Activated HGF-c-Met Axis in Head and Neck Cancer

Abstract
:
1. Introduction
2. Brief HGF/c-Met History
3. Role of HGF/c-Met Signaling in Homeostasis
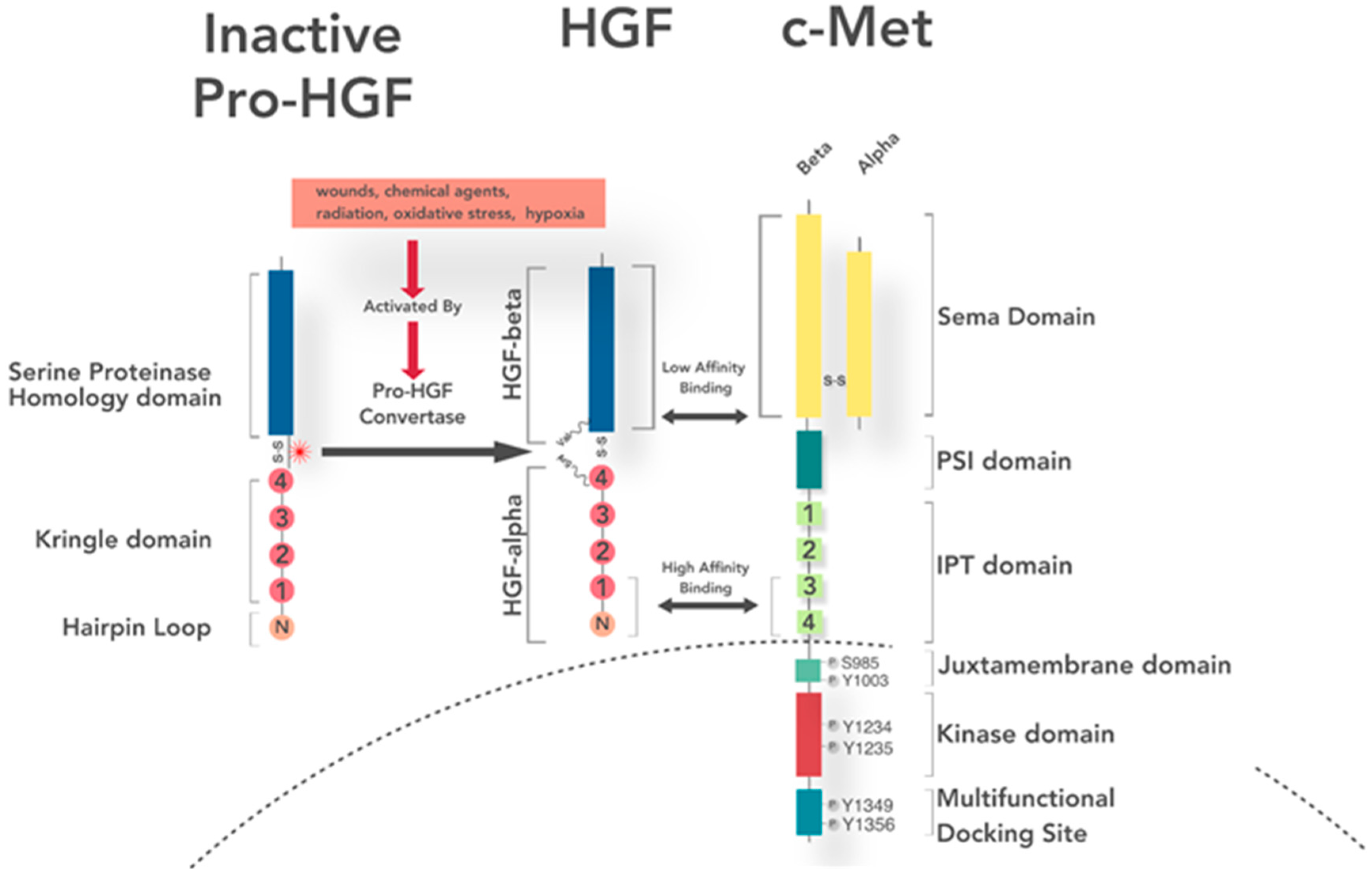
4. Ligand Expression, Secretion, and Activation
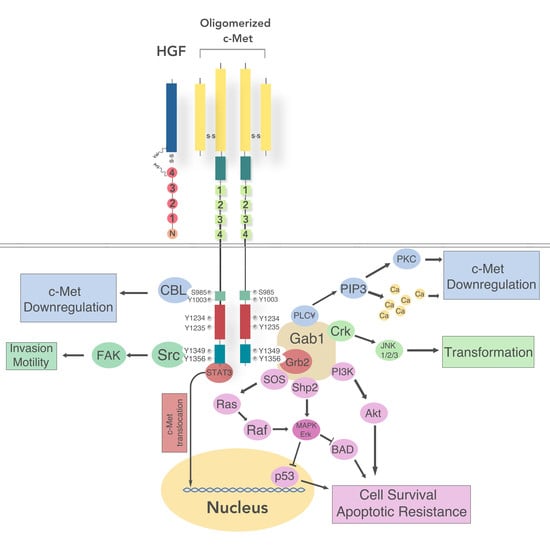
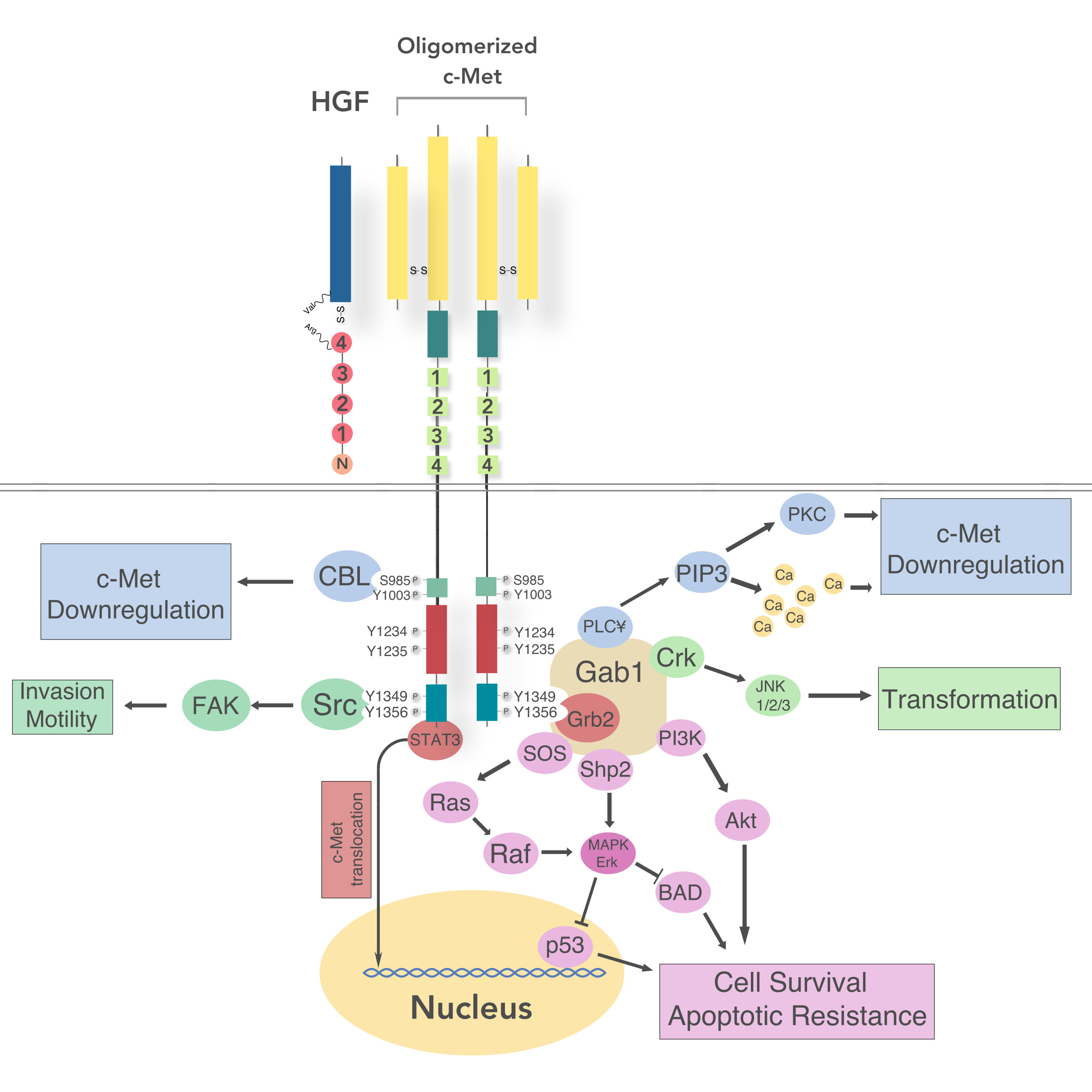
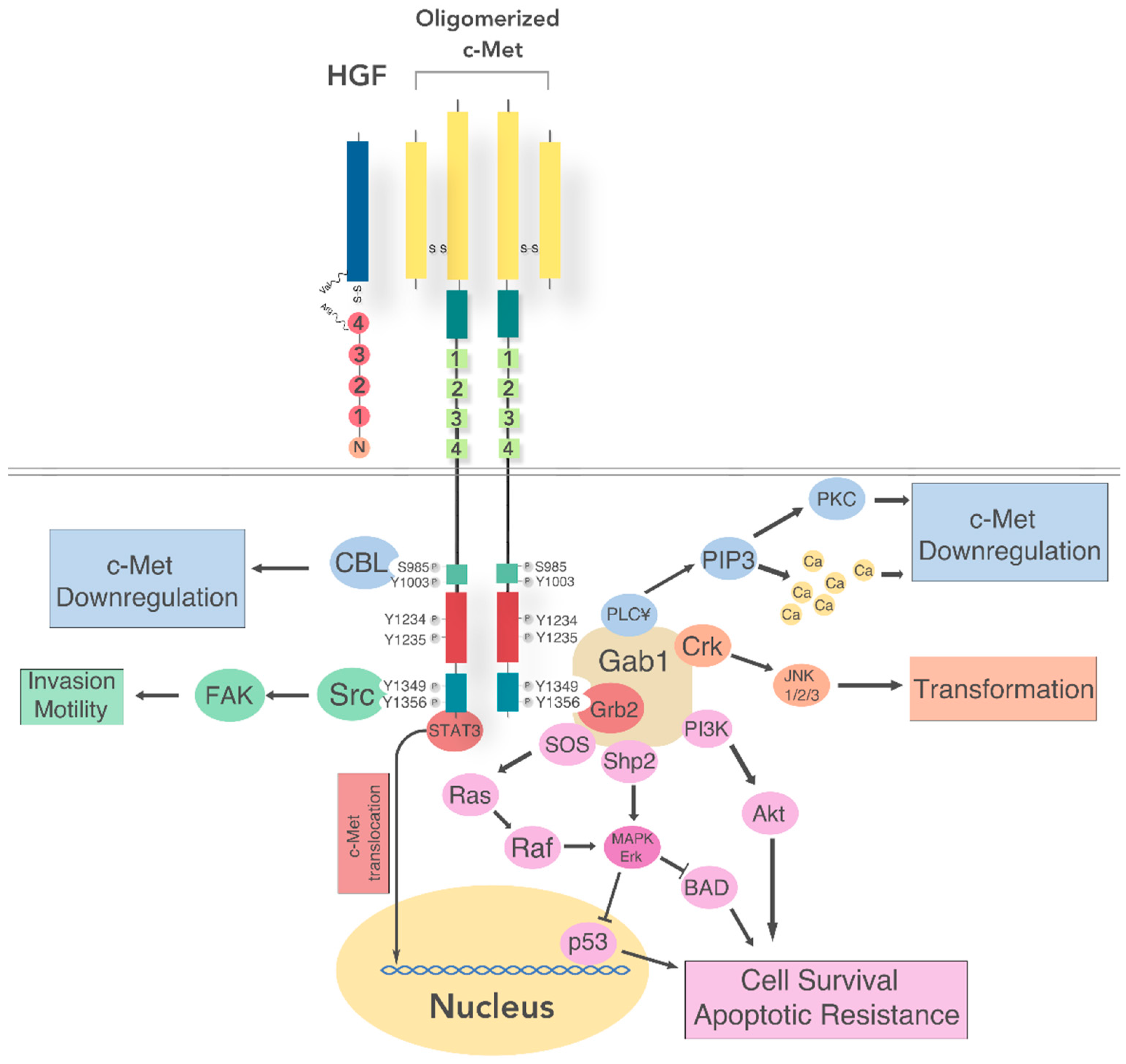
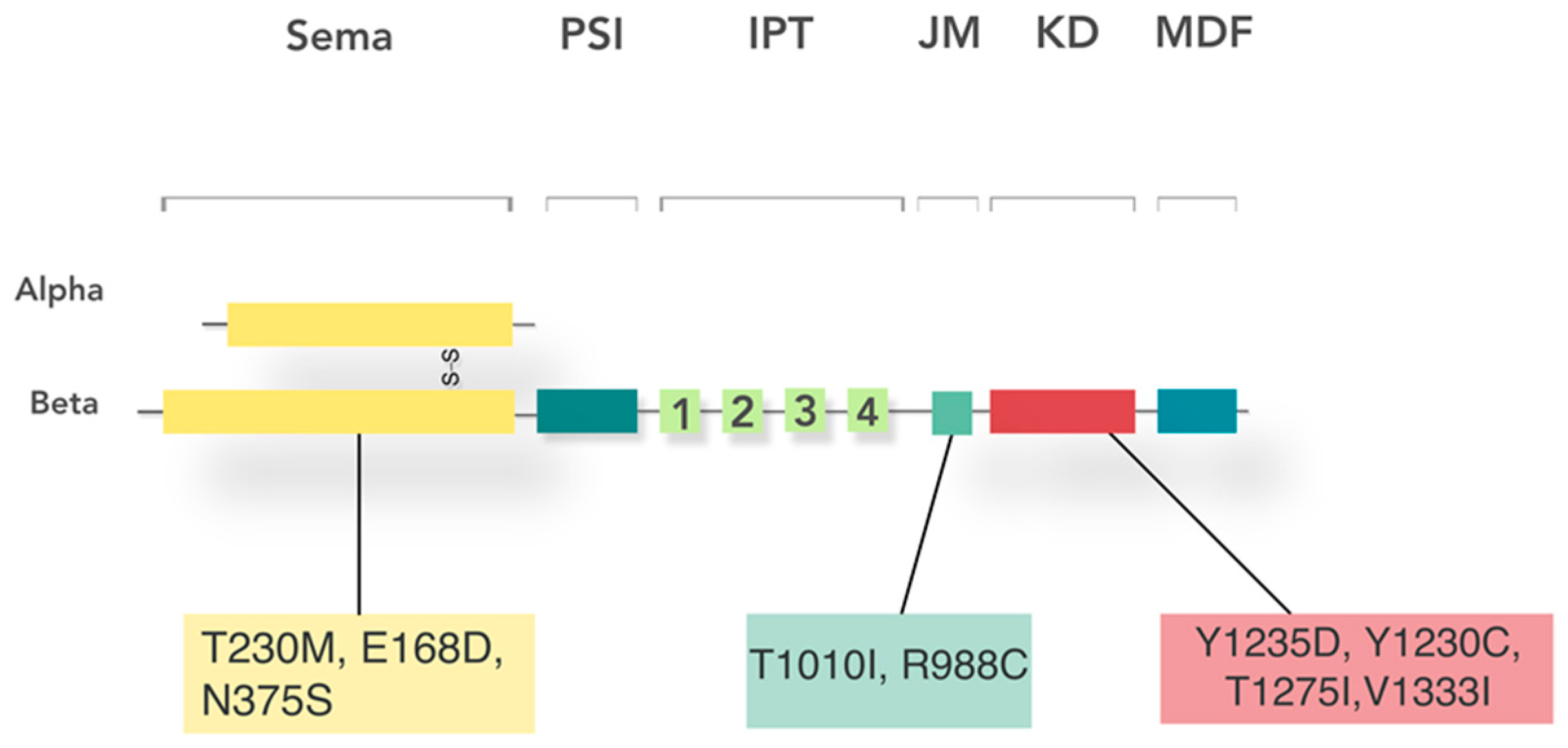
5. Receptor Structure and Activation
5.1. Intracellular Signaling
5.2. Regulation of c-Met Signaling
6. Aberrant Functions in HNSCC
6.1. c-MET Drives Tumorigenesis in HNSCC
6.2. c-MET Drives Metastasis in HNSCC
7. c-MET Pathway Crosstalk in HNSCC
7.1. Wnt/β-Catenin
7.2. c-SRC
7.3. TGF-β
7.4. EGFR
8. c-MET Contribution to Therapeutic Resistance in HNSCC
9. Co-Therapies in HNSCC Targeting c-Met
9.1. Crizotinib (PF2341066)
9.2. Capmatinib (INC-280)
9.3. Golvatinib (E7050)
9.4. Foretinib
9.5. Ficlatuzimab
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal Growth Factor Receptor |
| c-MET | Met Tyrosine Kinase Receptor, mesenchymal epithelial transition factor |
| HGF | Hepatocyte Growth Factor |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human Papilloma Virus |
| EGF | Epidermal Growth Factor |
| mAB | monoclonal antibody |
| TME | Tumor Microenvironment |
| SF | Scatter Factor |
| HSPG | Heparan Sulfate Proteoglycan |
References
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Baxi, S.; Fury, M.; Ganly, I.; Rao, S.; Pfister, D.G. Ten years of progress in head and neck cancers. J. Natl. Compr. Cancer Netw. 2012, 10, 806–810. [Google Scholar] [CrossRef]
- Li, Y.C.; Chang, J.T.; Chiu, C.; Lu, Y.C.; Li, Y.L.; Chiang, C.H.; You, G.R.; Lee, L.Y.; Cheng, A.J. Areca nut contributes to oral malignancy through facilitating the conversion of cancer stem cells. Mol. Carcinog. 2016, 55, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Galbiatti, A.L.; Padovani-Junior, J.A.; Maniglia, J.V.; Rodrigues, C.D.; Pavarino, E.C.; Goloni-Bertollo, E.M. Head and neck cancer: Causes, prevention and treatment. Braz. J. Otorhinolaryngol. 2013, 79, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar] [PubMed]
- Madoz-Gurpide, J.; Zazo, S.; Chamizo, C.; Casado, V.; Carames, C.; Gavin, E.; Cristobal, I.; Garcia-Foncillas, J.; Rojo, F. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G.J.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.-S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.A.; Kim, E.K.; Heo, S.J.; Cho, B.C.; Kim, H.R.; Chung, J.M.; Yoon, S.O. Alteration status and prognostic value of MET in head and neck squamous cell carcinoma. J. Cancer 2016, 7, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-N.; Guo, X.; Cao, B.; Kort, E.J.; Lee, C.-C.; Chen, J.; Wang, L.-M.; Mai, W.-Y.; Min, H.-Q.; Hong, M.-H.; et al. Met Protein Expression Level Correlates with Survival in Patients with Late-stage Nasopharyngeal Carcinoma. Cancer Res. 2002, 62, 589–596. [Google Scholar] [PubMed]
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459. [Google Scholar] [CrossRef]
- Nakamura, T.; Teramoto, H.; Ichihara, A. Purification and characterization of a growth factor from rat platelets for mature parenchymal hepatocytes in primary cultures. Proc. Natl. Acad. Sci. USA 1986, 83, 6489–6493. [Google Scholar] [CrossRef] [PubMed]
- Gohda, E.; Tsubouchi, H.; Nakayama, H.; Hirono, S.; Sakiyama, O.; Takahashi, K.; Miyazaki, H.; Hashimoto, S.; Daikuhara, Y. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J. Clin. Investig. 1988, 81, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, R.; Michalopoulos, G. Purification and biological characterization of human hepatopoietin A, a polypeptide growth factor for hepatocytes. Cancer Res. 1989, 49, 3314–3320. [Google Scholar] [PubMed]
- Asami, O.; Ihara, I.; Shimidzu, N.; Shimizu, S.; Tomita, Y.; Ichihara, A.; Nakamura, T. Purification and characterization of hepatocyte growth factor from injured liver of carbon tetrachloride-treated rats. J. Biochem. 1991, 109, 8–13. [Google Scholar] [PubMed]
- Stoker, M.; Perryman, M. An epithelial scatter factor released by embryo fibroblasts. J. Cell Sci. 1985, 77, 209–223. [Google Scholar] [PubMed]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; Behrens, J.; Vandekerckhove, J.; Birchmeier, W. Scatter factor: Molecular characteristics and effect on the invasiveness of epithelial cells. J. Cell Biol. 1990, 111, 2097–2108. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T.; Takehara, T.; Tsuji, T.; Ohsato, K.; Matsumoto, K.; Nakamura, T. Scatter factor from human embryonic lung fibroblasts is probably identical to hepatocyte growth factor. Biochem. Biophys. Res. Commun. 1991, 180, 765–773. [Google Scholar] [CrossRef]
- Furlong, R.A.; Takehara, T.; Taylor, W.G.; Nakamura, T.; Rubin, J.S. Comparison of biological and immunochemical properties indicates that scatter factor and hepatocyte growth factor are indistinguishable. J. Cell Sci. 1991, 100, 173–177. [Google Scholar] [PubMed]
- Hartmann, G.; Naldini, L.; Weidner, K.M.; Sachs, M.; Vigna, E.; Comoglio, P.M.; Birchmeier, W. A functional domain in the heavy chain of scatter factor/hepatocyte growth factor binds the c-Met receptor and induces cell dissociation but not mitogenesis. Proc. Natl. Acad. Sci. USA 1992, 89, 11574–11578. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; Arakaki, N.; Hartmann, G.; Vandekerckhove, J.; Weingart, S.; Rieder, H.; Fonatsch, C.; Tsubouchi, H.; Hishida, T.; Daikuhara, Y. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Ferracini, R.; Longati, P.; Gandino, L.; Prat, M.; Comoglio, P.M. The tyrosine kinase encoded by the MET proto-oncogene is activated by autophosphorylation. Mol. Cell. Biol. 1991, 11, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991, 6, 501–504. [Google Scholar] [PubMed]
- Gherardi, E.; Stoker, M. Hepatocyte growth factor—Scatter factor: Mitogen, motogen, and met. Cancer Cells 1991, 3, 227–232. [Google Scholar] [PubMed]
- Bussolino, F.; Di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 1992, 119, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Bodescot, M.; Blair, D.; Dunn, J.; Nakamura, T.; Mizuno, K.; Park, M.; Chan, A.; Aaronson, S.; Vande Woude, G.F. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol. Cell. Biol. 1992, 12, 5152–5158. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Zhen, Z.; Medico, E.; Gaudino, G.; Galimi, F.; Comoglio, P.M. Transfer of motogenic and invasive response to scatter factor/hepatocyte growth factor by transfection of human MET protooncogene. Proc. Natl. Acad. Sci. USA 1993, 90, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Tajima, H.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor has potent anti-proliferative activity in various tumor cell lines. FEBS Lett. 1991, 291, 229–232. [Google Scholar] [CrossRef]
- Higashio, K.; Shima, N.; Goto, M.; Itagaki, Y.; Nagao, M.; Yasuda, H.; Morinaga, T. Identity of a tumor cytotoxic factor from human fibroblasts and hepatocyte growth factor. Biochem. Biophys. Res. Commun. 1990, 170, 397–404. [Google Scholar] [CrossRef]
- Shima, N.; Itagaki, Y.; Nagao, M.; Yasuda, H.; Morinaga, T.; Higashio, K. A fibroblast-derived tumor cytotoxic factor/F-TCF (hepatocyte growth factor/HGF) has multiple functions in vitro. Cell Biol. Int. Rep. 1991, 15, 397–408. [Google Scholar] [CrossRef]
- Shima, N.; Nagao, M.; Ogaki, F.; Tsuda, E.; Murakami, A.; Higashio, K. Tumor cytotoxic factor/hepatocyte growth factor from human fibroblasts: Cloning of its cDNA, purification and characterization of recombinant protein. Biochem. Biophys. Res. Commun. 1991, 180, 1151–1158. [Google Scholar] [CrossRef]
- Shima, N.; Higashio, K. [Structure and biological property of fibroblast-derived tumor cytotoxic factor (F-TCF)]. Nihon Rinsho 1992, 50, 1962–1966. [Google Scholar] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Cooper, G.M. ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol. Cell. Biol. 1987, 7, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Park, M.; Vande Woude, G.F. Characterization of the rearranged tpr-met oncogene breakpoint. Mol. Cell. Biol. 1987, 7, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.A.; Park, M. Dimerization mediated through a leucine zipper activates the oncogenic potential of the met receptor tyrosine kinase. Mol. Cell. Biol. 1993, 13, 6711–6722. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Cooper, C.S.; Schmidt, M.; O’Brien, S.J.; Blair, D.G.; Vande Woude, G.F. Mechanism of met oncogene activation. Cell 1986, 45, 895–904. [Google Scholar] [CrossRef]
- Ponzetto, C.; Giordano, S.; Peverali, F.; Della Valle, G.; Abate, M.L.; Vaula, G.; Comoglio, P.M. c-met is amplified but not mutated in a cell line with an activated met tyrosine kinase. Oncogene 1991, 6, 553–559. [Google Scholar] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Ferro, S.; Prat, M.; Bongarzone, I.; Pilotti, S.; Belfiore, A.; Costantino, A.; Vigneri, R.; Pierotti, M.A.; et al. Overexpression of the c-MET/HGF receptor gene in human thyroid carcinomas. Oncogene 1992, 7, 2549–2553. [Google Scholar] [PubMed]
- Liu, C.; Park, M.; Tsao, M.S. Overexpression of c-met proto-oncogene but not epidermal growth factor receptor or c-erbB-2 in primary human colorectal carcinomas. Oncogene 1992, 7, 181–185. [Google Scholar] [PubMed]
- Matsumoto, K.; Horikoshi, M.; Rikimaru, K.; Enomoto, S. A study of an in vitro model for invasion of oral squamous cell carcinoma. J. Oral Pathol. Med. 1989, 18, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Matsumoto, K.; Nakamura, T.; Kramer, R.H. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J. Biol. Chem. 1994, 269, 31807–31813. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Sala, V.; Gatti, S.; Crepaldi, T. Cellular and molecular mechanisms of HGF/Met in the cardiovascular system. Clin. Sci. 2015, 129, 1173–1193. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.T.; Fan, C.M. c-MET regulates myoblast motility and myocyte fusion during adult skeletal muscle regeneration. PLoS ONE 2013, 8, e81757. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, C.E.; Zalata, A.; de Potter, C.R.; van Emmelo, J.; Comhaire, F.H. The receptor encoded by the human C-MET oncogene is expressed in testicular tissue and on human spermatozoa. Mol. Hum. Reprod. 1996, 2, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Li, P.P.; Madhavan, R.; Peng, H.B. Differential regulation of axonal growth and neuromuscular junction assembly by HGF/c-Met signaling. Dev. Dyn. 2012, 241, 1562–1574. [Google Scholar] [CrossRef] [PubMed]
- Montesano, R.; Matsumoto, K.; Nakamura, T.; Orci, L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 1991, 67, 901–908. [Google Scholar] [CrossRef]
- Fehlner-Gardiner, C.C.; Cao, H.; Jackson-Boeters, L.; Nakamura, T.; Elliott, B.E.; Uniyal, S.; Chan, B.M. Characterization of a functional relationship between hepatocyte growth factor and mouse bone marrow-derived mast cells. Differentiation 1999, 65, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Pena-Silva, R.A.; Chalouhi, N.; Wegman-Points, L.; Ali, M.; Mitchell, I.; Pierce, G.L.; Chu, Y.; Ballas, Z.K.; Heistad, D.; Hasan, D. Novel role for endogenous hepatocyte growth factor in the pathogenesis of intracranial aneurysms. Hypertension 2015, 65, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Loreto, C.; Caltabiano, R.; Musumeci, G.; Caltabiano, C.; Greco, M.G.; Leonardi, R. Hepatocyte growth factor receptor, c-Met, in human embryo salivary glands. An immunohistochemical study. Anat. Histol. Embryol. 2010, 39, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Amano, O.; Matsumoto, K.; Nakamura, T.; Iseki, S. Expression and localization of hepatocyte growth factor in rat submandibular gland. Growth Factors 1994, 10, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.V.; Pepper, M.S.; Nakamura, T.; Orci, L.; Montesano, R. Hepatocyte growth factor stimulates extensive development of branching duct-like structures by cloned mammary gland epithelial cells. J. Cell Sci. 1995, 108, 413–430. [Google Scholar] [PubMed]
- Yang, Y.; Spitzer, E.; Meyer, D.; Sachs, M.; Niemann, C.; Hartmann, G.; Weidner, K.M.; Birchmeier, C.; Birchmeier, W. Sequential requirement of hepatocyte growth factor and neuregulin in the morphogenesis and differentiation of the mammary gland. J. Cell Biol. 1995, 131, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Niemann, C.; Brinkmann, V.; Spitzer, E.; Hartmann, G.; Sachs, M.; Naundorf, H.; Birchmeier, W. Reconstitution of mammary gland development in vitro: Requirement of c-met and c-erbB2 signaling for branching and alveolar morphogenesis. J. Cell Biol. 1998, 143, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.; Stern, C.D.; Thery, C.; Ireland, G.W.; Aparicio, S.; Sharpe, M.J.; Gherardi, E. A role for HGF/SF in neural induction and its expression in Hensen’s node during gastrulation. Development 1995, 121, 813–824. [Google Scholar] [PubMed]
- Ebens, A.; Brose, K.; Leonardo, E.D.; Hanson, M.G., Jr.; Bladt, F.; Birchmeier, C.; Barres, B.A.; Tessier-Lavigne, M. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 1996, 17, 1157–1172. [Google Scholar] [CrossRef]
- Wong, V.; Glass, D.J.; Arriaga, R.; Yancopoulos, G.D.; Lindsay, R.M.; Conn, G. Hepatocyte growth factor promotes motor neuron survival and synergizes with ciliary neurotrophic factor. J. Biol. Chem. 1997, 272, 5187–5191. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Livet, J.; Pollock, R.A.; Garces, A.; Arce, V.; deLapeyriere, O.; Henderson, C.E. Hepatocyte growth factor (HGF/SF) is a muscle-derived survival factor for a subpopulation of embryonic motoneurons. Development 1997, 124, 2903–2913. [Google Scholar] [PubMed]
- Johnson, M.; Koukoulis, G.; Matsumoto, K.; Nakamura, T.; Iyer, A. Hepatocyte growth factor induces proliferation and morphogenesis in nonparenchymal epithelial liver cells. Hepatology 1993, 17, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Basler, K.; Hafen, E. Control of photoreceptor cell fate by the sevenless protein requires a functional tyrosine kinase domain. Cell 1988, 54, 299–311. [Google Scholar] [PubMed]
- Chabot, B.; Stephenson, D.A.; Chapman, V.M.; Besmer, P.; Bernstein, A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 1988, 335, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, E.; Godecke, A.; Walter, B.; Bladt, F.; Birchmeier, C. Transient and locally restricted expression of the ros1 protooncogene during mouse development. EMBO J. 1991, 10, 3693–3702. [Google Scholar] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; Fennelly, D.; Rafferty, M. Common critical pathways in embryogenesis and cancer. Acta Oncol. 2006, 45, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Chollet-Martin, S.; Crestani, B.; Delarche, C.; El Benna, J.; Boutten, A.; Andrieu, V.; Durand, G.; Gougerot-Pocidalo, M.-A.; Aubier, M.; et al. Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood 2002, 99, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Kakimoto, K.; Bandow, K.; Lowenstein, C.J.; Daikuhara, Y.; Matsuguchi, T. Mature Hepatocyte Growth Factor/Scatter Factor on the Surface of Human Granulocytes Is Released by a Mechanism Involving Activated Factor Xa. J. Immunol. 2006, 176, 6945–6953. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Hagiya, M.; Nishizawa, T.; Seki, T.; Shimonishi, M.; Shimizu, S.; Nakamura, T. Deduced primary structure of rat hepatocyte growth factor and expression of the mRNA in rat tissues. Proc. Natl. Acad. Sci. USA 1990, 87, 3200–3204. [Google Scholar] [CrossRef] [PubMed]
- Schwall, R.H.; Chang, L.Y.; Godowski, P.J.; Kahn, D.W.; Hillan, K.J.; Bauer, K.D.; Zioncheck, T.F. Heparin induces dimerization and confers proliferative activity onto the hepatocyte growth factor antagonists NK1 and NK2. J. Cell Biol. 1996, 133, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Kataoka, H. Mechanisms of Hepatocyte Growth Factor Activation in Cancer Tissues. Cancers 2014, 6, 1890–1904. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Qin, L.; Shimomura, T.; Kondo, J.; Matsumoto, K.; Denda, K.; Kitamura, N. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J. Biol. Chem. 1997, 272, 27558–27564. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, T.; Denda, K.; Kitamura, A.; Kawaguchi, T.; Kito, M.; Kondo, J.; Kagaya, S.; Qin, L.; Takata, H.; Miyazawa, K.; et al. Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J. Biol. Chem. 1997, 272, 6370–6376. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Ann. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Delehedde, M.; Lyon, M.; Sergeant, N.; Rahmoune, H.; Fernig, D.G. Proteoglycans: Pericellular and cell surface multireceptors that integrate external stimuli in the mammary gland. J. Mammary Gland Biol. Neoplasia 2001, 6, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Lander, A.D.; Nie, Q.; Wan, F.Y. Do morphogen gradients arise by diffusion? Dev. Cell 2002, 2, 785–796. [Google Scholar] [CrossRef]
- Garner, O.B.; Bush, K.T.; Nigam, K.B.; Yamaguchi, Y.; Xu, D.; Esko, J.D.; Nigam, S.K. Stage-Dependent Regulation of Mammary Ductal Branching by Heparan Sulfate and HGF-cMet Signaling. Dev. Biol. 2011, 355, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Prospero, T.; Brinkmann, V.; Ozcelik, Ö.; Winter, G.; Hepple, J.; Batley, S.; Bladt, F.; Sachs, M.; Birchmeier, C.; et al. Engineered mutants of HGF/SF with reduced binding to heparan sulphate proteoglycans, decreased clearance and enhanced activity in vivo. Curr. Biol. 1998, 8, 125–135. [Google Scholar] [CrossRef]
- Gross-Cohen, M.; Feld, S.; Doweck, I.; Neufeld, G.; Hasson, P.; Arvatz, G.; Barash, U.; Naroditsky, I.; Ilan, N.; Vlodavsky, I. Heparanase 2 attenuates head and neck tumor vascularity and growth. Cancer Res. 2016, 76, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Feld, S.; Cohen-Kaplan, V.; Shteingauz, A.; Gross, M.; Arvatz, G.; Naroditsky, I.; Ilan, N.; Doweck, I.; Vlodavsky, I. Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. J. Biol. Chem. 2010, 285, 28010–28019. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Qian, Y.; Zhou, X.; Lu, H.; Ramacciotti, E.; Zhang, L. Chemically oversulfated glycosaminoglycans are potent modulators of contact system activation and different cell signaling pathways. J. Biol. Chem. 2010, 285, 22966–22975. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Basilico, C.; Cavassa, S.; Pennacchietti, S.; Risio, M.; Naldin, L.; Comoglio, P.M.; Michieli, P. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J. Clin. Investig. 2004, 114, 1418–1432. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Bardelli, A.; Follenzi, A.; Galimi, F.; Comoglio, P.M. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J. Biol. Chem. 1995, 270, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Faletto, D.L.; Tsarfaty, I.; Kmiecik, T.E.; Gonzatti, M.; Suzuki, T.; Vande Woude, G.F. Evidence for non-covalent clusters of the c-met proto-oncogene product. Oncogene 1992, 7, 1149–1157. [Google Scholar] [PubMed]
- Gonzatti-Haces, M.; Park, M.; Dean, M.; Blair, D.G.; Vande Woude, G.F. The human met oncogene is a member of the tyrosine kinase family. Princess Takamatsu Symp. 1986, 17, 221–232. [Google Scholar] [PubMed]
- Chan, A.M.; King, H.W.; Deakin, E.A.; Tempest, P.R.; Hilkens, J.; Kroezen, V.; Edwards, D.R.; Wills, A.J.; Brookes, P.; Cooper, C.S. Characterization of the mouse met proto-oncogene. Oncogene 1988, 2, 593–599. [Google Scholar] [PubMed]
- Giordano, S.; Ponzetto, C.; Di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Arnesano, A.; Galluzzo, M.; Comoglio, P.M.; Michieli, P. A high affinity hepatocyte growth factor-binding site in the immunoglobulin-like region of Met. J. Biol. Chem. 2008, 283, 21267–21277. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Bardelli, A.; Ponzetto, C.; Naldini, L.; Comoglio, P.M. Tyrosines1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994, 9, 49–57. [Google Scholar] [PubMed]
- Pelicci, G.; Giordano, S.; Zhen, Z.; Salcini, A.E.; Lanfrancone, L.; Bardelli, A.; Panayotou, G.; Waterfield, M.D.; Ponzetto, C.; Pelicci, P.G.; et al. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene 1995, 10, 1631–1638. [Google Scholar] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Weidner, K.M.; Di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guzman, M.; Dolfi, F.; Zeh, K.; Vuori, K. Met-induced JNK activation is mediated by the adapter protein Crk and correlates with the Gab1-Crk signaling complex formation. Oncogene 1999, 18, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- Sakkab, D.; Lewitzky, M.; Posern, G.; Schaeper, U.; Sachs, M.; Birchmeier, W.; Feller, S.M. Signaling of hepatocyte growth factor/scatter factor (HGF) to the small GTPase Rap1 via the large docking protein Gab1 and the adapter protein CRKL. J. Biol. Chem. 2000, 275, 10772–10778. [Google Scholar] [CrossRef] [PubMed]
- Graziani, A.; Gramaglia, D.; Cantley, L.C.; Comoglio, P.M. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J. Biol. Chem. 1991, 266, 22087–22090. [Google Scholar] [PubMed]
- Boccaccio, C.; Ando, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Graziani, A.; Gramaglia, D.; dalla Zonca, P.; Comoglio, P.M. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J. Biol. Chem. 1993, 268, 9165–9168. [Google Scholar] [PubMed]
- Fixman, E.D.; Fournier, T.M.; Kamikura, D.M.; Naujokas, M.A.; Park, M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J. Biol. Chem. 1996, 271, 13116–13122. [Google Scholar] [CrossRef] [PubMed]
- Furge, K.A.; Zhang, Y.W.; Vande Woude, G.F. Met receptor tyrosine kinase: Enhanced signaling through adapter proteins. Oncogene 2000, 19, 5582–5589. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell. Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Comoglio, P.M.; Trusolino, L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J. Cell Biol. 2006, 175, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Parker, P.J. Receptor trafficking controls weak signal delivery: A strategy used by c-Met for STAT3 nuclear accumulation. J. Cell Biol. 2008, 182, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Mak, H.H.; Peschard, P.; Lin, T.; Naujokas, M.A.; Zuo, D.; Park, M. Oncogenic activation of the Met receptor tyrosine kinase fusion protein, Tpr-Met, involves exclusion from the endocytic degradative pathway. Oncogene 2007, 26, 7213–7221. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Urbe, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Machide, M.; Hashigasako, A.; Matsumoto, K.; Nakamura, T. Contact inhibition of hepatocyte growth regulated by functional association of the c-Met/hepatocyte growth factor receptor and LAR protein-tyrosine phosphatase. J. Biol. Chem. 2006, 281, 8765–8772. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Paliouras, G.N.; Abella, J.V.; Dube, N.; Monast, A.; Tremblay, M.L.; Park, M. Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J. Biol. Chem. 2008, 283, 34374–34383. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.W.; Lingen, M.W.; et al. The MET Receptor Tyrosine Kinase Is a Potential Novel Therapeutic Target for Head and Neck Squamous Cell Carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef] [PubMed]
- Ferracini, R.; Di Renzo, M.F.; Scotlandi, K.; Baldini, N.; Olivero, M.; Lollini, P.; Cremona, O.; Campanacci, M.; Comoglio, P.M. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995, 10, 739–749. [Google Scholar] [PubMed]
- Scotlandi, K.; Baldini, N.; Oliviero, M.; Di Renzo, M.F.; Martano, M.; Serra, M.; Manara, M.C.; Comoglio, P.M.; Ferracini, R. Expression of Met/hepatocyte growth factor receptor gene and malignant behavior of musculoskeletal tumors. Am. J. Pathol. 1996, 149, 1209–1219. [Google Scholar] [PubMed]
- Ferracini, R.; Olivero, M.; Di Renzo, M.F.; Martano, M.; De Giovanni, C.; Nanni, P.; Basso, G.; Scotlandi, K.; Lollini, P.L.; Comoglio, P.M. Retrogenic expression of the MET proto-oncogene correlates with the invasive phenotype of human rhabdomyosarcomas. Oncogene 1996, 12, 1697–1705. [Google Scholar] [PubMed]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and c-Met Participate in Paracrine Tumorigenic Pathways in Head and Neck Squamous Cell Cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef] [PubMed]
- Ghadjar, P.; Blank-Liss, W.; Simcock, M.; Hegyi, I.; Beer, K.T.; Moch, H.; Aebersold, D.M.; Zimmer, Y. MET Y1253D-activating point mutation and development of distant metastasis in advanced head and neck cancers. Clin. Exp. Metastasis 2009, 26, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1995, 1, 147–154. [Google Scholar] [PubMed]
- Takeo, S.; Arai, H.; Kusano, N.; Harada, T.; Furuya, T.; Kawauchi, S.; Oga, A.; Hirano, T.; Yoshida, T.; Okita, K.; et al. Examination of oncogene amplification by genomic DNA microarray in hepatocellular carcinomas: Comparison with comparative genomic hybridization analysis. Cancer Genet. Cytogenet. 2001, 130, 127–132. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Dong, S.M.; Kim, S.Y.; Na, E.Y.; Shin, M.S.; Pi, J.H.; Kim, B.J.; Bae, J.H.; Hong, Y.K.; Lee, K.S.; et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999, 59, 307–310. [Google Scholar] [PubMed]
- Aebersold, D.M.; Landt, O.; Berthou, S.; Gruber, G.; Beer, K.T.; Greiner, R.H.; Zimmer, Y. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene 2003, 22, 8519–8523. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, S.; Gkountakos, A.; Carbognin, L.; Scarpa, A.; Tortora, G.; Bria, E. MET exon 14 juxtamembrane splicing mutations: Clinical and therapeutical perspectives for cancer therapy. Ann. Transl. Med. 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Ruco, L.P.; Ranalli, T.; Marzullo, A.; Bianco, P.; Prat, M.; Comoglio, P.M.; Baroni, C.D. Expression of Met protein in thyroid tumours. J. Pathol. 1996, 180, 266–270. [Google Scholar] [CrossRef]
- Di Renzo, M.F.; Olivero, M.; Katsaros, D.; Crepaldi, T.; Gaglia, P.; Zola, P.; Sismondi, P.; Comoglio, P.M. Overexpression of the Met/HGF receptor in ovarian cancer. Int. J. Cancer 1994, 58, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Tapper, J.; Kettunen, E.; El-Rifai, W.; Seppala, M.; Andersson, L.C.; Knuutila, S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet. Cytogenet. 2001, 128, 1–6. [Google Scholar] [CrossRef]
- Di Renzo, M.F.; Poulsom, R.; Olivero, M.; Comoglio, P.M.; Lemoine, N.R. Expression of the Met/hepatocyte growth factor receptor in human pancreatic cancer. Cancer Res. 1995, 55, 1129–1138. [Google Scholar] [PubMed]
- Humphrey, P.A.; Zhu, X.; Zarnegar, R.; Swanson, P.E.; Ratliff, T.L.; Vollmer, R.T.; Day, M.L. Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am. J. Pathol. 1995, 147, 386–396. [Google Scholar] [PubMed]
- Natali, P.G.; Prat, M.; Nicotra, M.R.; Bigotti, A.; Olivero, M.; Comoglio, P.M.; Di Renzo, M.F. Overexpression of the met/HGF receptor in renal cell carcinomas. Int. J. Cancer 1996, 69, 212–217. [Google Scholar] [CrossRef]
- Tavian, D.; De Petro, G.; Benetti, A.; Portolani, N.; Giulini, S.M.; Barlati, S. u-PA and c-MET mRNA expression is co-ordinately enhanced while hepatocyte growth factor mRNA is down-regulated in human hepatocellular carcinoma. Int. J. Cancer 2000, 87, 644–649. [Google Scholar] [CrossRef]
- Taniguchi, K.; Yonemura, Y.; Nojima, N.; Hirono, Y.; Fushida, S.; Fujimura, T.; Miwa, K.; Endo, Y.; Yamamoto, H.; Watanabe, H. The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c-met), autocrine motility factor receptor, and urokinase-type plasminogen activator receptor. Cancer 1998, 82, 2112–2122. [Google Scholar] [CrossRef]
- Porte, H.; Triboulet, J.P.; Kotelevets, L.; Carrat, F.; Prevot, S.; Nordlinger, B.; DiGioia, Y.; Wurtz, A.; Comoglio, P.; Gespach, C.; et al. Overexpression of stromelysin-3, BM-40/SPARC, and MET genes in human esophageal carcinoma: Implications for prognosis. Clin. Cancer Res. 1998, 4, 1375–1382. [Google Scholar] [PubMed]
- Camp, R.L.; Rimm, E.B.; Rimm, D.L. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer 1999, 86, 2259–2265. [Google Scholar] [CrossRef]
- Wielenga, V.J.; van der Voort, R.; Taher, T.E.; Smit, L.; Beuling, E.A.; van Krimpen, C.; Spaargaren, M.; Pals, S.T. Expression of c-Met and heparan-sulfate proteoglycan forms of CD44 in colorectal cancer. Am. J. Pathol. 2000, 157, 1563–1573. [Google Scholar] [CrossRef]
- Morello, S.; Olivero, M.; Aimetti, M.; Bernardi, M.; Berrone, S.; Di Renzo, M.F.; Giordano, S. MET receptor is overexpressed but not mutated in oral squamous cell carcinomas. J. Cell. Physiol. 2001, 189, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Hays, J.L.; Watowich, S.J. Oligomerization-induced modulation of TPR-MET tyrosine kinase activity. J. Biol. Chem. 2003, 278, 27456–27463. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.D.; Kornberg, L.J. Overexpression of scatter factor and its receptor (c-met) in oral squamous cell carcinoma. Laryngoscope 1998, 108, 1413–1417. [Google Scholar] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, M.; Rong, S.; Vande Woude, G.F. Enhanced tumorigenicity and invasion-metastasis by hepatocyte growth factor/scatter factor-met signalling in human cells concomitant with induction of the urokinase proteolysis network. Mol. Cell. Biol. 1996, 16, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G.; Davies, G.; Martin, T.A.; Parr, C.; Watkins, G.; Mansel, R.E.; Mason, M.D. The potential lymphangiogenic effects of hepatocyte growth factor/scatter factor in vitro and in vivo. Int. J. Mol. Med. 2005, 16, 723–728. [Google Scholar] [PubMed]
- Kajiya, K.; Hirakawa, S.; Ma, B.; Drinnenberg, I.; Detmar, M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005, 24, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Gherardi, E.; Sellers, L.A.; Wood, J.M.; Sasisekharan, R.; Fan, T.P. Hepatocyte growth factor/scatter factor can induce angiogenesis independently of vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Goldson, T.M.; Han, Y.; Knight, K.B.; Weiss, H.L.; Resto, V.A. Clinicopathological predictors of lymphatic metastasis in HNSCC: Implications for molecular mechanisms of metastatic disease. J. Exp. Ther. Oncol. 2010, 8, 211–221. [Google Scholar] [PubMed]
- Choe, J.-Y.; Yun, J.Y.; Nam, S.-J.; Kim, J.E. Expression of c-Met Is Different along the Location and Associated with Lymph Node Metastasis of Head and Neck Carcinoma. Korean J. Pathol. 2012, 46, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Cortesina, G.; Martone, T.; Galeazzi, E.; Olivero, M.; De Stefani, A.; Bussi, M.; Valente, G.; Comoglio, P.M.; Di Renzo, M.F. Staging of head and neck squamous cell carcinoma using the MET oncogene product as marker of tumor cells in lymph node metastases. Int. J. Cancer 2000, 89, 286–292. [Google Scholar] [CrossRef]
- Galeazzi, E.; Olivero, M.; Gervasio, F.C.; De Stefani, A.; Valente, G.; Comoglio, P.M.; Di Renzo, M.F.; Cortesina, G. Detection of MET oncogene/hepatocyte growth factor receptor in lymph node metastases from head and neck squamous cell carcinomas. Eur. Arch. Otorhinolaryngol. 1997, 254 (Suppl. 1), S138–S143. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Hill, K.S.; Gaziova, I.; Sastry, S.K.; Qui, S.; Szaniszlo, P.; Fennewald, S.; Resto, V.A.; Elferink, L.A. Silencing Met receptor tyrosine kinase signaling decreased oral tumor growth and increased survival of nude mice. Oral Oncol. 2014, 50, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Shen, C.-J.; Chan, S.-H.; Lee, C.-T.; Huang, W.-C.; Tsai, J.-P.; Chen, B.-K. Oleic acid-induced ANGPTL4 enhances head and neck squamous cell carcinoma anoikis resistance and metastasis via up-regulation of fibronectin. Cancer Lett. 2017, 386, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Chen, S.; You, Z.; Yang, F.; Carey, T.E.; Saims, D.; Wang, C.Y. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J. Biol. Chem. 2002, 277, 25203–25208. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Settleman, J.; Quinlan, M.P. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008, 19, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, S.; Duan, S.Z.; Zhang, L.; Zhou, H.; Hu, Y.; Zhou, X.; Shi, C.; Zhou, R.; Zhang, Z. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res. 2014, 74, 7546–7559. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; He, G.; Lui, V.W.; Thomas, S.; Henry, C.; Gubish, C.T.; Joyce, S.; Quesnelle, K.M.; Siegfried, J.M.; Grandis, J.R. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin. Cancer Res. 2013, 19, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Peng, S.; Saigal, B.; Williams, M.D.; Johnson, F.M. Distinct interactions between c-Src and c-Met in mediating resistance to c-Src inhibition in head and neck cancer. Clin. Cancer Res. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-ß Signaling in Fibroblasts Modulates the Oncogenic Potential of Adjacent Epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Hoot, K.E.; Oka, M.; Han, G.; Bottinger, E.; Zhang, Q.; Wang, X.J. HGF upregulation contributes to angiogenesis in mice with keratinocyte-specific Smad2 deletion. J. Clin. Investig. 2010, 120, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Fertig, E.; Ozawa, H.; Hatakeyama, H.; Considine, M.; Perez, J.; Ochs, M.; Chung, C.H. Abstract 1889: Smad4 inactivation correlates with EMT and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Res. 2012, 72, 1889. [Google Scholar] [CrossRef]
- Spector, N.L.; Xia, W.; Burris, H., 3rd; Hurwitz, H.; Dees, E.C.; Dowlati, A.; O’Neil, B.; Overmoyer, B.; Marcom, P.K.; Blackwell, K.L.; et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Moasser, M.M. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br. J. Cancer 2007, 97, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Burbridge, M.F.; Gastaldi, S.; Galimi, F.; Torti, D.; Medico, E.; Giordano, S.; Corso, S.; Rolland-Valognes, G.; Lockhart, B.P.; et al. Only a Subset of Met-Activated Pathways Are Required to Sustain Oncogene Addiction. Sci. Signal. 2009, 2, ra80. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, H.; Liu, Y.; Anderson, A.; Peterson, J.; Greger, J.; Martin, A.M.; Gilmer, T.M. Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells. Mol. Cancer Ther. 2011, 10, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Stabile, L.P.; Gubish, C.T.; Gooding, W.E.; Grandis, J.R.; Siegfried, J.M. Dual blockade of EGFR and c-Met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin. Cancer Res. 2011, 17, 4425–4438. [Google Scholar] [CrossRef] [PubMed]
- Tepper, S.R.; Zuo, Z.; Khattri, A.; Heß, J.; Seiwert, T.Y. Growth factor expression mediates resistance to EGFR inhibitors in head and neck squamous cell carcinomas. Oral Oncol. 2016, 56, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef] [PubMed]
- Rubin Grandis, J.; Melhem, M.F.; Gooding, W.E.; Day, R.; Holst, V.A.; Wagener, M.M.; Drenning, S.D.; Tweardy, D.J. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J. Natl. Cancer Inst. 1998, 90, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Soulieres, D.; Senzer, N.N.; Vokes, E.E.; Hidalgo, M.; Agarwala, S.S.; Siu, L.L. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J. Clin. Oncol. 2004, 22, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal—Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, A.M.; Galoforo, S.; Thibodeau, B.J.; Ahmed, S.; Nirmal, S.; Akervall, J.; Wilson, G.D. Crizotinib Fails to Enhance the Effect of Radiation in Head and Neck Squamous Cell Carcinoma Xenografts. Anticancer Res. 2015, 35, 5973–5982. [Google Scholar] [PubMed]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y.; et al. A Novel Kinase Inhibitor, INCB28060, Blocks c-MET-Dependent Signaling, Neoplastic Activities, and Cross-Talk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Y.; Wang, K.; Gao, Y.; Han, J.; Cui, B.; Gong, P. Discovery of novel 4-(2-fluorophenoxy)quinoline derivatives bearing 4-oxo-1,4-dihydrocinnoline-3-carboxamide moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2013, 21, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Z.; Dai, W.-S.; Zhu, H.-C.; Song, H.-M.; Yang, X.; Wang, Y.-D.; Min, H.; Lu, Q.; Liu, S.; Sun, X.-C.; et al. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. J. Cancer 2017, 8, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.; Sarantopoulos, J.; Kallender, H.; McCallum, S.; Keer, H.N.; Blumenschein, G. Phase II trial of single-agent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2013, 31, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kandl, C.; Hamilton, C.D.; Shnayder, Y.; Tsue, T.T.; Kakarala, K.; Ledgerwood, L.; Sun, X.S.; Huang, H.J.; Girod, D.; et al. Mitigation of tumor-associated fibroblast-facilitated head and neck cancer progression with anti-hepatocyte growth factor antibody ficlatuzumab. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 1133–1139. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Class | IC50/Ki | Co-Treatments | Stage of Development in HNSCC | |
|---|---|---|---|---|---|
| c-Met | Cell | ||||
| Crizotinib | Ia | 2.0 nM (Ki) | 4.1–4.7 μM | Gefitinib | Preclinical |
| Radiation | Preclinical | ||||
| Capmatinib | Ib | 0.13 nM (IC50) | 1.2–12.4 nM | Cetuximab | Phase Ib/II (NCT02205398) |
| Golvatinib | II | 14 nM (IC50) | 6.2 nM–4.3 μM | Cetuximab | Phase I/II (NCT01332266) |
| Cisplatin and Capecitabine | Phase I/II * (NCT01355302) | ||||
| Foretinib | II | 1.16 nM (IC50) | 0.61–0.79 μM | Erlotinib | Preclinical |
| Radiation | Preclinical | ||||
| N/A | Phase I/II * (NCT00725764) | ||||
| Ficlatuzumab | Monoclonal Antibody | N/A | Cetuximab | Phase I (NCT02277197) | |
| Crizotinib | Golvatinib |
|---|---|
 |  |
| Capmatinib | Foretinib |
 |  |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met Axis in Head and Neck Cancer. Cancers 2017, 9, 169. https://doi.org/10.3390/cancers9120169
Arnold L, Enders J, Thomas SM. Activated HGF-c-Met Axis in Head and Neck Cancer. Cancers. 2017; 9(12):169. https://doi.org/10.3390/cancers9120169
Chicago/Turabian StyleArnold, Levi, Jonathan Enders, and Sufi Mary Thomas. 2017. "Activated HGF-c-Met Axis in Head and Neck Cancer" Cancers 9, no. 12: 169. https://doi.org/10.3390/cancers9120169
APA StyleArnold, L., Enders, J., & Thomas, S. M. (2017). Activated HGF-c-Met Axis in Head and Neck Cancer. Cancers, 9(12), 169. https://doi.org/10.3390/cancers9120169