
Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment

Abstract
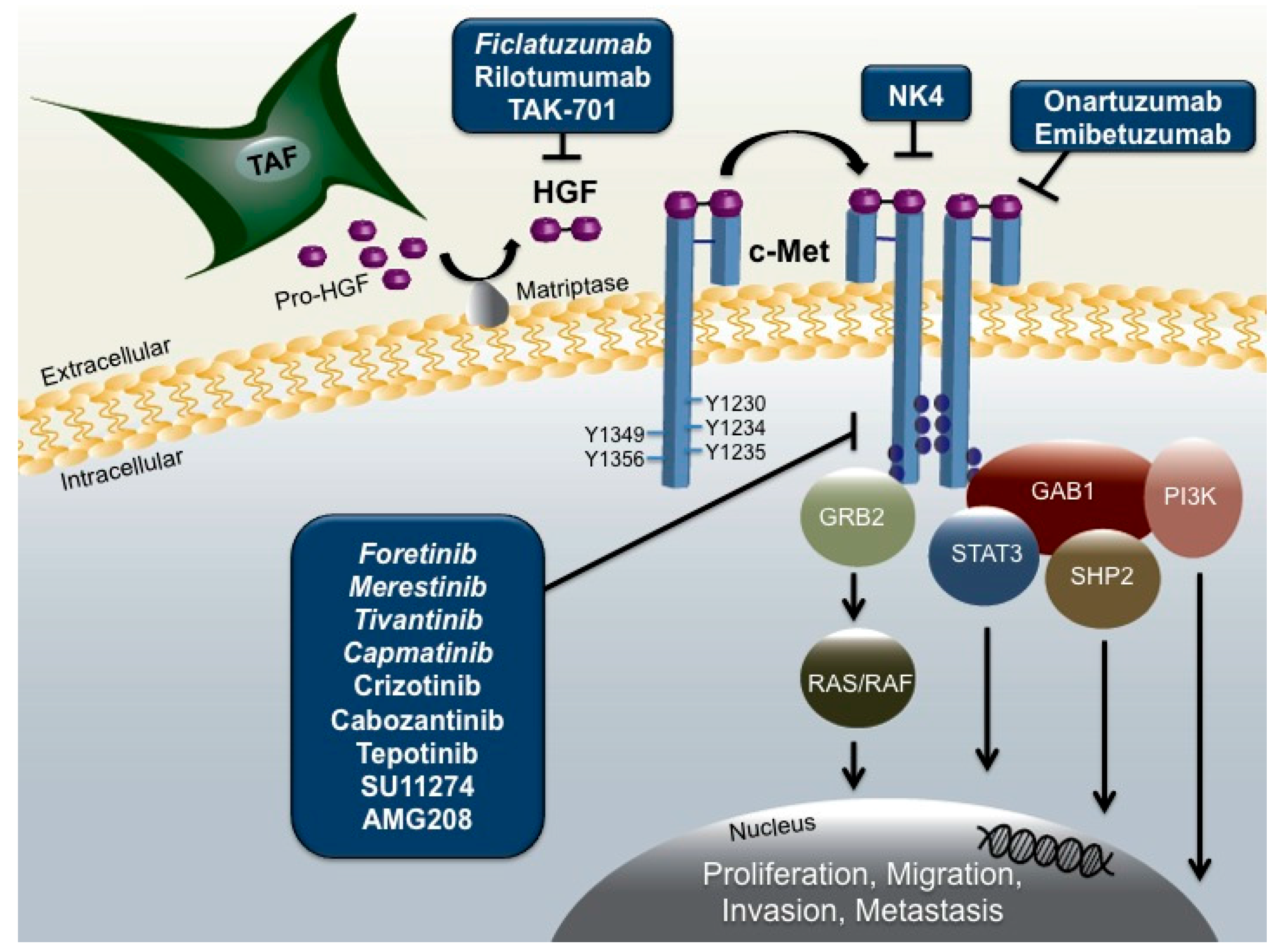
:1. Introduction
2. HGF/c-Met Signaling in HNSCC
3. HGF/c-Met Pathway Alterations in HNSCC
4. HGF/c-Met Pathway and HNSCC Progression
5. Relation of the HGF/c-Met Pathway to HNSCC Outcome
6. Role of HGF/c-Met Signaling as a Mechanism of Resistance to EGFR-Targeted Therapies
7. Targeting the HGF/c-Met Pathway in HNSCC
7.1. HGF/c-Met Targeted Therapies
7.2. Preclinical Models for HGF/c-Met Targeted Therapies
7.3. Clinical Trials in HNSCC Targeting the HGF/c-MET Pathway
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [PubMed]
- Pai, S.I.; Westra, W.H. Molecular pathology of head and neck cancer: Implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. 2009, 4, 49–70. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Eng, C. Epidemiology, staging, and screening of head and neck cancer. Cancer Treat. Res. 2003, 114, 15–60. [Google Scholar] [PubMed]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High incidence of head and neck squamous cell carcinoma in patients with fanconi anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Torre, L.A.; Jemal, A. Global trends of lung cancer mortality and smoking prevalence. Transl. Lung Cancer Res. 2015, 4, 327–338. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Colevas, A.D. Systemic therapy for metastatic or recurrent squamous cell carcinoma of the head and neck. J. Natl. Compr. Cancer Netw. 2015, 13, e37–e48. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar] [PubMed]
- Pomerantz, R.G.; Grandis, J.R. The role of epidermal growth factor receptor in head and neck squamous cell carcinoma. Curr. Oncol. Rep. 2003, 5, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Rubin Grandis, J.; Melhem, M.F.; Barnes, E.L.; Tweardy, D.J. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer 1996, 78, 1284–1292. [Google Scholar] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Serebriiskii, I.G.; Dunbrack, R.L., Jr.; Robinson, M.K.; Burtness, B.; Golemis, E.A. Protein-intrinsic and signaling network-based sources of resistance to EGFR- and ERBB family-targeted therapies in head and neck cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2011, 14, 260–279. [Google Scholar] [CrossRef] [PubMed]
- Zandberg, D.P.; Strome, S.E. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2014, 50, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Fuereder, T. Immunotherapy for head and neck squamous cell carcinoma. Memo 2016, 9, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet. Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: Results from the phase IB KEYNOTE-012 expansion cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.C.; Chan, A.T. Novel therapeutic target for head and neck squamous cell carcinoma: HGF-met signaling pathway. Anti-Cancer Drugs 2011, 22, 665–673. [Google Scholar] [CrossRef] [PubMed]
- De Herdt, M.J.; Baatenburg de Jong, R.J. HGF and c-Met as potential orchestrators of invasive growth in head and neck squamous cell carcinoma. Front. Biosci. J. Virtual Libr. 2008, 13, 2516–2526. [Google Scholar] [CrossRef]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Bhola, N.E.; Grandis, J.R. HGF/MET signaling in head and neck cancer: Impact on the tumor microenvironment. Clin. Cancer Res. 2016, 22, 4005–4013. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Kim, J.; Kahng, H.; Choi, E.C. Change of E-cadherin by hepatocyte growth factor and effects on the prognosis of hypopharyngeal carcinoma. Ann. Surg. Oncol. 2007, 14, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Kim, D.H.; Park, H.R.; Shin, H.S.; Kwon, J.H.; Lee, D.J.; Kim, J.H.; Cho, S.J.; Nam, E.S. Frequent hepatocyte growth factor overexpression and low frequency of c-Met gene amplification in human papillomavirus-negative tonsillar squamous cell carcinoma and their prognostic significances. Hum. Pathol. 2014, 45, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Nakamura, T. Roles of HGF as a pleiotropic factor in organ regeneration. Exs 1993, 65, 225–249. [Google Scholar] [PubMed]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Szabo, R.; Rasmussen, A.L.; Moyer, A.B.; Kosa, P.; Schafer, J.M.; Molinolo, A.A.; Gutkind, J.S.; Bugge, T.H. c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene 2011, 30, 2003–2016. [Google Scholar] [CrossRef] [PubMed]
- Vadnais, J.; Nault, G.; Daher, Z.; Amraei, M.; Dodier, Y.; Nabi, I.R.; Noel, J. Autocrine activation of the hepatocyte growth factor receptor/Met tyrosine kinase induces tumor cell motility by regulating pseudopodial protrusion. J. Biol. Chem. 2002, 277, 48342–48350. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of Met protooncogene cdna has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Ferracini, R.; Longati, P.; Naldini, L.; Vigna, E.; Comoglio, P.M. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J. Biol. Chem. 1991, 266, 19558–19564. [Google Scholar] [PubMed]
- Zhen, Z.; Giordano, S.; Longati, P.; Medico, E.; Campiglio, M.; Comoglio, P.M. Structural and functional domains critical for constitutive activation of the HGF-receptor (Met). Oncogene 1994, 9, 1691–1697. [Google Scholar] [PubMed]
- Weidner, K.M.; Di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Madoz-Gurpide, J.; Zazo, S.; Chamizo, C.; Casado, V.; Carames, C.; Gavin, E.; Cristobal, I.; Garcia-Foncillas, J.; Rojo, F. Activation of Met pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.; Lingen, M.W.; et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef] [PubMed]
- Morello, S.; Olivero, M.; Aimetti, M.; Bernardi, M.; Berrone, S.; Di Renzo, M.F.; Giordano, S. Met receptor is overexpressed but not mutated in oral squamous cell carcinomas. J. Cell. Physiol. 2001, 189, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.A.; Kim, E.K.; Heo, S.J.; Cho, B.C.; Kim, H.R.; Chung, J.M.; Yoon, S.O. Alteration status and prognostic value of Met in head and neck squamous cell carcinoma. J. Cancer 2016, 7, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the Met oncogene are selected during metastatic spread of human hnsc carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Ghadjar, P.; Blank-Liss, W.; Simcock, M.; Hegyi, I.; Beer, K.T.; Moch, H.; Aebersold, D.M.; Zimmer, Y. Met Y1253D-activating point mutation and development of distant metastasis in advanced head and neck cancers. Clin. Exp. Metastasis 2009, 26, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, D.M.; Landt, O.; Berthou, S.; Gruber, G.; Beer, K.T.; Greiner, R.H.; Zimmer, Y. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene 2003, 22, 8519–8523. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Pilotto, S.; Gkountakos, A.; Carbognin, L.; Scarpa, A.; Tortora, G.; Bria, E. Met exon 14 juxtamembrane splicing mutations: Clinical and therapeutical perspectives for cancer therapy. Ann. Transl. Med. 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.D.; Kornberg, L.J. Overexpression of scatter factor and its receptor (c-Met) in oral squamous cell carcinoma. Laryngoscope 1998, 108, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Cao, B.; Law, S.; Xie, Y.; Lee, P.Y.; Cheung, L.; Chen, Y.; Huang, X.; Chan, H.M.; Zhao, P.; et al. Hepatocyte growth factor promotes cancer cell migration and angiogenic factors expression: A prognostic marker of human esophageal squamous cell carcinomas. Clin. Cancer Res. 2005, 11, 6190–6197. [Google Scholar] [CrossRef] [PubMed]
- Koontongkaew, S.; Amornphimoltham, P.; Yapong, B. Tumor-stroma interactions influence cytokine expression and matrix metalloproteinase activities in paired primary and metastatic head and neck cancer cells. Cell Biol. Int. 2009, 33, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck 2014, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. Emt and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Frixen, U.H.; Behrens, J.; Sachs, M.; Eberle, G.; Voss, B.; Warda, A.; Lochner, D.; Birchmeier, W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991, 113, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, J.; Lin, X.; Wang, X. E-cadherin expression and prognosis of head and neck squamous cell carcinoma: Evidence from 19 published investigations. OncoTargets Ther. 2016, 9, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, S.; von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/EGR-1-mediated upregulation of snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Jiang, W.G. Association of the HGF/SF receptor, c-Met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem. Biophys. Res. Commun. 1999, 261, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, E.L.; Matrisian, L.M. Matrix metalloproteases in head and neck cancer. Head Neck 2006, 28, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Kamata, N.; Fujimoto, R.; Tsutsumi, S.; Tomonari, M.; Taki, M.; Hosokawa, H.; Nagayama, M. Increased invasion and matrix metalloproteinase-2 expression by snail-induced mesenchymal transition in squamous cell carcinomas. Int. J. Oncol. 2003, 22, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Johnson, N.W.; Gao, J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int. J. Oncol. 2010, 37, 663–668. [Google Scholar] [PubMed]
- Lui, V.W.; Wong, E.Y.; Ho, K.; Ng, P.K.; Lau, C.P.; Tsui, S.K.; Tsang, C.M.; Tsao, S.W.; Cheng, S.H.; Ng, M.H.; et al. Inhibition of c-Met downregulates TIGAR expression and reduces nadph production leading to cell death. Oncogene 2011, 30, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.Y.; Wong, S.C.; Chan, C.M.; Lam, E.K.; Ho, L.Y.; Lau, C.P.; Au, T.C.; Chan, A.K.; Tsang, C.M.; Tsao, S.W.; et al. Tp53-induced glycolysis and apoptosis regulator promotes proliferation and invasiveness of nasopharyngeal carcinoma cells. Oncol. Lett. 2015, 9, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Malhotra, P.S.; Thomas, G.R.; Ondrey, F.G.; Duffey, D.C.; Smith, C.W.; Enamorado, I.; Yeh, N.T.; Kroog, G.S.; Rudy, S.; et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin. Cancer Res. 1999, 5, 1369–1379. [Google Scholar] [PubMed]
- Eisma, R.J.; Spiro, J.D.; Kreutzer, D.L. Role of angiogenic factors: Coexpression of interleukin-8 and vascular endothelial growth factor in patients with head and neck squamous carcinoma. Laryngoscope 1999, 109, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Chen, Z.; Li, Z.Y.; Yeh, N.T.; Bancroft, C.C.; Van Waes, C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001, 61, 5911–5918. [Google Scholar] [PubMed]
- Worden, B.; Yang, X.P.; Lee, T.L.; Bagain, L.; Yeh, N.T.; Cohen, J.G.; Van Waes, C.; Chen, Z. Hepatocyte growth factor/scatter factor differentially regulates expression of proangiogenic factors through EGR-1 in head and neck squamous cell carcinoma. Cancer Res. 2005, 65, 7071–7080. [Google Scholar] [CrossRef] [PubMed]
- Ninck, S.; Reisser, C.; Dyckhoff, G.; Helmke, B.; Bauer, H.; Herold-Mende, C. Expression profiles of angiogenic growth factors in squamous cell carcinomas of the head and neck. Int. J. Cancer 2003, 106, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Mamelle, G.; Pampurik, J.; Luboinski, B.; Lancar, R.; Lusinchi, A.; Bosq, J. Lymph node prognostic factors in head and neck squamous cell carcinomas. Am. J. Surg. 1994, 168, 494–498. [Google Scholar] [CrossRef]
- Noguchi, M.; Kido, Y.; Kubota, H.; Kinjo, H.; Kohama, G. Prognostic factors and relative risk for survival in N1–3 oral squamous cell carcinoma: A multivariate analysis using Cox’s hazard model. Br. J. Oral Maxillofac. Surg. 1999, 37, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Moon, S.K.; Bae, J.H.; Lee, J.H.; Han, J.H.; Kim, K.; Choi, E.C. Expression of hepatocyte growth factor and c-Met in hypopharyngeal squamous cell carcinoma. Acta Oto-Laryngol. 2006, 126, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Wang, J.T.; Chang, Y.F.; Liu, B.Y.; Wang, Y.P.; Sun, A.; Chiang, C.P. Expression of hepatocyte growth factor and c-Met protein is significantly associated with the progression of oral squamous cell carcinoma in Taiwan. J. Oral Pathol. Med. 2004, 33, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Cortesina, G.; Martone, T.; Galeazzi, E.; Olivero, M.; De Stefani, A.; Bussi, M.; Valente, G.; Comoglio, P.M.; Di Renzo, M.F. Staging of head and neck squamous cell carcinoma using the Met oncogene product as marker of tumor cells in lymph node metastases. Int. J. Cancer 2000, 89, 286–292. [Google Scholar] [CrossRef]
- Galeazzi, E.; Olivero, M.; Gervasio, F.C.; De Stefani, A.; Valente, G.; Comoglio, P.M.; Di Renzo, M.F.; Cortesina, G. Detection of Met oncogene/hepatocyte growth factor receptor in lymph node metastases from head and neck squamous cell carcinomas. Eur. Arch. Oto-Rhino-Laryngol. 1997, 254, S138–S143. [Google Scholar] [CrossRef]
- Baschnagel, A.M.; Williams, L.; Hanna, A.; Chen, P.Y.; Krauss, D.J.; Pruetz, B.L.; Akervall, J.; Wilson, G.D. c-Met expression is a marker of poor prognosis in patients with locally advanced head and neck squamous cell carcinoma treated with chemoradiation. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Farina, A.; Rubini, C.; Coccia, E.; Capogreco, M.; Colella, G.; Leonardi, R.; Campisi, G.; Carinci, F. Effect of c-Met expression on survival in head and neck squamous cell carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2006, 27, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Alexander, D.; Reinert, S.; Hoffmann, J. Prognostic value of c-Met expression in oral squamous cell carcinoma. Exp. Ther. Med. 2010, 1, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Sawatsubashi, M.; Sasatomi, E.; Mizokami, H.; Tokunaga, O.; Shin, T. Expression of c-Met in laryngeal carcinoma. Virchows Arch. Int. J. Pathol. 1998, 432, 331–335. [Google Scholar] [CrossRef]
- Endo, K.; Shirai, A.; Furukawa, M.; Yoshizaki, T. Prognostic value of cell motility activation factors in patients with tongue squamous cell carcinoma. Hum. Pathol. 2006, 37, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, D.M.; Kollar, A.; Beer, K.T.; Laissue, J.; Greiner, R.H.; Djonov, V. Involvement of the hepatocyte growth factor/scatter factor receptor c-Met and of Bcl-XL in the resistance of oropharyngeal cancer to ionizing radiation. Int. J. Cancer 2001, 96, 41–54. [Google Scholar] [CrossRef]
- Lacroix, L.; Post, S.F.; Valent, A.; Melkane, A.E.; Vielh, P.; Egile, C.; Castell, C.; Larois, C.; Micallef, S.; Saulnier, P.; et al. Met genetic abnormalities unreliable for patient selection for therapeutic intervention in oropharyngeal squamous cell carcinoma. PLoS ONE 2014, 9, e84319. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Wang, D.; Magliocca, K.R.; Hu, Z.; Nannapaneni, S.; Kim, S.; Chen, Z.; Sun, S.Y.; Shin, D.M.; Saba, N.F.; et al. Human papillomavirus oncoprotein e6 upregulates c-Met through p53 downregulation. Eur. J. Cancer 2016, 65, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Lee, J.S.; Kang, S.O.; Bae, J.H.; Hong, S.P.; Kahng, H. Serum hepatocyte growth factor as a marker of tumor activity in head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Duffy, S.; Teknos, T.; Islam, M.; Chen, Z.; Albert, P.S.; Wolf, G.; Van Waes, C. Nuclear factor-kappab-related serum factors as longitudinal biomarkers of response and survival in advanced oropharyngeal carcinoma. Clin. Cancer Res. 2007, 13, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Kawamata, H.; Omotehara, F.; Nakashiro, K.; Kimura-Yanagawa, T.; Hino, S.; Begum, N.M.; Hoque, M.O.; Yoshida, H.; Sato, M.; et al. Role of HGF/c-Met system in invasion and metastasis of oral squamous cell carcinoma cells in vitro and its clinical significance. Int. J. Cancer 2001, 93, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S. The role of human papillomavirus infection in head and neck cancers. Ann. Oncol. 2010, 21, vii243–vii245. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Reeves, T.D.; Hill, E.G.; Armeson, K.E.; Gillespie, M.B. Cetuximab therapy for head and neck squamous cell carcinoma: A systematic review of the data. Otolaryngol. Head Neck Surg. 2011, 144, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Herbst, R.S.; Leon, X.; Amellal, N.; Baselga, J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer 2008, 112, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Rabinowits, G.; Haddad, R.I. Overcoming resistance to EGFR inhibitor in head and neck cancer: A review of the literature. Oral Oncol. 2012, 48, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Storkel, S.; Kerr, K.M.; Van Cutsem, E.; Pirker, R.; Hirsch, F.R.; Vermorken, J.B.; von Heydebreck, A.; Esser, R.; Celik, I.; et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: Analysis of data from the extreme and crystal studies. Eur. J. Cancer 2013, 49, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Mesia, R.; Rivera, F.; Remenar, E.; Hitt, R.; Erfan, J.; Rottey, S.; Kawecki, A.; Zabolotnyy, D.; Benasso, M.; et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: Extreme study. Ann. Oncol. 2011, 22, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; He, G.; Lui, V.W.; Thomas, S.; Henry, C.; Gubish, C.T.; Joyce, S.; Quesnelle, K.M.; Siegfried, J.M.; Grandis, J.R. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin. Cancer Res. 2013, 19, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Krumbach, R.; Schuler, J.; Hofmann, M.; Giesemann, T.; Fiebig, H.H.; Beckers, T. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: Activation of Met as one mechanism for drug resistance. Eur. J. Cancer 2011, 47, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Stabile, L.P.; Gubish, C.T.; Gooding, W.E.; Grandis, J.R.; Siegfried, J.M. Dual blockade of egfr and c-Met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin. Cancer Res. 2011, 17, 4425–4438. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Yamada, Y.; Furuta, K.; Honma, Y.; Iwasa, S.; Takashima, A.; Kato, K.; Hamaguchi, T.; Shimada, Y. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-egfr antibody treatment in kras wild-type metastatic colorectal cancer. Br. J. Cancer 2014, 110, 2716–2727. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takeuchi, S.; Kita, K.; Bando, H.; Nakamura, T.; Matsumoto, K.; Yano, S. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J. Thorac. Oncol. 2012, 7, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kimura, T.; Kudoh, S.; Mitsuoka, S.; Watanabe, T.; Suzumura, T.; Tachibana, K.; Noguchi, M.; Yano, S.; Hirata, K. Reaction of plasma hepatocyte growth factor levels in non-small cell lung cancer patients treated with EGFR-TKIs. Int. J. Cancer 2011, 129, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Bewley, A.F.; Sperry, S.M.; Montone, K.T.; Gimotty, P.A.; Rasanen, K.; Facompre, N.D.; Weinstein, G.S.; Nakagawa, H.; Diehl, J.A.; et al. EGFR inhibition promotes an aggressive invasion pattern mediated by mesenchymal-like tumor cells within squamous cell carcinomas. Mol. Cancer Ther. 2013, 12, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, S.; Duan, S.Z.; Zhang, L.; Zhou, H.; Hu, Y.; Zhou, X.; Shi, C.; Zhou, R.; Zhang, Z. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res. 2014, 74, 7546–7559. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in Ros1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, H.; Liu, Y.; Anderson, A.; Peterson, J.; Greger, J.; Martin, A.M.; Gilmer, T.M. Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells. Mol. Cancer Ther. 2011, 10, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Jeay, S.; Li, Y.; Chen, C.R.; France, D.S.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Leggett, D.S.; Li, C.J. Arq 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol. Cancer Ther. 2010, 9, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.H.; Yang, L.Y.; Cao, Z.Y.; Qian, Y. Tivantinib (arq-197) exhibits anti-tumor activity with down-regulation of fak in oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2015, 457, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Calles, A.; Kwiatkowski, N.; Cammarata, B.K.; Ercan, D.; Gray, N.S.; Janne, P.A. Tivantinib (arq 197) efficacy is independent of Met inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 2015, 9, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Nisa, L.; Aebersold, D.M.; Giger, R.; Zimmer, Y.; Medova, M. Biological, diagnostic and therapeutic relevance of the Met receptor signaling in head and neck cancer. Pharmacol. Ther. 2014, 143, 337–349. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, M.; Cappuzzo, F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biol. Targets Ther. 2013, 7, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kandl, C.; Hamilton, C.D.; Shnayder, Y.; Tsue, T.T.; Kakarala, K.; Ledgerwood, L.; Sun, X.S.; Huang, H.J.; Girod, D.; et al. Mitigation of tumor-associated fibroblast-facilitated head and neck cancer progression with anti-hepatocyte growth factor antibody ficlatuzumab. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006, 66, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Rothstein, M.E.; Keohavong, P.; Jin, J.; Yin, J.; Land, S.R.; Dacic, S.; Luong, T.M.; Kim, K.J.; Dulak, A.M.; et al. Therapeutic targeting of human hepatocyte growth factor with a single neutralizing monoclonal antibody reduces lung tumorigenesis. Mol. Cancer Ther. 2008, 7, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Coon, V.; Laukert, T.; Pedone, C.A.; Laterra, J.; Kim, K.J.; Fults, D.W. Molecular therapy targeting sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol. Cancer Ther. 2010, 9, 2627–2636. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, W.; Okamoto, I.; Tanaka, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Yamaguchi, H.; Arao, T.; Nishio, K.; Fukuoka, M.; et al. Tak-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived hgf in non-small cell lung cancer with an EGFR mutation. Mol. Cancer Ther. 2010, 9, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Rothstein, M.E.; Keohavong, P.; Lenzner, D.; Land, S.R.; Gaither-Davis, A.L.; Kim, K.J.; Kaminski, N.; Siegfried, J.M. Targeting of both the c-Met and egfr pathways results in additive inhibition of lung tumorigenesis in transgenic mice. Cancers 2010, 2, 2153–2170. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J.; et al. Monovalent antibody design and mechanism of action of onartuzumab, a Met antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R.; et al. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent Met activation and tumor growth. Clin. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Nakamura, T. HGF-MET cascade, a key target for inhibiting cancer metastasis: The impact of NK4 discovery on cancer biology and therapeutics. Int. J. Mol. Sci. 2013, 14, 888–919. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Klieser, W. Three-dimensional cell cultures: From molecular mechanisms to clinical applications. Am. J. Physiol. 1997, 273, C1109–C1123. [Google Scholar] [PubMed]
- Mery, B.; Rancoule, C.; Guy, J.B.; Espenel, S.; Wozny, A.S.; Battiston-Montagne, P.; Ardail, D.; Beuve, M.; Alphonse, G.; Rodriguez-Lafrasse, C.; et al. Preclinical models in HNSCC: A comprehensive review. Oral Oncol. 2017, 65, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Rong, S.; Bodescot, M.; Blair, D.; Dunn, J.; Nakamura, T.; Mizuno, K.; Park, M.; Chan, A.; Aaronson, S.; Vande Woude, G.F. Tumorigenicity of the Met proto-oncogene and the gene for hepatocyte growth factor. Mol.Cell. Biol. 1992, 12, 5152–5158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Su, Y.; Lanning, N.; Gustafson, M.; Shinomiya, N.; Zhao, P.; Cao, B.; Tsarfaty, G.; Wang, L.M.; Hay, R.; et al. Enhanced growth of human met-expressing xenografts in a new strain of immunocompromised mice transgenic for human hepatocyte growth factor/scatter factor. Oncogene 2005, 24, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Francone, T.D.; Landmann, R.G.; Chen, C.T.; Sun, M.Y.; Kuntz, E.J.; Zeng, Z.; Dematteo, R.P.; Paty, P.B.; Weiser, M.R. Novel xenograft model expressing human hepatocyte growth factor shows ligand-dependent growth of c-Met-expressing tumors. Mol. Cancer Ther. 2007, 6, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Lyker, J.S.; Land, S.R.; Dacic, S.; Zamboni, B.A.; Siegfried, J.M. Transgenic mice overexpressing hepatocyte growth factor in the airways show increased susceptibility to lung cancer. Carcinogenesis 2006, 27, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.N.; Holsinger, F.C.; Jasser, S.A.; Bekele, B.N.; Fidler, I.J. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin. Cancer Res. 2002, 8, 293–298. [Google Scholar] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Keysar, S.B.; Astling, D.P.; Anderson, R.T.; Vogler, B.W.; Bowles, D.W.; Morton, J.J.; Paylor, J.J.; Glogowska, M.J.; Le, P.N.; Eagles-Soukup, J.R.; et al. A patient tumor transplant model of squamous cell cancer identifies pi3k inhibitors as candidate therapeutics in defined molecular bins. Mol. Oncol. 2013, 7, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.; Sarantopoulos, J.; Kallender, H.; McCallum, S.; Keer, H.N.; Blumenschein, G., Jr. Phase II trial of single-agent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2013, 31, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Perez, K.; Safran, H.; Ruth He, A.; Giles, J.; Hu, T.; Moser, B.; Turner, P.; Walgren, R.; Plimack, E. Interim results of a first-in-human phase 1 study of the oral Met kinase inhibitor, ly2801653, in patients with advanced cancer. In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research, San Diego, CA, USA, 5–9 April 2014. [Google Scholar]


{kind=link}
| Clinicopathological Correlations with Elevated c-Met Expression | HNSCC Sample Site/Size | Reference |
|---|---|---|
| Lymph node metastasis | Larynx (n = 82) | Sawatsubashi 1998 (ref. [79]) |
| Decreased local failure-free survival | Oropharynx (n = 97) | Aebersold 2001 (ref. [81]) |
| Decreased disease-free survival | ||
| Decreased overall survival | ||
| Higher tumor staging | Oral Cavity (n = 93) | Chen 2004 (ref. [73]) |
| Lymph node metastasis | ||
| Clinical staging | ||
| Decreased overall survival rate | Oral Cavity (n = 84) | Lo Muzio 2006 (ref. [77]) |
| Lymph node metastasis | Hypopharynx (n = 40) | Kim 2006 (ref. [72]) |
| Higher tumor stage | Tongue (n = 99) | Endo 2006 (ref. [80]) |
| Lymph node metastasis | ||
| Clinical Stage | ||
| Local recurrence | ||
| Distant metastatic recurrence | ||
| Lower tumor staging | Oral Cavity (n = 211) | Freudlsperger 2010 (ref. [78]) |
| Worse disease-free survival in HPV-negative patients | Oropharynx (n = 70) | Baschnagel 2014 (ref. [76]) |
| Larynx (n = 27) | ||
| Hypopharynx (n = 7) | ||
| Oral Cavity (n = 3) | ||
| Decreased progression-free survival * Decreased overall survival * | Oral Cavity (n = 7) | Madoz-Gurpide 2015 (ref. [40]) |
| Oropharynx (n = 7) | ||
| Hypopharynx (n = 6) | ||
| Larynx (n = 12) | ||
| Occult (n = 1) | ||
| Higher tumor staging | Oropharynx (n = 78) | Qian 2016 (ref. [83]) |
| HPV-positive status |
| Clinicopathological Correlations with Elevated Serum/Tumoral HGF | HNSCC Sample Site/Size | Reference |
|---|---|---|
| Elevated serum HGF correlates with cancer burden | Oral Cavity (n = 31) | Uchida 2001 (ref. [86]) |
| Elevated tumoral HGF correlates with metastasis | ||
| Elevated tumoral HGF correlates with lymph node metastasis and pathologic stage | Hypopharynx (n = 40) | Kim 2006 (ref. [72]) |
| Elevated serum HGF correlates with higher tumor staging | Oral Cavity (n = 22) | Kim 2007 (ref. [84]) |
| Larynx (n = 21) | ||
| Oropharynx (n = 16) | ||
| Hypopharynx (n = 14) | ||
| Maxilla (n = 5) | ||
| Longitudinal increases of serum HGF correlate with decreased cause-specific survival | Oropharynx (n = 30) | Allen 2007 (ref. [85]) |
| Drug | Primary Molecular Targets | Stage in Clinical Development |
|---|---|---|
| c-Met TKIs | ||
| Crizotinib (PF 2341066) | c-Met/ALK/ROS-1 | FDA approved for ALK-Positive/ROS-1 rearrangement-positive NSCLC Preclinical: HNSCC |
| Foretinib (GSK 1363089) | c-Met/VEGFR2 | Phase II: R/M breast cancer, papillary renal-cell carcinoma, NSCLC, metastatic gastric cancer, R/M HNSCC |
| Tivantinib (ARQ 197) | c-Met | Phase III: hepatocellular carcinoma Phase II: HNSCC |
| SU11274 | c-Met | Preclinical: HNSCC and NSCLC |
| Tepotinib (EMD 1214603) | c-Met | Phase II: NSCLC |
| AMG 208 | c-Met/VEGF | Phase I: advanced solid tumors |
| Cabozantinib (XL 184) | c-Met/VEGFRs/AXL | FDA approved for medullary thyroid cancer and advanced renal cell carcinoma patients with prior angiogenic therapy |
| HGF Antibodies | ||
| Ficlatuzumab (AV-299) | HGF | Phase II: NSCLC Phase Ib: HNSCC |
| Rilotumumab (AMG 102) | HGF | Phase II/III: combined with erlotinib in recurrent stage IV squamous cell lung cancer Preclinical: glioblastoma |
| TAK-701 (L2G7) | HGF | Phase I: advanced solid tumors Preclinical: HNSCC |
| c-Met Antibodies | ||
| Onartuzumab (MetMab) | c-Met | Phase III: in combination with oxaliplatin in metastatic gastroesophageal cancer and in combination with erlotinib in advanced NSCLC |
| Emibetuzumab (LY 2875358) | c-Met | Phase II: NSCLC, advanced gastric cancer |
| HGF Antagonists | ||
| NK4 | c-Met | Preclinical: gallbladder, pancreatic, myeloma carcinomas |
| Clinical Trial | Phase | HGF/c-Met Agent | Other Agents | Setting/Status |
|---|---|---|---|---|
| Single agent | ||||
| NCT00725764 | II | Foretinib (GSK1363089) | - | R/M/Completed [131] |
| NCT01285037 | I | Merestinib (LY2801653) | - | R/M/Ongoing |
| Dual agent | ||||
| NCT01696955 | II | Tivantinib (ARQ 197) | Cetuximab | c-Met positive; R/M/Ongoing |
| NCT02205398 | Ib | Capmatinib (INC280) | Cetuximab | R/M/Ongoing |
| NCT02277197 | Ib | Ficlatuzumab (AV-299) | Cetuximab | R/M/Ongoing |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothenberger, N.J.; Stabile, L.P. Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers 2017, 9, 39. https://doi.org/10.3390/cancers9040039
Rothenberger NJ, Stabile LP. Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers. 2017; 9(4):39. https://doi.org/10.3390/cancers9040039
Chicago/Turabian StyleRothenberger, Natalie J., and Laura P. Stabile. 2017. "Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment" Cancers 9, no. 4: 39. https://doi.org/10.3390/cancers9040039
APA StyleRothenberger, N. J., & Stabile, L. P. (2017). Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers, 9(4), 39. https://doi.org/10.3390/cancers9040039