
EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer


Abstract
:1. Introduction
2. Epidermal Growth Factor Receptor Family
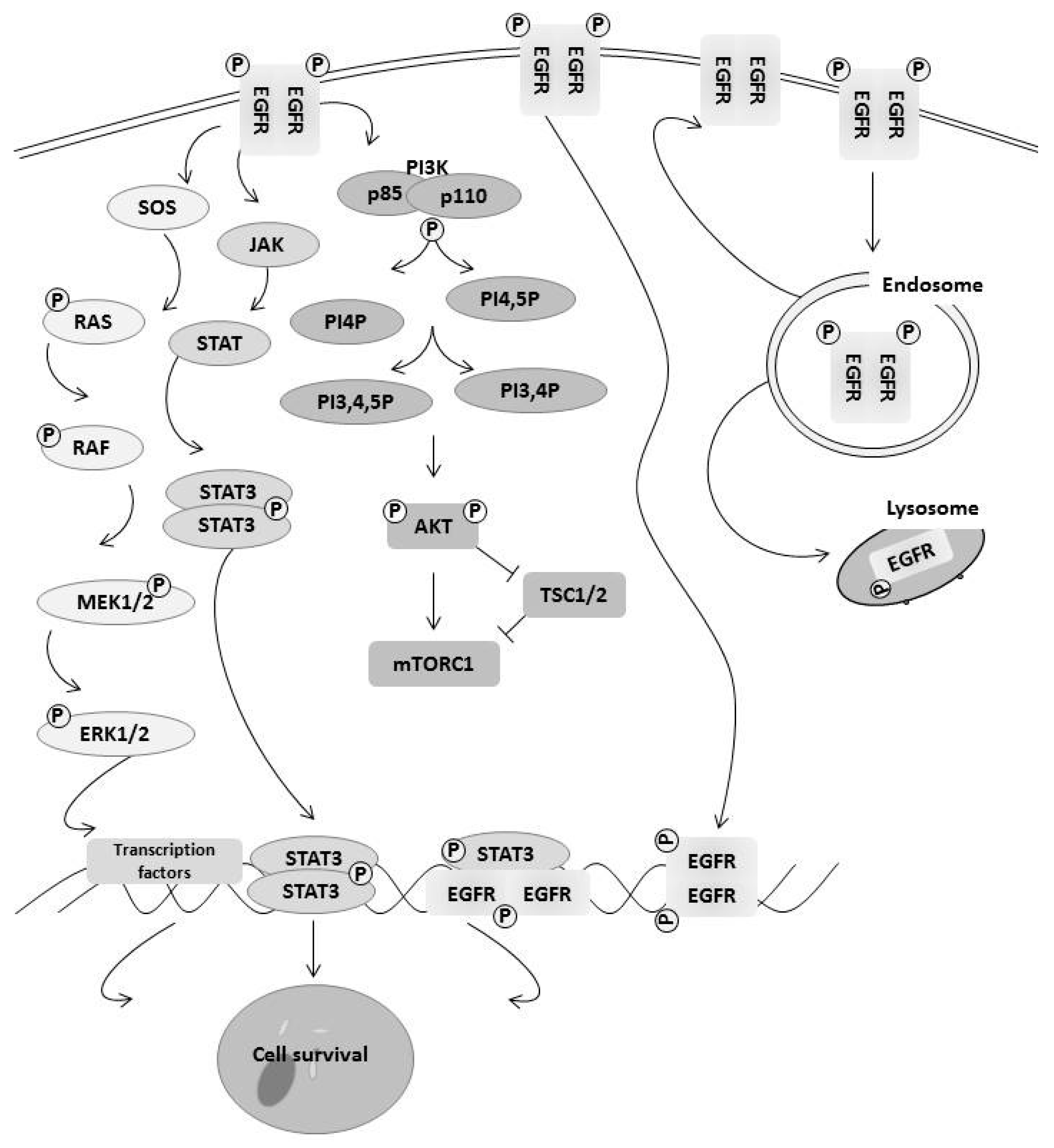
3. How EGFR Family Members Regulate Cell Survival
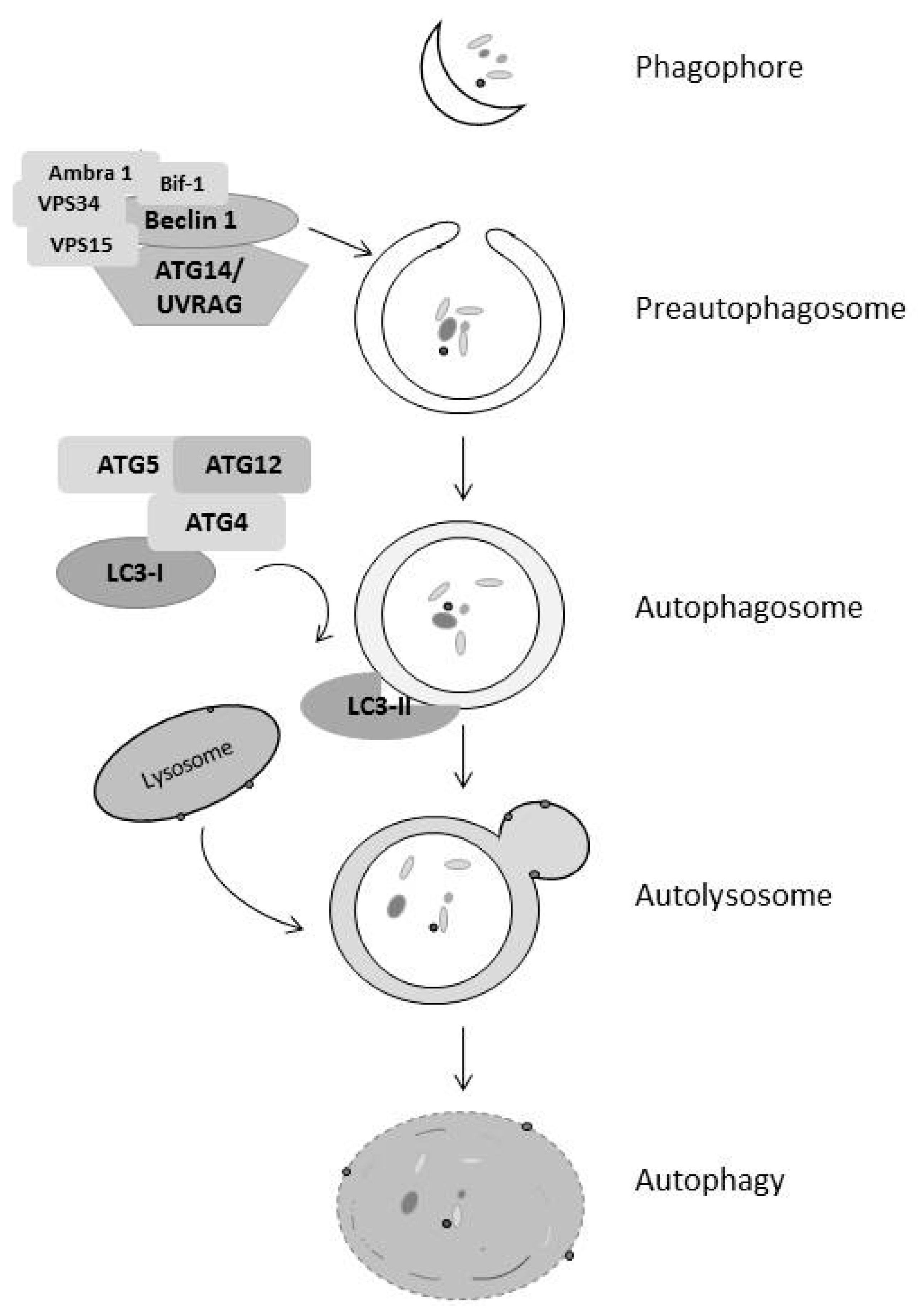
4. Autophagy Regulates Both Cell Survival and Death
5. Autophagy and Cancer
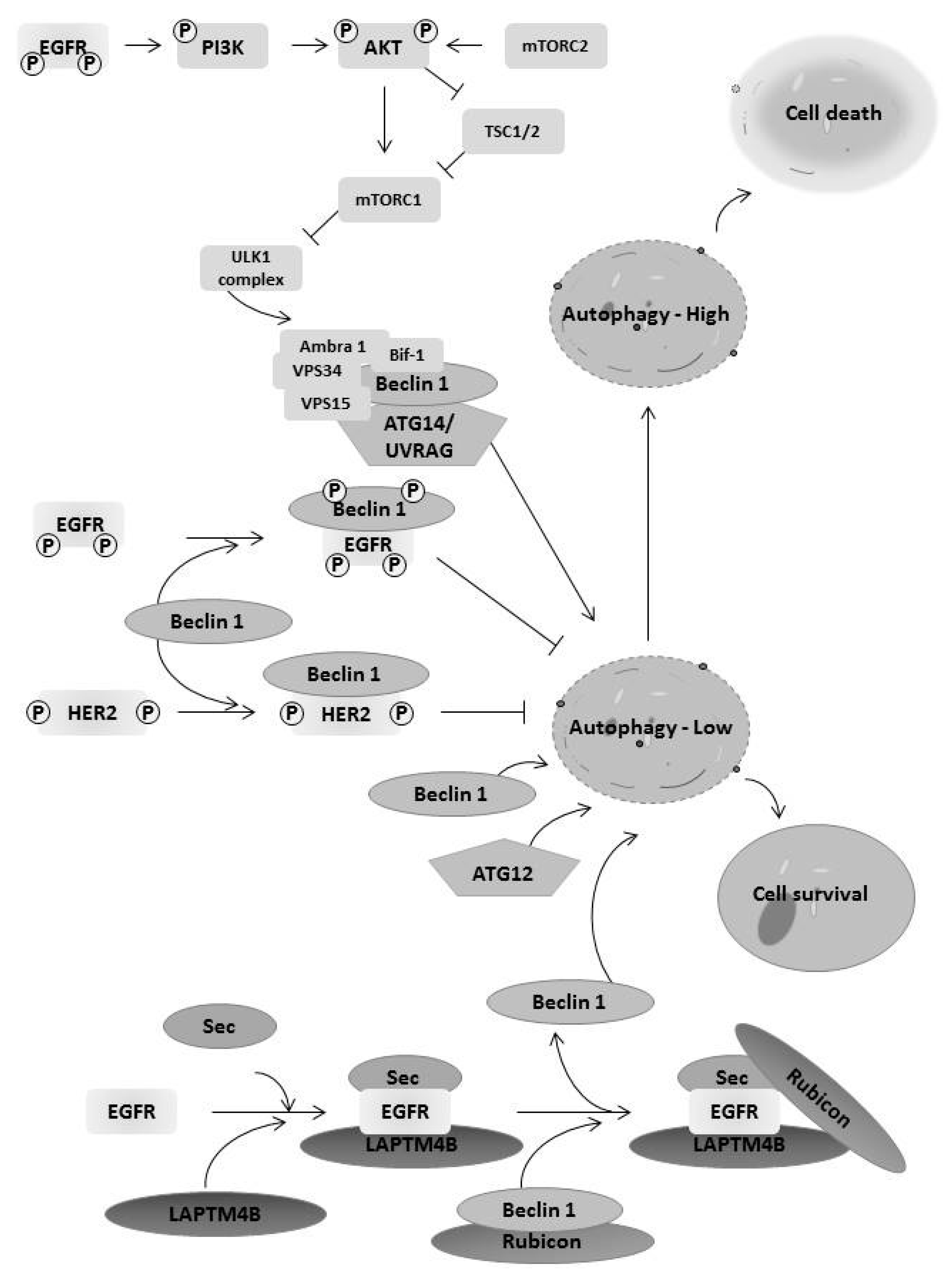
6. EGFR Family Members Regulates Autophagy
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Rouschop, K.M. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2014, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Henson, E.S.; Gibson, S.B. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: implications for cancer therapy. Cell Signal 2006, 18, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Epidermal growth factor signaling in transformed cells. Int. Rev. Cell Mol. Biol. 2015, 314, 1–41. [Google Scholar] [PubMed]
- Normanno, N.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; De Luca, A.; Caponigro, F.; Salomon, D.S. The ErbB receptors and their ligands in cancer: an overview. Curr. Drug Targets 2005, 6, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, G.; Staaf, J.; Vallon-Christersson, J.; Ringner, M.; Holm, K.; Hegardt, C.; Gunnarsson, H.; Fagerholm, R.; Strand, C.; Agnarsson, B.A.; et al. Genomic subtypes of breast cancer identified by array-comparative genomic hybridization display distinct molecular and clinical characteristics. Breast Cancer Res.: BCR. 2010, 12, R42. [Google Scholar] [CrossRef] [PubMed]
- Amann, J.; Kalyankrishna, S.; Massion, P.P.; Ohm, J.E.; Girard, L.; Shigematsu, H.; Peyton, M.; Juroske, D.; Huang, Y.; Stuart, S.J.; et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65, 226–235. [Google Scholar] [PubMed]
- Pedersen, M.W.; Meltorn, M.; Damstrup, L.; Poulsen, H.S. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann. Oncol. 2001, 12, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Izumi, Y.; Xu, L.; di Tomaso, E.; Fukumura, D.; Jain, R.K. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature 2002, 416, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Longva, K.E.; Pedersen, N.M.; Haslekas, C.; Stang, E.; Madshus, I.H. Herceptin-induced inhibition of ErbB2 signaling involves reduced phosphorylation of Akt but not endocytic down-regulation of ErbB2. Int. J. Cancer 2005, 116, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Luque-Cabal, M.; Garcia-Teijido, P.; Fernandez-Perez, Y.; Sanchez-Lorenzo, L.; Palacio-Vazquez, I. Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lyu, H.; Huang, J.; Liu, B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol. cancer 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. HGF-MET in cancer progression and biomarker discovery. Cancer Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Tokudome, N.; Sugihara, T.; Takahashi, S.; Hatake, K. Does lapatinib, a small-molecule tyrosine kinase inhibitor, constitute a breakthrough in the treatment of breast cancer? Breast Cancer 2007, 14, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Browne, B.C.; O'Brien, N.; Duffy, M.J.; Crown, J.; O'Donovan, N. HER-2 signaling and inhibition in breast cancer. Curr. Cancer Drug Targets 2009, 9, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Moy, B.; Goss, P.E. Lapatinib: current status and future directions in breast cancer. Oncologist 2006, 11, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, B.; Zaal, E.A.; Zecha, J.; Wu, W.; Berkers, C.R.; Kuster, B.; Lemeer, S. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kancha, R.K.; von Bubnoff, N.; Bartosch, N.; Peschel, C.; Engh, R.A.; Duyster, J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE 2011, 6, e26760. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; De Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Nahta, R.; Hung, M.C.; Esteva, F.J. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004, 64, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, G.; Miao, X.B.; Deng, X.B.; Wu, Y.; Liu, Y.; Jin, Z.R.; Li, X.Q.; Liu, Q.Z.; Sun, D.X.; et al. Cancer stem-like cell properties are regulated by EGFR/AKT/beta-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2027–2041. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. PNAS 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Dassonville, O.; Bozec, A.; Fischel, J.L.; Milano, G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors. Similarities and differences. Crit. Rev. Oncol. Hematol. 2007, 62, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Maiello, M.R.; Campiglio, M.; Napolitano, M.; Mancino, M.; Carotenuto, A.; Viglietto, G.; Menard, S. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell. Physiol. 2006, 207, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Booy, E.P.; Henson, E.S.; Gibson, S.B. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 2011, 30, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Sakimura, A.; Park, C.M.; Tomaru, R.; Tanaka, T.; Ozawa, T.; Zhou, Y.; Narita, K.; Kishi, H.; Muraguchi, A.; et al. Feedback control of ErbB2 via ERK-mediated phosphorylation of a conserved threonine in the juxtamembrane domain. Sci. Rep. 2016, 6, 31502. [Google Scholar] [CrossRef] [PubMed]
- Lux, M.P.; Fasching, P.A.; Schrauder, M.G.; Hein, A.; Jud, S.M.; Rauh, C.; Beckmann, M.W. The PI3K Pathway: Background and Treatment Approaches. Breast Care 2016, 11, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Nandini, D.; Pradip, D.; Brian, L.J. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Di Fiore, P.P.; Sigismund, S. Endocytic control of signaling at the plasma membrane. Curr. Opin. Cell Biol. 2016, 39, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Debnath, J. Autophagy and tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; Lopez-Otin, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Henson, E.S.; Xiao, W.; Huang, D.; McMillan-Ward, E.M.; Israels, S.J.; Gibson, S.B. Tyrosine kinase receptor EGFR regulates the switch in cancer cells between cell survival and cell death induced by autophagy in hypoxia. Autophagy 2016, 12, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Noman, M.Z.; Arakelian, T.; Lequeux, A.; Chouaib, S.; Berchem, G.; Moussay, E.; Paggetti, J.; Janji, B. Hijacker of the Antitumor Immune Response: Autophagy Is Showing Its Worst Facet. Front Oncol. 2016, 6, 246. [Google Scholar] [CrossRef] [PubMed]
- Janji, B.; Viry, E.; Moussay, E.; Paggetti, J.; Arakelian, T.; Mgrditchian, T.; Messai, Y.; Noman, M.Z.; Van Moer, K.; Hasmim, M.; et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016, 7, 17591–17607. [Google Scholar] [CrossRef] [PubMed]
- Dragowska, W.H.; Weppler, S.A.; Wang, J.C.; Wong, L.Y.; Kapanen, A.I.; Rawji, J.S.; Warburton, C.; Qadir, M.A.; Donohue, E.; Roberge, M.; et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS ONE 2013, 8, e76503. [Google Scholar]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gao, S.; Wang, D.; Song, D.; Feng, Y. Colorectal cancer cells are resistant to anti-EGFR monoclonal antibody through adapted autophagy. Am. J. Transl. Res. 2016, 8, 1190–1196. [Google Scholar] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gogol, M.; Gaudenz, K.; Gerton, J.L. Improved transcription and translation with L-leucine stimulation of mTORC1 in Roberts syndrome. BMC Genomics 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Sobhakumari, A.; Schickling, B.M.; Love-Homan, L.; Raeburn, A.; Fletcher, E.V.; Case, A.J.; Domann, F.E.; Miller, F.J.; Simons, A.L. NOX4 mediates cytoprotective autophagy induced by the EGFR inhibitor erlotinib in head and neck cancer cells. Toxicol. Appl. Pharmacol. 2013, 272, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Y.; Wei, X.; Zhou, X.; Gong, C.; Zhang, T.; Jin, P.; Xu, S.; Ma, D.; Gao, Q. Co-targeting EGFR and Autophagy Impairs Ovarian Cancer Cell Survival during Detachment from the ECM. Curr. Cancer Drug Targets 2015, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.; Wu, M.Y.; Hwang, M.H.; Chang, Y.T.; Huang, H.J.; Lin, A.M.; Yang, J.C. Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells. PLoS ONE 2015, 10, e0119135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Du, T.; Dong, X.; Li, Z.; Wu, G.; Zhang, R. Autophagy inhibition facilitates erlotinib cytotoxicity in lung cancer cells through modulation of endoplasmic reticulum stress. Int. J. Oncol. 2016, 48, 2558–2566. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvuri, U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol. Ther. 2012, 13, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- So, K.S.; Kim, C.H.; Rho, J.K.; Kim, S.Y.; Choi, Y.J.; Song, J.S.; Kim, W.S.; Choi, C.M.; Chun, Y.J.; Lee, J.C. Autophagosome-mediated EGFR down-regulation induced by the CK2 inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung cancer cells with resistance by T790M. PLoS ONE 2014, 9, e114000. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Wang, T.; Chen, Y.; Tang, X.; Yuan, Y.; Zhao, Y. beta-Elemene enhances the efficacy of gefitinib on glioblastoma multiforme cells through the inhibition of the EGFR signaling pathway. Int. J. Oncol. 2016, 49, 1427–1436. [Google Scholar] [PubMed]
- Jutten, B.; Keulers, T.G.; Schaaf, M.B.; Savelkouls, K.; Theys, J.; Span, P.N.; Vooijs, M.A.; Bussink, J.; Rouschop, K.M. EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival. Radiother. Oncol. 2013, 108, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Song, W.; Zhang, W.; Chen, L.; Xi, Z.; Xin, Z.; Jiang, X. Mitochondrially localized EGFR is subjected to autophagic regulation and implicated in cell survival. Autophagy 2008, 4, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.W.; Law, B.Y.; Mok, S.W.; Leung, E.L.; Fan, X.X.; Coghi, P.S.; Zeng, W.; Leung, C.H.; Ma, D.L.; Liu, L.; et al. Autophagic degradation of epidermal growth factor receptor in gefitinib-resistant lung cancer by celastrol. Int. J. Oncol. 2016, 49, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Lu, C.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. Interaction between Her2 and Beclin-1 proteins underlies a new mechanism of reciprocal regulation. J. Biol. Chem. 2013, 288, 20315–20325. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jung, W.; Koo, J.S. Expression of autophagy-related markers beclin-1, light chain 3A, light chain 3B and p62 according to the molecular subtype of breast cancer. Histopathol. 2013, 62, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyas, E.; Lopez-Bonet, E.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Choutka, C.; Horlings, H.M.; Leung, S.; Baker, J.H.; Lebovitz, C.; Dragowska, W.H.; Go, N.E.; Bally, M.B.; Minchinton, A.I.; et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget 2016, 7, 66970–66988. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.E.; Reidel, S.I.; Bal de Kier Joffe, E.D.; Jasnis, M.A.; Fiszman, G.L. Autophagy Protects from Trastuzumab-Induced Cytotoxicity in HER2 Overexpressing Breast Tumor Spheroids. PLoS ONE 2015, 10, e0137920. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, L.; Qiao, H.; Han, T.; Chen, S.; Liu, X.; Jiang, R.; Wei, Y.; Feng, D.; Zhang, Y.; et al. Autophagy stimulates apoptosis in HER2-overexpressing breast cancers treated by lapatinib. J. Cell. Biochem. 2013, 114, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chi, C.W.; Su, W.C.; Huang, H.L. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget 2014, 5, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Fang, L.W.; Su, W.C.; Hsu, W.Y.; Yang, K.C.; Huang, H.L. Lapatinib induces autophagic cell death and differentiation in acute myeloblastic leukemia. Onco. Targets Ther. 2016, 9, 4453–4464. [Google Scholar] [PubMed]
- Bisaro, B.; Sciortino, M.; Colombo, S.; Leal, M.P.C.; Costamagna, A.; Castellano, I.; Montemurro, F.; Rossi, V.; Valabrega, G.; Turco, E.; et al. p130Cas scaffold protein regulates ErbB2 stability by altering breast cancer cell sensitivity to autophagy. Oncotarget 2016, 7, 4442–4453. [Google Scholar] [PubMed]
- Nunes, J.; Zhang, H.; Angelopoulos, N.; Chhetri, J.; Osipo, C.; Grothey, A.; Stebbing, J.; Giamas, G. ATG9A loss confers resistance to trastuzumab via c-Cbl mediated Her2 degradation. Oncotarget 2016, 7, 27599–27612. [Google Scholar] [PubMed]
- Chiacchiera, F.; Grossi, V.; Cappellari, M.; Peserico, A.; Simonatto, M.; Germani, A.; Russo, S.; Moyer, M.P.; Resta, N.; Murzilli, S.; et al. Blocking p38/ERK crosstalk affects colorectal cancer growth by inducing apoptosis in vitro and in preclinical mouse models. Cancer Lett. 2012, 324, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Lai, F.J.; Sheu, H.M.; Lin, Y.S.; Chang, T.H.; Jan, M.S.; Chen, S.M.; Hsu, P.C.; Huang, T.T.; Huang, T.C.; et al. WWOX suppresses autophagy for inducing apoptosis in methotrexate-treated human squamous cell carcinoma. Cell Death Dis. 2013, 4, e792. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I.; Donati, V.; Gaudio, E.; Nicoloso, M.S.; Sundvall, M.; Korhonen, A.; Lundin, J.; Isola, J.; Sudol, M.; Joensuu, H.; et al. Association of Wwox with ErbB4 in breast cancer. Cancer Res. 2007, 67, 9330–9336. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Therapy | Target | Autophagy | Disease Site |
|---|---|---|---|
| lapatinib | EGFR/ErbB2 | Induction | breast cancer |
| rapamycin | mTOR | Induction | renal cancer |
| cetuximab | EGFR | Induction | colon cancer/head and neck cancer |
| trastuzumab | ErbB2 | Induction | gastric cancer/breast cancer |
| neratinib | EGFR/ErbB2 | Unknown | Not FDA approved |
| afatinib (BIBW2992) | ErbB2 | Unknown | lung cancer |
| pertuzumab | ErbB2 | Unknown | breast cancer |
| gefitinib | EGFR | Induction | lung cancer |
| panitumumab | EGFR | Induction | colon cancer |
| erlotinib | EGFR | Induction | lung and colon cancer |
| AG1478 | EGFR | Inhibition | ovarian |
| β-elemene | ATG-5 | Induction | gastric cancer |
| CX-4945 | Casein Kinase 2 | Induction | Not FDA approved |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henson, E.; Chen, Y.; Gibson, S. EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers 2017, 9, 27. https://doi.org/10.3390/cancers9040027
Henson E, Chen Y, Gibson S. EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers. 2017; 9(4):27. https://doi.org/10.3390/cancers9040027
Chicago/Turabian StyleHenson, Elizabeth, Yongqiang Chen, and Spencer Gibson. 2017. "EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer" Cancers 9, no. 4: 27. https://doi.org/10.3390/cancers9040027
APA StyleHenson, E., Chen, Y., & Gibson, S. (2017). EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers, 9(4), 27. https://doi.org/10.3390/cancers9040027