2.1. Catalyst Characterization
A summary of the measured physico-chemical properties of the catalysts is shown in
Table 1. For all the samples, the actual platinum content and Co/Al atomic ratio were very close to the nominal value. Pt-containing samples presented a larger specific surface area (S
BET) and pore volume (V
pore) than CoAl. The addition of Pt seemed to shift the pore size (d
pore) of the xPt/CoAl samples towards higher values. After reduction, S
BET of the resultant catalysts decreased while d
pore increased, except for 0.3Pt/Al, which preserved its textural properties. Pore size distribution, calculated from the nitrogen adsorption–desorption isotherms (
Figure S1, Supplementary Materials), revealed that both the calcined and reduced samples were mainly mesoporous materials.
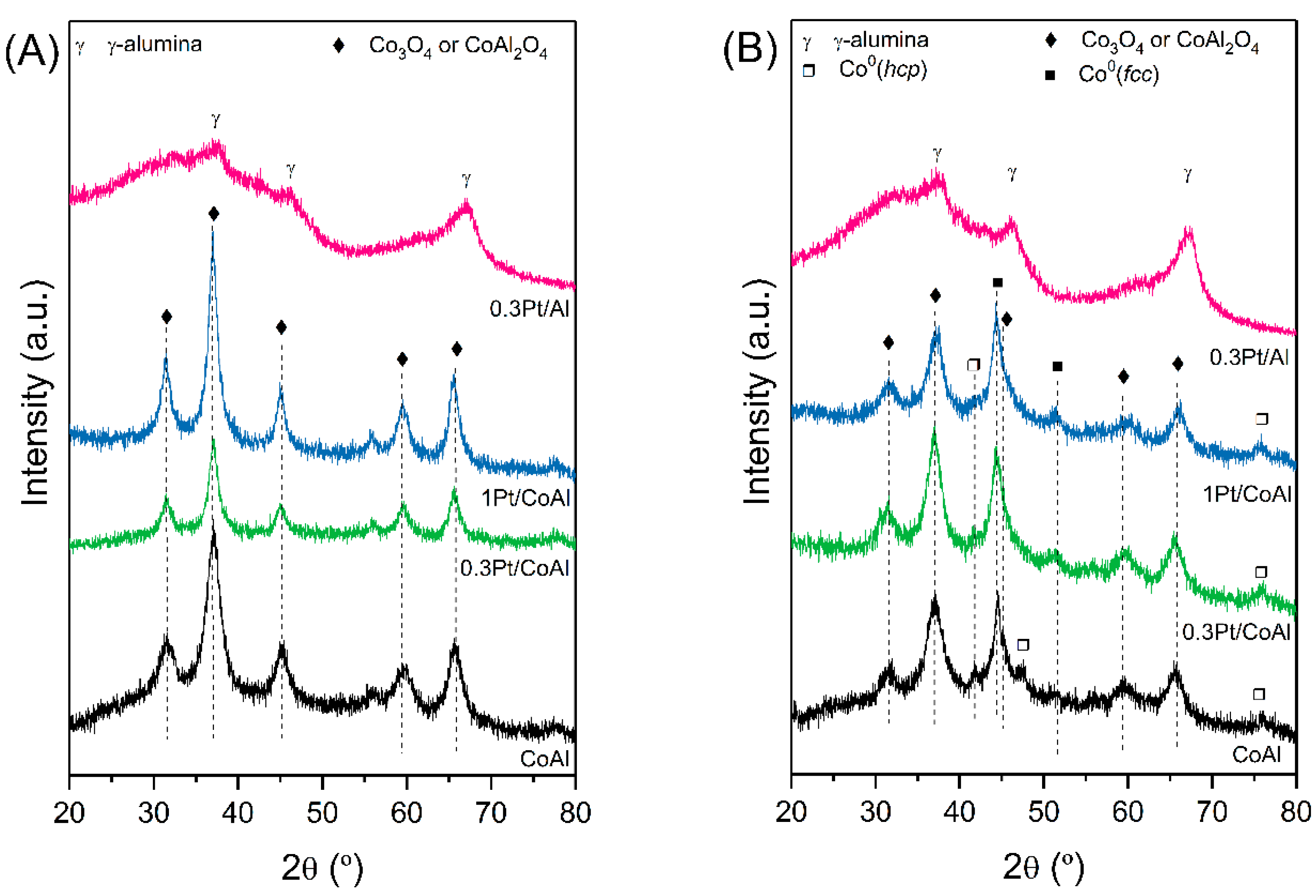
The phase composition and morphology of the samples was evaluated by XRD, and the obtained diffraction patterns are shown in
Figure 1. Calcined CoAl and xPt/CoAl samples (
Figure 1A) presented the characteristic diffraction peaks of cobalt aluminate (PDF 00-044-0160) and cobalt oxide (PDF 00-042-1467) spinels. Both species, with cubic crystal system and Fd3m space group, have almost the same diffraction angles and only could be distinguished by their different lattice parameters (smaller for Co
3O
4). The lattice parameter (a) of the calcined spinel phase was calculated from the Bragg equation, which holds for the
fcc structure:
The calculated parameter for the bare CoAl sample was 8.042 Å, which was lower than the lattice constant of standard stoichiometric CoAl2O4 (8.104 Å). This could be due to the non-stoichiometric composition of CoAl, caused by the higher Co/Al mole ratio (0.634) compared to the corresponding stoichiometric one. After the addition of Pt, the lattice parameter increased slightly, proportionally to platinum content, suggesting that Pt strongly interacted with the support. The average crystallite size of the spinel phase of the calcined samples was estimated by means of the Scherrer equation and indicated an increase of 26% with platinum addition. No characteristic peak of Pt species was observed, probably because of their high dispersion and low loadings. Catalyst 0.3Pt/Al showed characteristic peaks of γ-alumina, with a clear baseline elevation, indicative of its amorphous nature.
Regarding the reduced assays (
Figure 1B), cobalt-containing catalysts showed diffraction peaks ascribed to metallic Co, prevailing in the
fcc phase with respect to the
hcp phase. In addition, the aforementioned cobalt spinel phase could be still identified. However, as for the calcined samples, no peak in metallic Pt was detected. After Pt loadings, metallic cobalt crystallite size decreased by half, suggesting that Pt acts as a structural promoter which decreases metallic Co particle size (
Table 1).
H
2-TPR experiments were carried out in order to gain information about reducible species. The H
2-TPR profiles of all the prepared catalysts are displayed in
Figure 2 (due to its low intensity, the signal from 0.3Pt/Al was five-fold magnified). The monometallic 0.3Pt/Al sample showed three reduction peaks, of low intensity. The two low temperature peaks were related to the reduction of platinum species (PtOx). The peak at 227 °C was attributed to the reduction of platinum species with low interaction with the alumina, and the one at 375 °C to platinum species in close interaction with the support. The high temperature peak, at around 660 °C, was due to the reduction of alumina, assisted by the hydrogen spillover on Pt
0 [
25]. For the CoAl sample, the reduction peaks at 292 and 413 °C came from the Co
3+ → Co
2+ reduction, the first of surface cobalt cations without interaction with the support and the latter from the Co
3+ species in close interaction with the support. The peak at 594 °C was ascribed to Co
2+ → Co
0 reduction. Finally, the peak at 783 °C corresponded to the reduction of Co ions as cobalt aluminate (CoAl
2O
4) [
16]. Regarding bimetallic xPt/CoAl samples, these presented four reduction peaks. The H
2 uptake at low temperatures largely exceeded the theoretical hydrogen consumption of platinum species (
Table 2). Based on this, the peaks centered at 192 °C (0.3Pt/CoAl) and 157 °C (1Pt/CoAl) were ascribed to the reduction of PtO
x species alongside that of platinum-promoted (via hydrogen spillover) Co
3+ → Co
2+ reduction. As can be seen, the addition of Pt caused two different behaviors depending on its loading. On one hand, at the lowest loading (0.3 wt.%), the presence of Pt decreased the Co
2+ → Co
0 reduction temperature from 594 to 563 °C and that of cobalt ions in the spinel phase from 783 to 736 °C. Nevertheless, by increasing the Pt loading to 1 wt.%, the reduction temperature of the aforementioned cobalt species increased by 74 and 84 °C, respectively. Thus, it could be concluded that the addition of 0.3 wt.% Pt improved the reducibility of both metal species (Pt and Co), as deduced from the increased low temperature hydrogen consumption and the amount of available metallic Co with respect to reference CoAl (
Table 2). This promotional effect could be attributed to a strong Pt–Co interaction. The smaller size of Co
0 in the bimetallic samples, as estimated by XRD, could be also involved. As expected, the amount of available metallic Pt sites increased proportionally to Pt loading—that is, Pt dispersion remained practically unchanged (as seen in
Table 2).
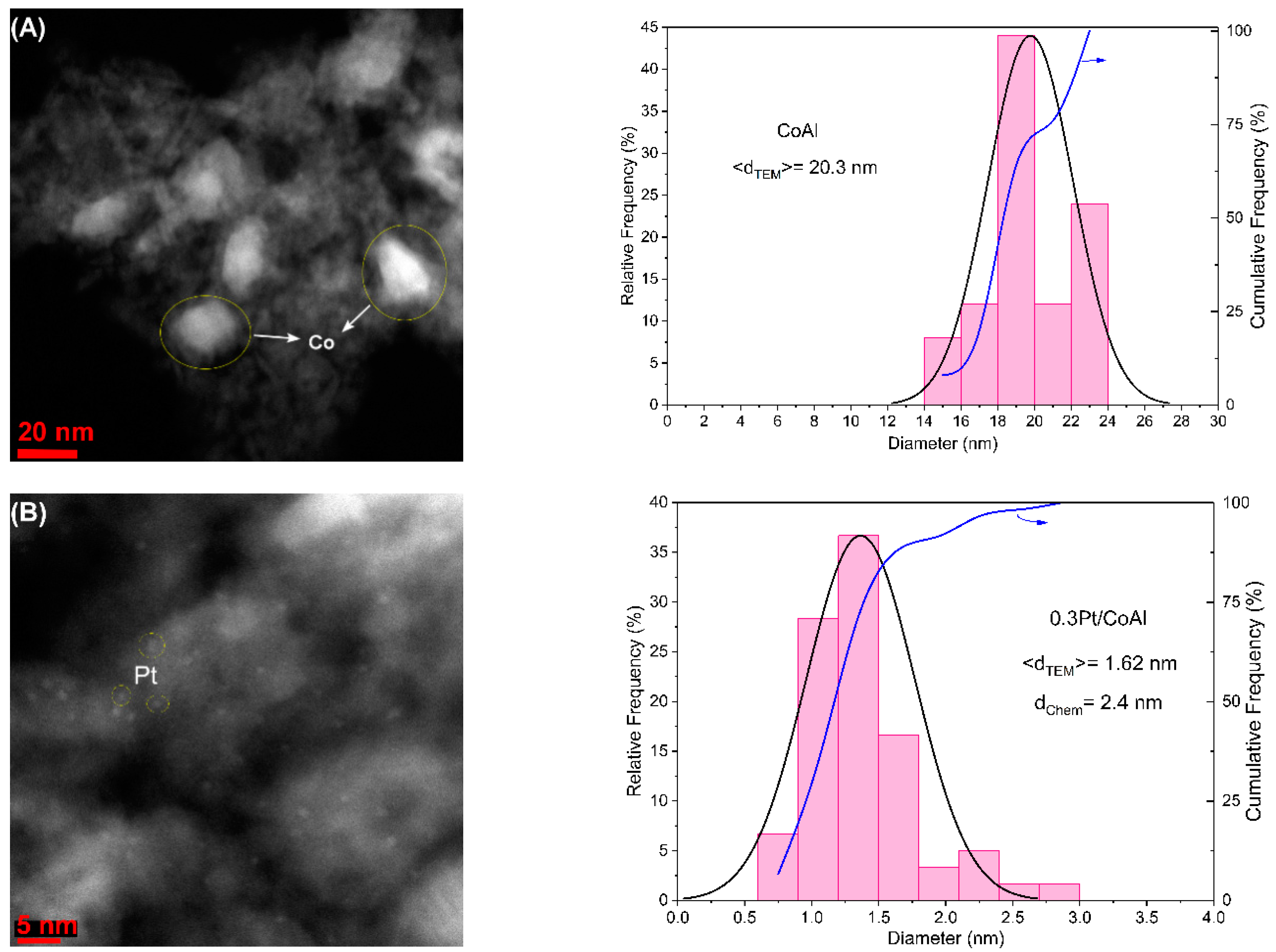
The morphology of the reduced samples was investigated by scanning transmission electron microscopy (STEM) techniques.
Figure 3 shows their corresponding STEM micrographs and the resulting particle size distribution histograms. The surface morphology of cobalt aluminate support is basically preserved after Pt addition. The analysis revealed the presence of well-dispersed metallic particles of homogeneous size, especially in Pt-containing catalysts. The overall particle size of metallic cobalt nanoparticles in the CoAl sample was in the range of 14–24 nm, leading to an average size of 20.3 nm, with a cube/cuboid shape. On the other hand, Pt nanoparticles were of near-spherical shape, with a much smaller size, in concordance with the absence of diffraction peaks in XRD. Pt nanoparticles ranged between 0.6 and 3.0 nm, with an average size of 1.62 nm for 0.3Pt/CoAl and 1.88 nm for 1Pt/CoAl. Pt nanoparticles were homogeneously dispersed onto larger Co nanoparticles (comparable with those observed in the CoAl catalyst), as can be seen in
Figure 3B,C. For the monometallic 0.3Pt/Al sample, most of the nanoparticles were uniform, with an average size of 1.84 nm. The energy dispersive X-ray spectra (EDX) analysis (
Figure S2, Supplementary Materials) confirmed the metallic composition of the Pt nanoparticles.
Platinum particle size was also calculated from H2 chemisorption, and a mean diameter of about 2.2 nm was measured for all the samples, which was slightly larger than the STEM calculated value. This discrepancy could be due to a limited hydrogen adsorption capacity due to strong Pt–Co interactions and likely alloy formation. The large discrepancy in the 0.3Pt/CoAl catalyst suggested a stronger Pt–Co interaction, in agreement with H2-TPR. Pt dispersion was around 62% for the 1Pt/CoAl and 0.3Pt/Al samples and slightly lower (58%) in catalyst 0.3Pt/CoAl. We hypothesize that the dispersion measured by H2 chemisorption for 0.3Pt/CoAl was under-estimated due to the high Co–Pt interaction. In any case, it is worth noting the good agreement between the STEM and H2 chemisorption measured Pt dispersion data.
The density of the surface basic sites of the reduced catalysts was evaluated by thermogravimetry, measuring the sample mass gain upon CO
2 chemisorption. As reported in
Table 2, the CoAl sample had the highest density of surface basic sites, with 1.90 µmol
CO2/m
2. The xPt/CoAl series presented around 30%–40% lower density than monometallic CoAl. Among them, density slightly increased at 1 wt.% Pt loading, whereas it was the lowest for catalyst 0.3Pt/Al (0.32 µmol
CO2/m
2).
The effect of Pt addition on the distribution of Co species lying on the near-surface of the calcined and reduced samples was investigated by XPS. The binding energies (BE) of the Al, Co and Pt species and the surface Co/Al atomic ratios are reported in
Table 3. The Co 2p and Al 2p X-ray photoelectron spectra for the reduced samples are shown in
Figure 4 (Pt 4d
5/2 spectra are given in
Figure S3, Supplementary Materials). Prior to reduction, binding energies of Al 2p and Co 2p
3/2 were significantly higher in CoAl than in Pt-impregnated catalysts. The higher values can be due to the strong interaction between aluminum and cobalt [
26]. Upon reduction, all samples showed the characteristic Co 2p doublet (Co 2p
3/2 and Co 2p
1/2) due to spin-orbit splitting. The spectra were deconvoluted into Co
0, Co
2+ and shake up satellites; however, no peaks could be assigned to Co
3+, probably due to the self-reduction in the high vacuum chamber. Co 2p peaks of the xPt/CoAl samples shifted to higher binding energies compared to parent CoAl. This upshift can be attributed to the electron transfer from less electronegative Co to Pt atoms that decreased electron density at the Co site [
27]. The Al 2p peak at the binding energy of 74.2 eV corresponded to Al cations placed at the octahedral sites [
28]. As the strong Al 2p peak overlaps with the most prominent platinum electron line (Pt 4f), the Pt 4d
5/2 was analyzed instead. Although limited due to the low platinum content, the spectra of Pt 4d region showed peaks located at 314.0 ± 0.2 eV for the Pt-containing catalysts, which can be ascribed to the Pt
0 phase [
29].
The overall surface Co/Al atomic ratio, calculated from Co 2p to Al 2p peak intensities (
Table 3), decreased in comparison to bulk composition (evaluated by ICP-AES). This surface enrichment on Al
3+ ions was attributed to the lower surface free energy of Al as compared to Co [
30]. Upon reduction, the Co/Al atomic ratio on surface decreased, probably due to the incorporation of metallic species into the porous structure [
31], which would also explain the decrease in pore volume. Furthermore, it is interesting to note the higher Co/Al values measured for the bimetallic assays (as compared to parent CoAl), regardless of their calcined or reduced state.
2.2. Liquid-Phase WGS Activity in the Absence of Hydrogen (H2/CO = 0)
The WGS performance of the prepared catalysts was evaluated at 260 °C and 50 bar (which corresponded to 3.2 bar above saturation), with H
2O/CO = 15/1 mole ratio in the feedstream and a space-time of 76.8 kg
cat·s/mol
CO. The obtained CO conversion (X
CO), CO
2 yield (Y
CO2) and CH
4 yield (Y
CH4) at 10 h TOS are shown in
Figure 5. Among all those tested, the monometallic 0.3Pt/Al catalyst exhibited the lowest activity in the aqueous-phase WGS, with a 29.9% conversion of CO. The platinum-free monometallic CoAl catalyst was the most active assay, with 51.2% conversion. Bimetallic catalysts performed better; thereby, after the addition of 0.3% Pt to CoAl, CO conversion increased to 63.8%. Moreover, it was further improved by 1% Pt loading, which reached 97.1% conversion, very close to thermodynamic equilibrium (98.8%). It was evidenced that Pt loading on CoAl produced a promotional effect in the WGS activity and also confirmed the strong influence of the support on the catalytic behavior. The activation of the water molecule, a critical step in the WGS reaction, can occur either on the oxygen vacancies (reducible support) or on the metal particle. The H
2-TPR results indicated the very low reducibility of the 0.3Pt/Al catalysts, which suggested that the support did not provide sufficient oxygen vacancies for the activation of water molecules, as cobalt-containing catalysts did. Therefore, the WGS activity of the 0.3Pt/Al catalyst should be related mainly to the available Pt particles, with competitive adsorption of both CO and H
2O. The apparent reaction rate, normalized per Pt site (on the H
2 chemisorption basis), was calculated and was used to compare the WGS activity for both catalysts with similar Pt content (0.3%). The obtained values (0.31 s
−1 vs. 0.88 s
−1 for 0.3Pt/Al and 0.3Pt/CoAl, respectively) supported the hypothesis of the active participation of the support in the reaction mechanism. On the other hand, the apparent reaction rates normalized per Co site (based on H
2 chemisorption) for the CoAl and 0.3Pt/CoAl catalysts were very similar (around 0.16 s
−1) and increased to 0.27 s
−1 for the 1Pt/CoAl catalyst.
The acid/basic character of the surface can also affect the WGS catalyst activity [
12,
32]. Among our samples, the surface basic site density of 0.3Pt/Al (0.32 µmol
CO2/m
2) was lower than the cobalt-containing catalysts (1.16–1.90 µmol
CO2/m
2). The basic sites can polarize the water molecule and induce its dissociation to hydroxyl [
12], which is the kinetically limiting step in WGS. The excellent WGS catalytic performance of the Pt-Co ensemble was ascribed to its improved ability for water dissociation (by the alloyed cobalt surface) as well as by a lowered CO binding energy, in comparison to monometallic catalysts [
33].
Two general mechanisms are commonly accepted for the gas-phase WGS reaction over noble metals supported on oxides: (i) regenerative mechanism, where the labile oxygen from the support oxidizes CO adsorbed on the metal and the formed oxygen vacancy is re-oxidized by water; (ii) associative mechanism, where adsorbed CO reacts with terminal hydroxyl groups of the support to form a C-containing intermediate that is decomposed to CO
2 and H
2. Thus, both the dispersed metal and the support participate in the reaction.
Figure 6 depicts the schematic representation of the WGS reaction when water reacts from the vapor or liquid phase. Davda et al. [
10] contemplated the idea that WGS occurred in the gas bubbles formed within the liquid-phase reactor (scheme A), with the vapor water amount dictated by its liquid–vapor saturation. In scheme B, water reacts from the liquid phase and CO reacts from the gas phase, which implies that the reaction takes place at the gas–liquid–solid interface [
14]. In both schemes, the CO reactant and the obtained products were partially dissolved in liquid water, dictated by the vapor/solution equilibrium (Henry’s law). Partial pressures of water, CO and H
2, and the liquid/gas distribution of CO and H
2 for each experiment, are given in
Table S2, Supplementary Materials. As can be seen, the very high partial pressure of water, close to the operation pressure, has a large impact on the CO and H
2 partial pressures, which are very low.
Under the investigated operation conditions, all the catalysts showed high CO2 yield (ranging from 27.4% for 0.3Pt/Al to 95.7% for 1Pt/CoAl). Moreover, CO2 selectivity was above 90% for all the catalysts. Thus, the CO2 yields indicated a satisfactory product distribution, with mainly H2 and CO2 in the gas product stream. Methane was also detected, though to a much lesser extent. Methane yield increased as follows: 0.3Pt/Al (0.3%) < 0.3Pt/CoAl (0.9%) < CoAl (1.5%) < 1Pt/CoAl (1.9%).
Two mechanisms are usually proposed for CO hydrogenation: (i) associative, where carbonyl combines with H
ads followed by C–O bond cleavage, and (ii) dissociative, where C–O bond breaking takes place directly on the metal, before the hydrogenation step [
34]. Under the investigated conditions (i.e., feedstream containing only CO and water), the negligible hydrogenation of CO was associated with the low availability of the in-situ produced hydrogen that, in fact, competed for the metal sites. Moreover, not all the hydrogen produced in the gas phase was readily available for CO hydrogenation, as it was partly dissolved in the aqueous phase. The Henry’s constants in
Section 3.2 indicate the higher solubility of hydrogen gas as compared to CO. Nevertheless, the fraction of CO and H
2 dissolved in the liquid phase was four orders of magnitude lower than that remaining in the liquid phase (see
Table S2, Supplementary Materials). This was reflected in the large Y
CO2/Y
CH4 yields ratio for all the samples (
Table 4), between 30.8 (for CoAl) and 88.1 (for 0.3Pt/Al), indicating the superior CO hydrogenation activity of cobalt in a hydrogen-lean scenario. Further investigation was carried out to simulate real APR operation conditions (high H
2/CO ratio) and elucidate whether high hydrogen availability could overcome such CO hydrogenation limitations. The results are presented in the following section.
2.3. Liquid-Phase WGS Activity in the Presence of Hydrogen (H2/CO > 0)
In order to investigate the CO hydrogenation capacity of the catalysts under real APR conditions, H
2 was co-fed at different H
2/CO mole ratios (4/3, 7/3 and 12/3). The investigated H
2/CO ratios were established based on the stoichiometric ratio in a typical APR process.
Figure 7A displays the reached CO conversion as a function of the H
2/CO ratio for all the catalysts assayed, evaluated at 260 °C and 50 bar after 10 h TOS. For comparison purposes, the X
CO values attained for the hydrogen-free feedstream are also depicted (H
2/CO = 0). For all the catalysts, the achieved CO conversion was lower than that obtained in the absence of hydrogen in the feedstream. Additionally, a decreasing trend of Xco with H
2/CO was observed. The figure shows that bimetallic xPt/CoAl catalysts remained as the most active, independent of the H
2/CO ratio. The achieved CO conversion values varied as follows: 1Pt/CoAl >> 0.3Pt/CoAl >> CoAl > 0.3Pt/Al. It is interesting to note that the monometallic CoAl catalyst was the most sensitive to the feedstream composition (it had the largest negative slope in the figure). Catalyst CoAl showed the major drop, of around 50% (X
CO = 39.4% and dropped to 19.9% by passing from H
2/CO = 4/3 to 12/3). The decrease was quite similar for 0.3Pt/Al (29%) and 0.3Pt/CoAl (25%) catalysts. As noted, 1Pt/CoAl was the least sensitive to H
2/CO ratio, with only an 11% decrease in X
CO. A possible explanation of the negative effect of H
2/CO ratio on Xco could be the lower residence time of the gas phase (as H
2/CO increased) and the competition between H
2 and CO for the available metal sites. In addition, the higher H
2/CO ratio decreased the CO partial pressure, which also might negatively affect the reaction rate. At these reaction conditions, CO was converted by both the WGS and CO hydrogenation reactions. It is known that CO poses a negative reaction order in the methanation reaction, whereas it is slightly positive for H
2 [
35]. The apparent reaction order with respect to CO was positive for all the catalysts and ranged between 0.12 and 0.17 for the bimetallic catalysts and 0.57 for the monometallic CoAl catalyst, while the order with respect to H
2 was neatly negative (between −0.34 and −0.80 for bimetallic catalysts).
Regarding product distribution, unreacted H
2 and CO, together with produced CO
2, CH
4 and traces of ethane, were detected in the reactor outlet. The very low production of C
2+ alkanes indicated the low Fisher–Tropsch activity of our catalysts at these reaction conditions. Indeed, the production of methane was notably higher than under the hydrogen-free feed conditions, except for the 0.3Pt/Al catalysts, for which it slightly decreased. Several studies have shown that in the gas phase, the CO hydrogenation rate is higher than CO
2 hydrogenation, evidencing that high concentrations of CO in the reaction bed could inhibit the reaction of CO
2 [
34]. On this basis, we assumed that methane was only formed from CO hydrogenation. In this scenario, CO was consumed by both WGS and CO hydrogenation reactions.
Figure 7B shows the trend in the CO
2 and CH
4 yields with H
2/CO ratio. The CO
2 yield showed a similar decreasing trend to X
CO. As occurred for H
2/CO = 0 (hydrogen was not fed), the bimetallic xPt/CoAl catalysts remained the most selective to CO
2, independent of the H
2/CO ratio, while both monometallic catalysts showed similar values of Y
CO2, especially at H
2/CO ≥ 7/3.
Methane yield (Y
CH4) was used as representative of the CO hydrogenation activity. The results in
Figure 7B clearly evidence that hydrogenation activity was more pronounced for Co-containing catalysts, in agreement with the literature [
36]. Not in vain, cobalt is one of the most used catalysts for CO hydrogenation [
37]. On the opposite side, the 0.3Pt/Al catalyst showed very low Y
CH4. For example, at the highest H
2/CO ratio of 12/3, catalyst 0.3Pt/Al showed 0.29% of methane yield, whereas the rest of the investigated catalysts ranged in the 3.1%–3.8% interval. Among the cobalt-containing catalysts, the different behavior of Y
CH4 for the monometallic CoAl is noticeable when changing H
2/CO from 0 to 4/3—that is, for bimetallic catalysts, methane yield slightly decreased as H
2 availability increased, while for the CoAl catalyst, methane yield was boosted. This behavior could be related to the size of the cobalt particles, which was two-fold larger in the monometallic sample. Indeed, the hydrogenation of CO is known to be favored onto large cobalt particles [
38]. Note that, even in the case of more available hydrogen (higher H
2/CO), Y
CH4 reached a plateau for the bimetallic catalysts. The effect on Y
CH4 of the H
2/CO ratio change from 4/3 to 7/3 for cobalt-containing catalysts was also noticeable and could be related to the formation of cobalt carbide-like species at low hydrogen partial pressures [
39].
As previously noted, we assumed that the CO
2 hydrogenation rate was negligible with respect to CO hydrogenation. In addition, no carbon deposits were detected in the spent catalysts. Therefore, the yield ratio (Y
CO2/Y
CH4) could be assumed as representative of the selectivity to WGS reaction (instead of CO hydrogenation): higher Y
CO2/Y
CH4 indicated higher selectivity to WGS—that is, the CO was converted preferably by WGS rather than by CO hydrogenation.
Table 4 shows the obtained Y
CO2/Y
CH4 values. Regardless of the catalyst, selectivity to WGS decreased with increasing hydrogen concentration in the feed, as more hydrogen is available for hydrogenation. It is interesting to note that the bimetallic xPt/CoAl catalysts experienced the highest drop in Y
CO2/Y
CH4 values (84.4% and 64.9% for 0.3 and 1% Pt loading, respectively), as can be seen in
Table 4. Even so, they presented high WGS activity, only surpassed by the monometallic Pt sample.
Net hydrogen change (ΔH
2) has been defined as the difference in hydrogen molar flow between the reactor outlet and inlet: it can be either positive or negative depending on the net generation or consumption of hydrogen in the reaction system. In addition, the ΔH
2 parameter reflects the hydrogenation activity. CO hydrogenation (methanation) is a highly hydrogen-consuming reaction and pulls the ΔH
2 parameter to negative values. As can be seen in
Figure 8, for cobalt-containing catalysts, ΔH
2 decreased with the increase of H
2/CO, due to the hydrogenation capability of cobalt, boosted by the hydrogen excess. Among all catalysts, both bimetallic xPt/CoAl catalysts presented positive ΔH
2, independent of the H
2/CO used. The largest positive ΔH
2 corresponded to the bimetallic 1Pt/CoAl catalysts, in agreement with its highest activity and selectivity to the WGS reaction, with a maximum value of 33.9% for H
2/CO = 4/3. Catalyst 0.3Pt/Al presented slightly negative ΔH
2 at a low H
2/CO ratio (−1.02% at H
2/CO = 4/3), which turned slightly positive (+0.52%) for the highest hydrogen to CO mole ratio.
2.4. Effect of Temperature and Pressure
Both the reaction temperature and pressure have a strong influence on the polyol APR activity and product distribution [
40]. Whether the effect is related to reforming or to WGS is of great interest in order to understand the APR behavior. With this aim, further experiments were carried out with catalyst 0.3Pt/CoAl in order to investigate the influence of temperature and pressure on the WGS activity and with H
2 co-feeding.
Figure 9A shows the CO conversion achieved at 50 bar at different reaction temperatures, at H
2/CO = 12/3. The observed initial increase in the conversion (which lasted about 3 h) was due to unsteady operation due to the reaction protocol, where the temperature was ramped while holding pressure. Thereafter, the conversion of CO remained stable. Noticeably, the increase in the reaction temperature led to increased CO conversion values. However, the obtained values were far from the equilibrium (given in
Figure 9), indicating a kinetically controlled reaction regime under these operation conditions. At low reaction temperatures, the higher overpressure may play a negative role in the catalytic activity, as the product’s desorption from the catalyst surface was hindered [
41]. In addition, the water solubility of the reactants (CO and H
2) increased according to Henry´s law (H constants from
Section 3.2). The fact that these values barely varied with temperature (from 2.2 at 220 °C to 2.5 at 260 °C) suggested that solubility issues hardly affected the H
2/CO mole ratio in the gas phase (see
Table S2 in Supplementary Materials). According to
Table 4, the Y
CO2/Y
CH4 ratio increased with the operation temperature, i.e., it became more selective to WGS. This trend could be explained by two reasons: firstly, the thermodynamics of the CO hydrogenation reaction (highly exothermic reaction), which is highly favorable at lower temperatures; secondly, the increased partial pressure of water in the gas phase, which would shift the WGS equilibrium towards the products.
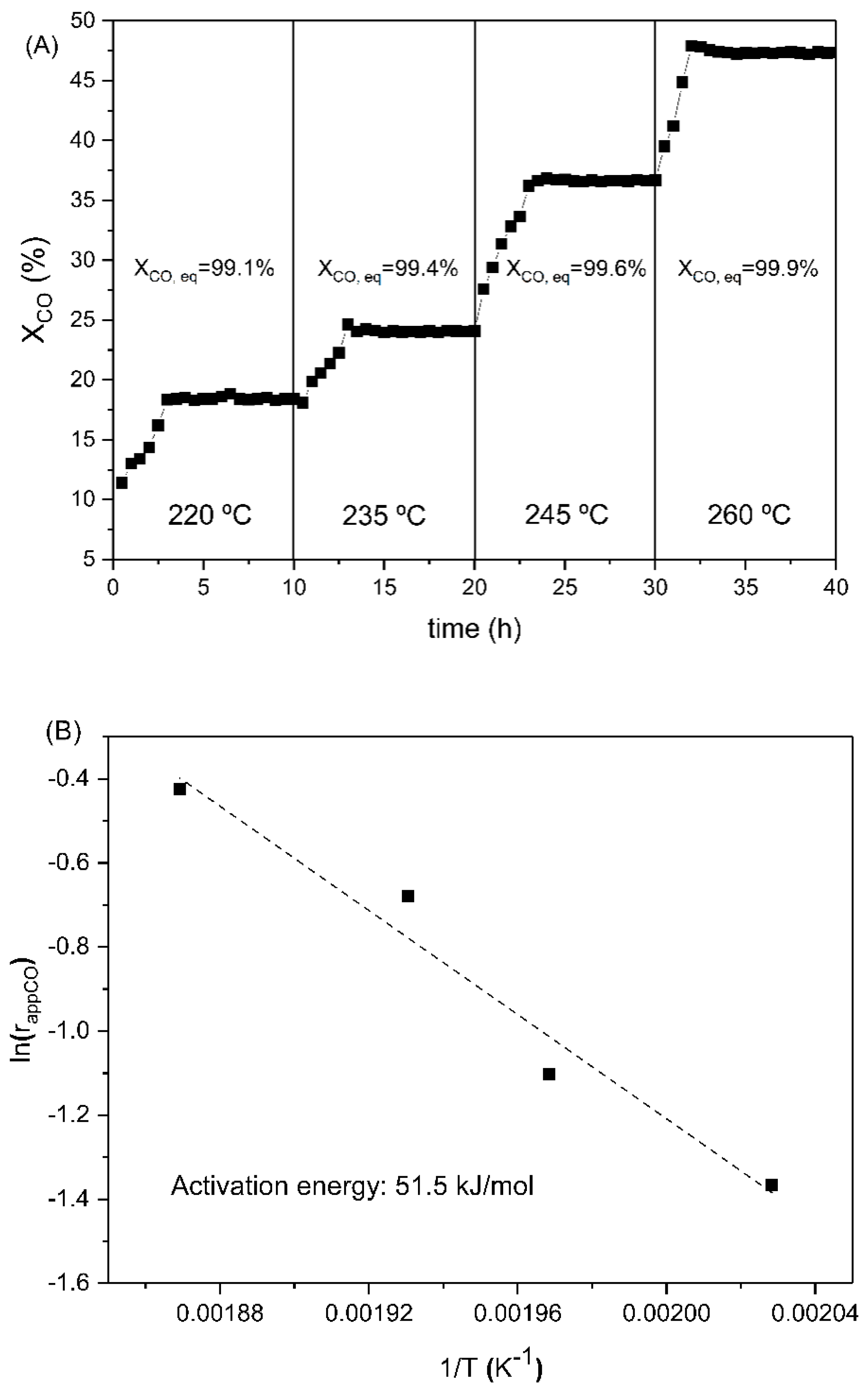
The apparent reaction rate normalized per Pt site (r
app,co) (on the H
2 chemisorption basis) was calculated for each reaction temperature, at 50 bar and H
2/CO = 12/3 in the feedstream. Obtained values are shown in
Table 5, and they revealed that r
app,co was strongly affected by the reaction temperature, as supported by the activation energy value of 51.5 kJ/mol calculated from the Arrhenius plot (
Figure 9B). To our knowledge, this is the first approach to the experimental determination of the apparent activation energy for the liquid-phase WGS. For comparison purposes, the apparent activation energy values reported in the literature for Pt catalysts are shown in
Table S1 (Supplementary Materials). It is of interest to note the inhibitory effect of hydrogen on the intrinsic reaction rate, as r
app,co decreased from 0.88 (for hydrogen-free feedstream) to 0.654 s
−1 (for H
2/CO = 12/3).
Figure 10 shows CO conversion at 220 °C for different reaction pressures, with H
2/CO = 12/3. The observed gap in the first 2.5 h of reaction at each pressure is because the reactor outlet flow valve was shut until the desired pressure was achieved. Notably, CO conversion increased with operation pressure, at a constant reaction temperature. For example, at 25 bar, the obtained CO conversion was 9.6% and two-fold increased at 50 bar. The achieved CO conversions were far from the equilibrium (which decreased from 99.2% at 25 bar to 89.0% at 50 bar), which was a result of the slow kinetics due to the low reaction temperature.
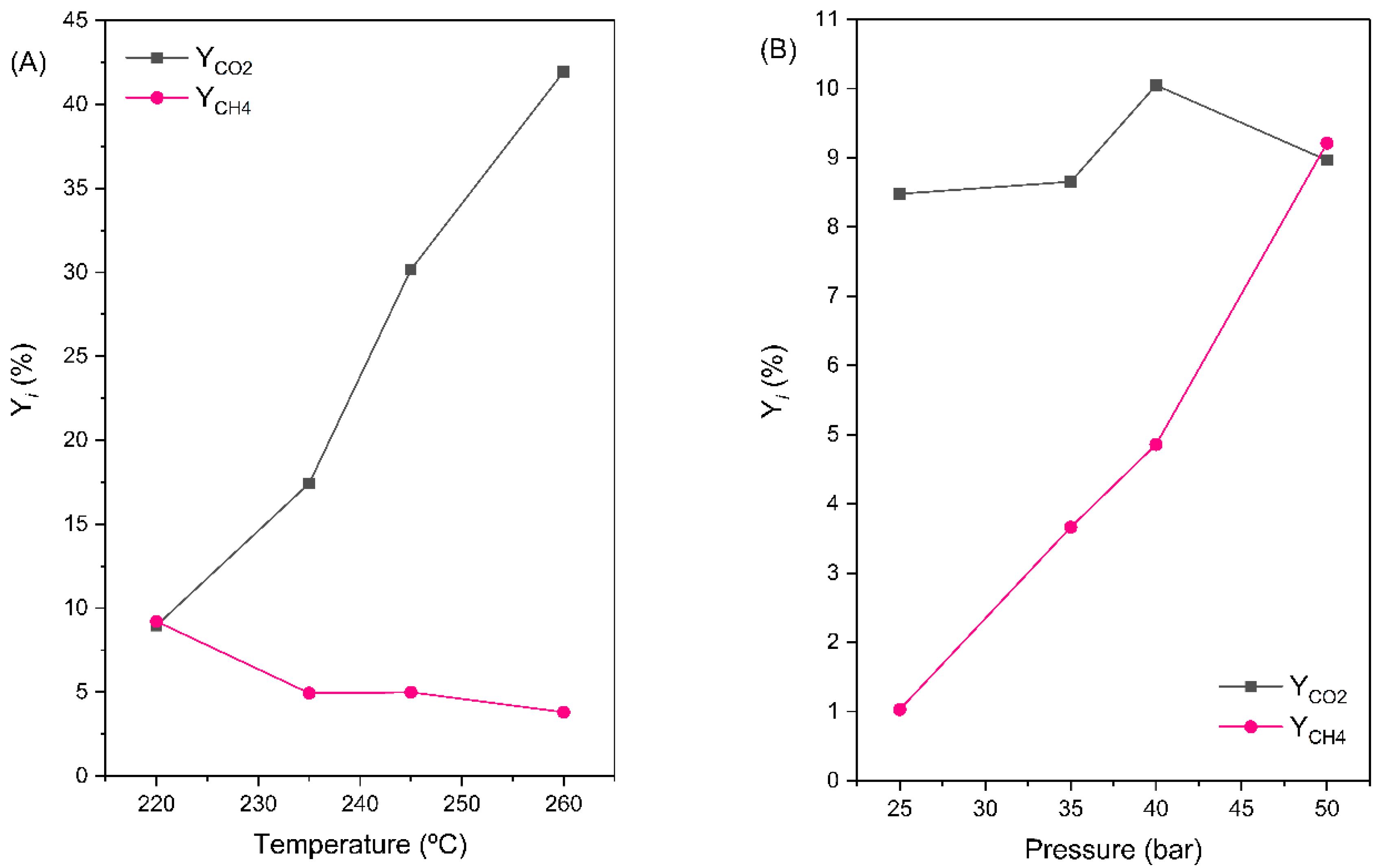
Product yields (Y
CO2 and Y
CH4) for 0.3Pt/CoAl at different temperatures (A) and pressure (B) values are displayed in
Figure 11. CO
2 yield turned out to be more sensitive to temperature than to pressure changes. For example, CO
2 yield increased by 95% by passing from 220 to 235 °C. In like manner, a further increase in temperature to 260 °C caused an increase of about 140%. On the contrary, methane yield hardly varied with temperature, though a slight tendency to decrease with increasing temperature could be deduced. This led to a substantial increase in Y
CO2/Y
CH4, with around a ten-fold increase in the 220 to 260 °C range. As a result, the increase in WGS activity with respect to hydrogenation activity with increasing temperature is conclusive (
Table 4). Regarding the influence of pressure on the product distribution,
Figure 11B shows that CO
2 yield slightly increased with pressure. Methane yield, however, increased ten-fold, from 25 to 50 bar. Therefore, the increased CO conversion at high pressure was due to a favored hydrogenation activity (i.e., ten-fold increase of Y
CH4 in the 25 to 50 bar range) rather than the WGS. Note that, at a constant temperature, the vapor pressure of water remained constant, thus the increase in the operating pressure was accomplished by increasing the partial pressures of the CO and H
2 (see
Table S2 in Supplementary Materials). Although the water solubility of hydrogen is twice that of CO, its effect on the available hydrogen in the gas phase was offset due to the high partial pressure of H
2 under the established reaction conditions. Moreover, the fact that CO can react via WGS and produce even more hydrogen would also increase the hydrogenation rate [
35]. The Y
CO2/Y
CH4 data in
Table 4 clearly shows that the higher the overpressure, the less selectivity towards WGS, which is the opposite effect of the temperature increase.
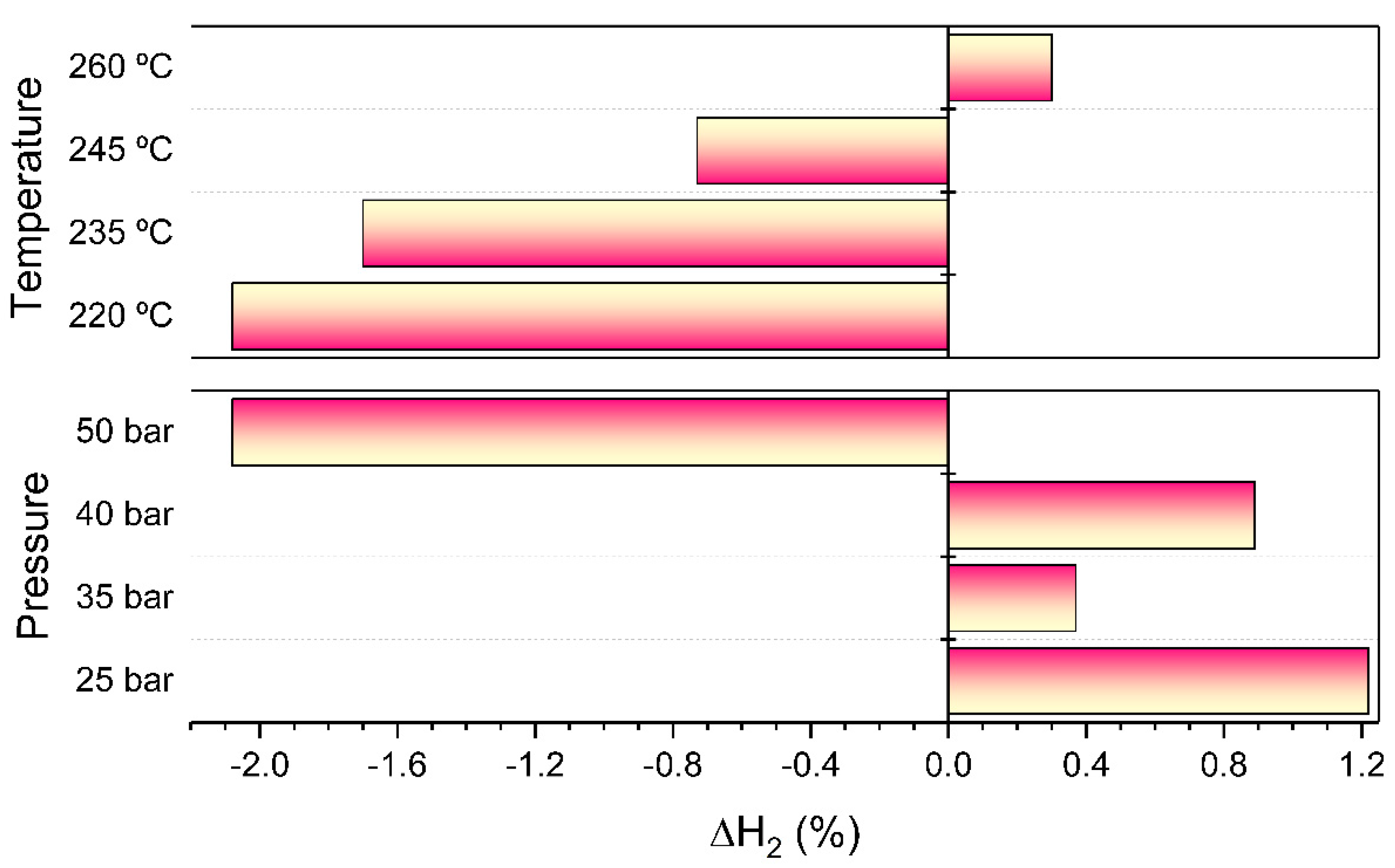
The evolution of the net hydrogen variation is shown in
Figure 12. A large net hydrogen consumption (∆H
2 < 0) was observed at low temperatures due to the high hydrogenation activity. It was verified that methane production decreased at increasing temperatures. Moreover, hydrogen consumption progressively decreased until a positive balance was obtained at the highest temperature screened. In agreement with the observed trend in the CO
2 and CH
4 yield, an increase in pressure favored selectivity towards CO hydrogenation. Hence, at the maximum working pressure, the rate of hydrogen consumption overtook that of production.

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}