos-Triguero, A., Willmann, D., Sum, M., Urban, S., Bauer, H., Rieder, A., Wang, S., Greschik, H., Metzger, E., & Schüle, R. (2024). Lysine Methyltransferase 9 (KMT9) Is an Actionable Target in Muscle-Invasive Bladder Cancer. Cancers, 16(8), 1532. https://doi.org/10.3390/cancers16081532