
Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
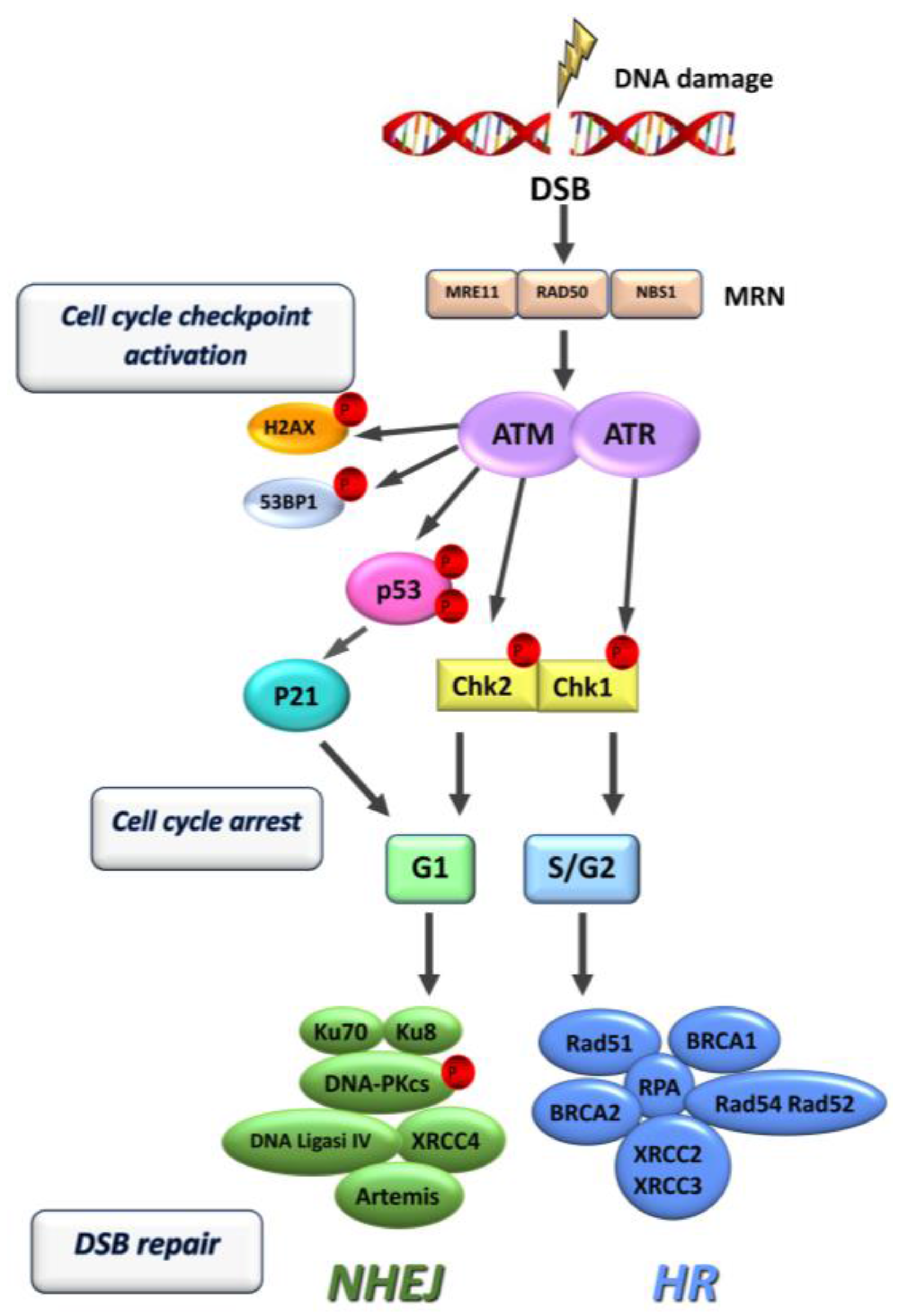
2. DNA Damage Response (DDR)
3. Glioma Stem Cells (GSCs) and Chemoresistance
4. Double Strand Break Repair
5. Resistance Mechanisms to Alkylating Agents
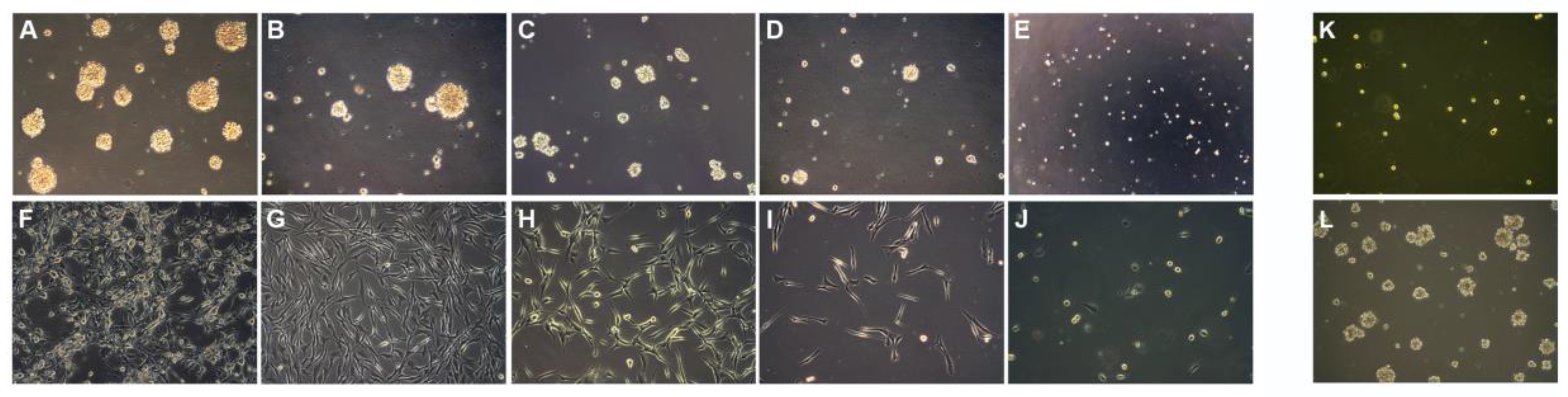
6. In Vitro Cytotoxicity Study on Neurospheres (NS) and Adherent Cell (AC) Cultures
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, Revised 4th ed.; IARC Press: Lyon, France, 2016; pp. 1–408. [Google Scholar]
- Minniti, G.; Muni, R.; Lanzetta, G.; Marchetti, P.; Enrici, R.M. Chemotherapy for glioblastoma: Current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009, 29, 5171–5184. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Schmalz, P.G.; Shen, M.J.; Park, J.K. Treatment resistance mechanisms of malignant glioma tumor stem cells. Cancers Basel 2011, 3, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.M.; Pinnell, N.; Wen, P.Y.; D’Andrea, A. Targeting DNA repair and the cell cycle in glioblastoma. J. Neuro-Oncol. 2012, 107, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Holland, E.C. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011, 71, 5945–5949. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Salmaggi, A.; Boiardi, A.; Gelati, M.; Russo, A.; Calatozzolo, C.; Ciusani, E.; Sciacca, F.L.; Ottolina, A.; Parati, E.A.; La Porta, C. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia 2006, 54, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Caldera, V.; Mellai, M.; Annovazzi, L.; Monzeglio, O.; Piazzi, A.; Schiffer, D. MGMT hypermethylation and MDR system in glioblastoma cancer stem cells. Cancer Genom. Proteom. 2012, 9, 171–178. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signaling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signaling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.F.; Wheeler, H.T.; McDonald, K.L. Contribution of DNA repair mechanisms to determining chemotherapy response in high-grade glioma. J. Clin. Neurosci. 2008, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Facchino, S.; Abdouh, M.; Bernier, G. Brain cancer stem cells: Current status on glioblastoma multiforme. Cancers Basel 2011, 3, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Fosmark, S.; Hellwege, S.; Beier, D.; Kristensen, B.W.; Beier, C.P. Chemoresistance and chemotherapy targeting stem-like cells in malignant glioma. Adv. Exp. Med. Biol. 2015, 853, 111–138. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells—Much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Bjerkvig, R.; Tysnes, B.B. DNA repair and cancer stem-like cells—Potential partners in glioma drug resistance? Cancer Treat. Rev. 2008, 34, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Mazzucco, M.; Mellai, M. The Microenvironment in Gliomas: Phenotypic Expressions. Cancers Basel 2015, 7, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Persano, L.; Pistollato, F.; Rampazzo, E.; Della Puppa, A.; Abbadi, S.; Frasson, C.; Volpin, F.; Indraccolo, S.; Scienza, R.; Basso, G. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death Dis. 2012, 3, e412. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Cassoni, P.; Valentini, M.C.; Mazzucco, M.; Mellai, M. Glioblastoma Stem Cells: Conversion or Reprogramming from Tumor non-Stem Cells? J. Stem Cell Res. Ther. 2015, 5, 11. [Google Scholar] [CrossRef]
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sakariassen, P.O.; Tsinkalovsky, O.; Immervoll, H.; Boe, S.O.; Svendsen, A.; Prestegarden, L.; Rosland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Ghods, A.J.; Irvin, D.; Liu, G.; Yuan, X.; Abdulkadir, I.R.; Tunici, P.; Konda, B.; Wachsmann-Hogiu, S.; Black, K.L.; Yu, J.S. Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem Cells 2007, 25, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and non stem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G. The bright and the dark sides of DNA repair in stem cells. J. Biomed. Biotechnol. 2010, 845396. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol. Cancer Res. 2009, 7, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G. Frontiers in targeting glioma stem cells. Eur. J. Cancer 2011, 47, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Vecchio, D.; Cappelli, E.; Ropolo, M.; Poggi, A.; Nozza, P.; Biassoni, R.; Mascelli, S.; Capra, V.; Kalfas, F.; et al. Characterization of glioma stem cells through multiple stem cell markers and their specific sensitization to double-strand break-inducing agents by pharmacological inhibition of ataxia telangiectasia mutated protein. Brain Pathol. 2012, 22, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R.; Ferguson, D.J.; Callaghan, R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br. J. Cancer 2005, 93, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Masunaga, S.; Ono, K.; Hori, H.; Suzuki, M.; Kinashi, Y.; Takagaki, M.; Kasai, S.; Nagasawa, H.; Uto, Y. Potentially lethal damage repair by total and quiescent tumor cells following various DNA-damaging treatments. Radiat. Med. 1999, 17, 259–264. [Google Scholar] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schriefer, B.; Brawanski, K.; Hau, P.; Weis, J.; Schulz, J.B.; Beier, C.P. Efficacy of clinically relevant temozolomide dosing schemes in glioblastoma cancer stem cell lines. J. Neuro-Oncol. 2012, 109, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Bleau, AM.; Holland, E.C. Chemotherapeutic treatment of gliomas increases the amount of cancer stem-like cells. Med. Sci. (Paris) 2009, 25, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Pallini, R.; Ricci-Vitiani, L.; Montano, N.; Mollinari, C.; Biffoni, M.; Cenci, T.; Pierconti, F.; Martini, M.; De Maria, R.; Larocca, L.M. Expression of the stem cell marker CD133 in recurrent glioblastoma and its value for prognosis. Cancer 2011, 117, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gónzalez, B.; Melguizo, C. Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef] [PubMed]
- Mihaliak, A.M.; Gilbert, C.A.; Li, L.; Daou, M.C.; Moser, R.P.; Reeves, A.; Cochran, B.H.; Ross, A.H. Clinically relevant doses of chemotherapy agents reversibly block formation of glioblastoma neurospheres. Cancer Lett. 2010, 296, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.A.; Daou, M.C.; Moser, R.P.; Ross, A.H. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010, 70, 6870–6879. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Kang, S.K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Kandenwein, J.A.; Karl, S.; Vellanki, S.H.; Braun, V.; Eramo, A.; Antoniadis, G.; Debatin, K.M.; Fulda, S. The pyridinylfuranopyrimidine inhibitor, PI-103, chemosensitizes glioblastoma cells for apoptosis by inhibiting DNA repair. Oncogene 2009, 28, 3586–3596. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a novel dual PI3K-mTOR inhibitor displays anti-glioma activity and reduces chemoresistance to temozolomide in human glioma cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.C.; Roos, W.P.; Jost, E.; Belohlavek, C.; Lennerz, V.; Schmidt, C.W.; Christmann, M.; Kaina, B. Temozolomide-and fotemustine-induced apoptosis in human malignant melanoma cells: Response related to MGMT, MMR, DSBs, and p53. Br. J. Cancer 2009, 100, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Rep. (Amst) 2009, 8, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Bolderson, E.; Richard, D.J.; Zhou, B.B.; Khanna, K.K. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin. Cancer Res. 2009, 15, 6314–6320. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Khalil, A.; McEwen, A.; Holmes, M.; Neill, S.; Povirk, L.F.; Valerie, K. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J. Biol. Chem. 2004, 279, 15402–15410. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Mori, E.; Takahashi, A. DNA double strand breaks: Their production, recognition, and repair in eukaryotes. Mutat. Res. 2009, 669, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Seol, H.J.; Yoo, H.Y.; Jin, J.; Joo, K.M.; Kong, D.S.; Yoon, S.J.; Yang, H.; Kang, W.; Lim, D.H.; Park, K.; et al. Prognostic implications of the DNA damage response pathway in glioblastoma. Oncol. Rep. 2011, 26, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.F.; de Tribolet, N.; Regli, L.; Wick, W.; Kouwenhoven, M.C.; et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; McEllin, B.; Camacho, C.V.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA double-strand break repair: A molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009, 69, 4252–4259. [Google Scholar] [CrossRef] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6 methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Takahashim, A.; Mori, E.; Ohnishi, K.; McKinnon, P.J.; Sakaki, T.; Nakase, H.; Ohnishi, T. DNA ligase IV as a new molecular target for temozolomide. Biochem. Biophys. Res. Commun. 2009, 387, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- D’Atri, S.; Tentori, L.; Lacal, P.M.; Graziani, G.; Pagani, E.; Benincasa, E.; Zambruno, G.; Bonmassar, E.; Jiricny, J. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol. Pharmacol. 1998, 54, 334–341. [Google Scholar] [PubMed]
- Günther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer. 2003, 88, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797. [Google Scholar] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gilbert, M.R.; Chakravarti, A. Chemoradiotherapy in malignant glioma: Standard of care and future directions. J. Clin. Oncol. 2007, 25, 4127–4136. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Erkkinen, M.G.; Nestler, U.; Stupp, R.; Mehta, M.; Aldape, K.; Gilbert, M.R.; Black, P.M.; Loeffler, J.S. Temozolomide-mediated radiation enhancement in glioblastoma: A report on underlying mechanisms. Clin. Cancer Res. 2006, 12, 4738–4746. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Thomé, M.P.; Bueno e Silva, M.M.; Pelegrini, A.L.; Ledur, P.F.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol abrogates the temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the temozolomide-induced senescence in glioma cells. BMC Cancer 2013, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.W.; Mymryk, J.S.; Cairncross, J.G. Pharmaceutical-mediated inactivation of p53 sensitizes U87MG glioma cells to BCNU and temozolomide. Int. J. Cancer 2005, 116, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kurose, A.; Ogawa, A.; Ogasawara, K.; Traganos, F.; Darzynkiewicz, Z.; Sawai, T. Diversity of DNA damage response of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol. Ther. 2009, 8, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Bjerkvig, R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev. Anticancer Ther. 2012, 12, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Bocangel, D.B.; Finkelstein, S.; Schold, S.C.; Bhakat, K.K.; Mitra, S.; Kokkinakis, D.M. Multifaceted resistance of gliomas to temozolomide. Clin. Cancer Res. 2002, 8, 2725–2734. [Google Scholar] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Langella, T.; Corbetta, C.; Pellegatta, S. Hypermutations in gliomas: A potential immunotherapy target. Discov. Med. 2017, 23, 113–120. [Google Scholar] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.; Le, D.T. Efficacy of PD-1 blockade in tumors with MMR deficiency. Immunotherapy 2016, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Shou, J.; Mullapudi, S.R.; Lang, F.F., Jr.; Rao, J.S.; Ali-Osman, F. Enforced expression of wild-type p53 curtails the transcription of the O(6)-methylguanine DNA methyltransferase gene in human tumor cells and enhances their sensitivity to alkylating agents. Clin. Cancer Res. 2001, 7, 1398–1409. [Google Scholar] [PubMed]
- Ito, M.; Ohba, S.; Gaensler, K.; Ronen, S.M.; Mukherjee, J.; Pieper, R.O. Early Chk1 phosphorylation is driven by temozolomide-induced, DNA double strand break- and mismatch repair-independent DNA damage. PLoS ONE 2013, 8, e62351. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Tsaryk, R.; Fabian, K.; Thacker, J.; Kaina, B. Xrcc2 deficiency sensitizes cells to apoptosis by MNNG and the alkylating anticancer drugs temozolomide, fotemustine and mafosfamide. Cancer Lett. 2006, 239, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Rad51 and BRCA2 – new molecular targets for sensitizing glioma cells to alkylating anticancer drugs. PLoS ONE 2011, 6, e27183. [Google Scholar] [CrossRef] [PubMed]
- Short, S.C.; Giampieri, S.; Worku, M.; Alcaide-German, M.; Sioftanos, G.; Bourne, S.; Lio, K.I.; Shaked-Rabi, M.; Martindale, C. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor derived cells. Neuro-Oncol. 2011, 13, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Kitange, G.J.; Gupta, S.K.; Joughin, B.A.; Chaim, I.A.; Mazzucato, P.; Lauffenburger, D.A.; Sarkaria, J.N.; Samson, L.D. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res. 2017, 77, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.W.; Ellsworth, R.K.; Kumar, R.; Fjerstad, K.; Martinez, J.; Nagel, R.B.; Eschbacher, J.; Stea, B. Rad51 protein expression and survival in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Roos, W.P.; Christmann, M.; Menck, C.F.; Kaina, B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007, 67, 11886–11895. [Google Scholar] [CrossRef] [PubMed]
- Ludlum, D.B. The chloroethylnitrosoureas: Sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest. 1997, 15, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Hennekes, F.; Bhatti, A.; Kaina, B. Chloroethylnitrosourea-induced cell death and genotoxicity: Cell cycle dependence and the role of DNA double-strand breaks, HR and NHEJ. Cell Cycle 2012, 11, 2606–2619. [Google Scholar] [CrossRef] [PubMed]
- Annovazzi, L.; Caldera, V.; Mellai, M.; Riganti, C.; Battaglia, L.; Chirio, D.; Melcarne, A.; Schiffer, D. The DNA damage/repair cascade in glioblastoma cell lines after chemotherapeutic agent treatment. Int. J. Oncol. 2015, 46, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Stan, A.C.; Casares, S.; Radu, D.; Walter, G.F.; Brumeanu, T.D. Doxorubicin-induced cell death in highly invasive human gliomas. Anticancer Res. 1999, 19, 941–950. [Google Scholar] [PubMed]
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005, 25, 3825–3831. [Google Scholar] [PubMed]
- Chang, S.M.; Kuhn, J.G.; Robins, H.I.; Schold, S.C.; Spence, A.M.; Berger, M.S.; Mehta, M.; Pollack, I.F.; Rankin, C.; Prados, M.D. A phase II study of paclitaxel in patients with recurrent malignant glioma using different doses depending upon the concomitant use of anticonvulsants: A North American brain tumor consortium report. Cancer 2001, 91, 417–422. [Google Scholar] [CrossRef]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef] [PubMed]
- Branham, M.T.; Nadin, S.B.; Vargas-Roig, L.M.; Ciocca, D.R. DNA damage induced by paclitaxel and DNA repair capability of peripheral blood lymphocytes as evaluated by the alkaline comet assay. Mutat. Res. 2004, 560, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.; Sun, G.; Doorly, M.; Wang, J.Y.; Schwartz, M.A. Modulation of DNA damage-induced apoptosis by cell adhesion is independently mediated by p53 and c-Abl. Proc. Natl. Acad. Sci. USA 2003, 100, 10281–10286. [Google Scholar] [CrossRef] [PubMed]
- Coutts, A.S.; Weston, L.; La Thangue, N.B. A transcription co-factor integrates cell adhesion and motility with the p53 response. Proc. Natl. Acad. Sci. USA 2009, 106, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Gallarate, M.; Peira, E.; Chirio, D.; Muntoni, E.; Biasibetti, E.; Capucchio, M.T.; Valazza, A.; Panciani, P.P.; Lanotte, M.; et al. Solid lipid nanoparticles for potential doxorubicin delivery in glioblastoma treatment: Preliminary in vitro studies. J. Pharm. Sci. 2014, 103, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Chirio, D.; Gallarate, M.; Peira, E.; Battaglia, L.; Muntoni, E.; Riganti, C.; Biasibetti, E.; Capucchio, M.T.; Valazza, A.; Panciani, P.; et al. Positive-charged solid lipid nanoparticles as paclitaxel drug delivery system in glioblastoma treatment. Eur. J. Pharm. Biopharm. 2014, 88, 746–758. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. https://doi.org/10.3390/cancers9060057
Annovazzi L, Mellai M, Schiffer D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers. 2017; 9(6):57. https://doi.org/10.3390/cancers9060057
Chicago/Turabian StyleAnnovazzi, Laura, Marta Mellai, and Davide Schiffer. 2017. "Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma" Cancers 9, no. 6: 57. https://doi.org/10.3390/cancers9060057
APA StyleAnnovazzi, L., Mellai, M., & Schiffer, D. (2017). Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers, 9(6), 57. https://doi.org/10.3390/cancers9060057