Transition Metal Carbides (TMCs) Catalysts for Gas Phase CO2 Upgrading Reactions: A Comprehensive Overview



Abstract
:1. Introduction
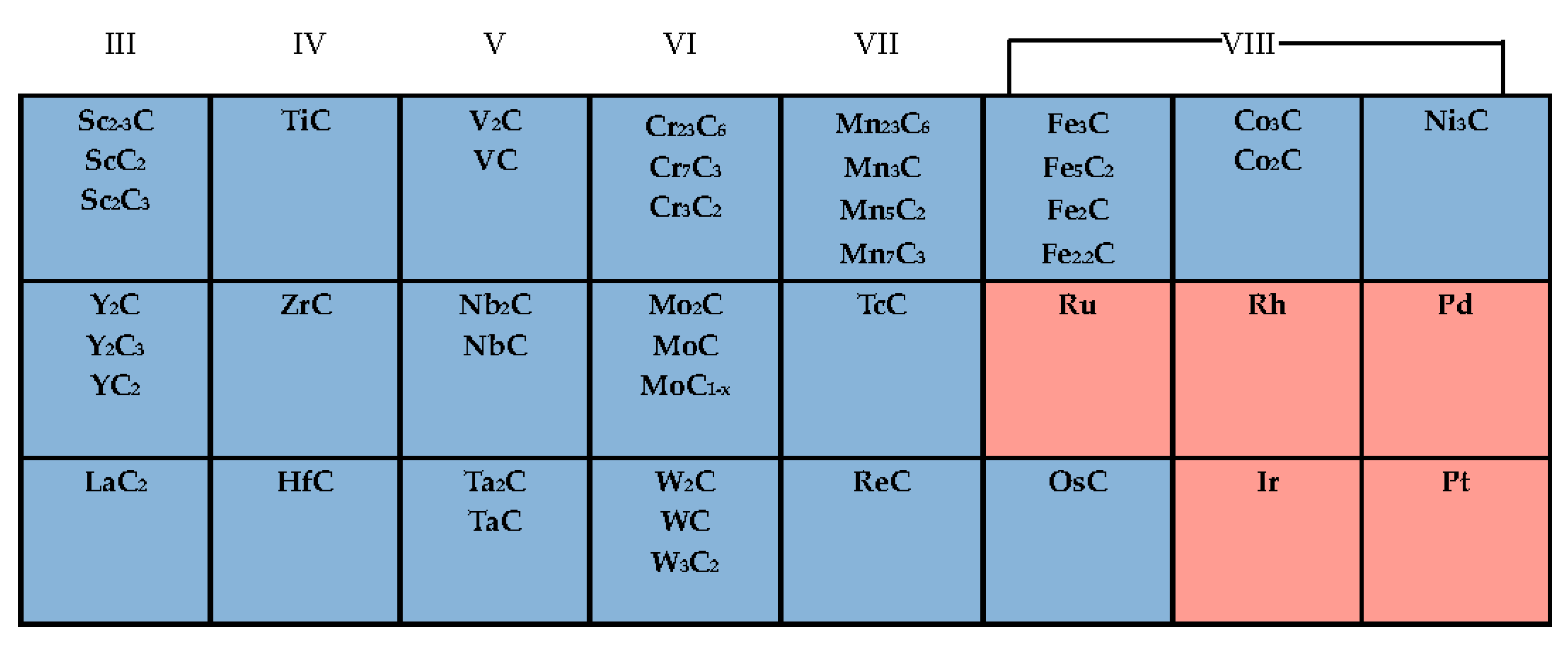
2. General Properties of Transition Metal Carbides (TMCs)
3. TMC Catalysts
3.1. Molybdenum Carbide
3.2. Tungsten Carbide
3.3. Iron Carbide
3.4. Titanium Carbide
3.5. Other Carbides
4. Limitations of TMCs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aressta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. J. Chem. Soc. Dalton Trans. 2007, 28, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem. Int. Ed. 2014, 53, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Tan, C.S. A review: CO2 utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef] [Green Version]
- Muradov, N. Industrial Utilization of CO2: A Win–Win Solution. In Liberating Energy from Carbon: Introduction to Decarbonization; Lecture Notes in Energy; Springer: New York, NY, USA, 2014; Volume 22. [Google Scholar] [CrossRef]
- Ettehadtavakkol, A.; Lake, L.W.; Bryant, S.L. CO2-EOR and storage design optimization. Int. J. Greenh. Gas Control 2014, 25, 79–92. [Google Scholar] [CrossRef]
- Nicot, J.P.; Duncan, I.J. Review: Common attributes of hydraulically fractured oil and gas production and CO2 geological sequestration. Greenh. Gases Sci. Technol. 2012, 2, 352–368. [Google Scholar] [CrossRef]
- Shi, Y.; Jia, Y.; Pan, W.; Huang, L.; Yan, J.; Zheng, R. Potential evaluation on CO2-EGR in tight and low-permeability reservoirs. Nat. Gas Ind. B 2017, 4, 311–318. [Google Scholar] [CrossRef]
- Lin, T.K.; Hsieh, B.Z. Prevention of seabed subsidence of class-1 gas hydrate deposits via CO2-EGR: A numerical study with coupled geomechanics-hydrate reaction-multiphase fluid flow model. Energies 2020, 13, 1579. [Google Scholar] [CrossRef] [Green Version]
- Bongole, K.; Sun, Z.; Yao, J.; Mehmood, A.; Yueying, W.; Mboje, J.; Xin, Y. Multifracture response to supercritical CO2-EGS and water-EGS based on thermo-hydro-mechanical coupling method. Int. J. Energy Res. 2019, 43, 7173–7196. [Google Scholar] [CrossRef]
- Zhang, F.Z.; Jiang, P.X.; Xu, R.N. System thermodynamic performance comparison of CO2-EGS and water-EGS systems. Appl. Therm. Eng. 2013, 61, 236–244. [Google Scholar] [CrossRef]
- Borgia, A.; Pruess, K.; Kneafsey, T.J.; Oldenburg, C.M.; Pan, L. Simulation of CO2-EGS in a fractured reservoir with salt precipitation. Energy Procedia 2013, 37, 6617–6624. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, A.; Khalilpour, K.R.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kent, R.A.; Yung, M.M.; Kuhn, J.N. Carbon dioxide conversion by reverse water-gas shift chemical looping on perovskite-type oxides. Ind. Eng. Chem. Res. 2014, 53, 5828–5837. [Google Scholar] [CrossRef]
- Verhelst, S.; Turner, J.W.; Sileghem, L.; Vancoillie, J. Methanol as a fuel for internal combustion engines. Prog. Energy Combust. Sci. 2019, 70, 43–88. [Google Scholar] [CrossRef] [Green Version]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Wang, F.; Wei, M.; Evans, D.G.; Duan, X. CeO2-based heterogeneous catalysts toward catalytic conversion of CO2. J. Mater. Chem. A 2016, 4, 5773–5783. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.; Jiang, Y.; Wu, X.; Xia, C.; Peng, R.; Lu, Y. CO2 Activation and Reduction on Pt-CeO2-Based Catalysts. J. Phys. Chem. C 2019, 123, 17092–17101. [Google Scholar] [CrossRef]
- Wang, W.; Qu, Z.; Song, L.; Fu, Q. CO2 hydrogenation to methanol over Cu/CeO2 and Cu/ZrO2 catalysts: Tuning methanol selectivity via metal-support interaction. J. Energy Chem. 2020, 40, 22–30. [Google Scholar] [CrossRef] [Green Version]
- le Saché, E.; Santos, J.L.; Smith, T.J.; Centeno, M.A.; Arellano-Garcia, H.; Odriozola, J.A.; Reina, T.R. Multicomponent Ni-CeO2 nanocatalysts for syngas production from CO2/CH4 mixtures. J. CO2 Util. 2018, 25, 68–78. [Google Scholar] [CrossRef]
- Stroud, T.; Smith, T.J.; le Saché, E.; Santos, J.L.; Centeno, M.A.; Arellano-Garcia, H.; Odriozola, J.A.; Reina, T.R. Chemical CO2 recycling via dry and bi reforming of methane using Ni-Sn/Al2O3 and Ni-Sn/CeO2-Al2O3 catalysts. Appl. Catal. B Environ. 2018, 224, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Rood, S.C.; Ahmet, H.B.; Gomez-Ramon, A.; Torrente-Murciano, L.; Reina, T.R.; Eslava, S. Enhanced ceria nanoflakes using graphene oxide as a sacrificial template for CO oxidation and dry reforming of methane. Appl. Catal. B Environ. 2019, 242, 358–368. [Google Scholar] [CrossRef]
- Centeno, M.A.; Reina, T.R.; Ivanova, S.; Laguna, O.H.; Odriozola, J.A. Au/CeO2 catalysts: Structure and CO oxidation activity. Catalysts 2016, 6, 158. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Pérez, L.; le Saché, E.; Jones, C.; Gu, S.; Arellano-Garcia, H.; Reina, T.R. Synthetic natural gas production from CO2 over Ni-x/CeO2-ZrO2(x = Fe, Co) catalysts: Influence of promoters and space velocity. Catal. Today 2017, 2, 10–15. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Patel, V.; le Saché, E.; Reina, T.R. CO2 methanation in the presence of methane: Catalysts design and effect of methane concentration in the reaction mixture. J. Energy Inst. 2020, 93, 415–424. [Google Scholar] [CrossRef]
- Shi, Z.; Tan, Q.; Wu, D. Enhanced CO2 hydrogenation to methanol over TiO2 nanotubes-supported CuO-ZnO-CeO2 catalyst. Appl. Catal. A Gen. 2019, 581, 58–66. [Google Scholar] [CrossRef]
- Liu, C.; Nauert, S.L.; Alsina, M.A.; Wang, D.; Grant, A.; He, K.; Weitz, E.; Nolan, M.; Gray, K.A.; Notestein, J.M. Role of surface reconstruction on Cu/TiO2 nanotubes for CO2 conversion. Appl. Catal. B Environ. 2019, 255, 117754. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Shah, M.; le Saché, E.; Reina, T.R. Improving Fe/Al2O3 catalysts for the reverse water-gas shift reaction: On the effect of cs as activity/selectivity promoter. Catalysts 2018, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Duyar, M.S.; Ramachandran, A.; Wang, C.; Farrauto, R.J. Kinetics of CO2 methanation over Ru/γ-Al2O3 and implications for renewable energy storage applications. J. CO2 Util. 2015, 12, 27–33. [Google Scholar] [CrossRef]
- Levy, R.B.; Boudart, M. Platinum-Like Behavior of Tungsten Carbide in Surface Catalysis. Science 1973, 181, 547–549. [Google Scholar] [CrossRef]
- Oshikawa, K.; Nagai, M.; Omi, S. Characterization of molybdenum carbides for methane reforming by TPR, XRD, and XPS. J. Phys. Chem. B 2001, 105, 9124–9131. [Google Scholar] [CrossRef]
- Prats, H.; Piñero, J.J.; Viñes, F.; Bromley, S.T.; Sayós, R.; Illas, F. Assessing the usefulness of transition metal carbides for hydrogenation reactions. Chem. Commun. 2019, 55, 12797–12800. [Google Scholar] [CrossRef] [PubMed]
- Qi, K.Z.; Wang, G.C.; Zheng, W.J. A first-principles study of CO hydrogenation into methane on molybdenum carbides catalysts. Surf. Sci. 2013, 614, 53–63. [Google Scholar] [CrossRef]
- Ramanathan, S.; Oyama, S.T. New catalysts for hydroprocessing: Transition metal carbides and nitrides. J. Phys. Chem. 1995, 99, 16365–16372. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Nørskov, J.K.; Barteau, M.A.; Chen, J.G. Trends in the chemical properties of early transition metal carbide surfaces: A density functional study. Catal. Today 2005, 105, 66–73. [Google Scholar] [CrossRef]
- Hwu, H.H.; Chen, J.G. Surface chemistry of transition metal carbides. Chem. Rev. 2005, 105, 185–212. [Google Scholar] [CrossRef]
- Stottlemyer, A.L.; Kelly, T.G.; Meng, Q.; Chen, J.G. Reactions of oxygen-containing molecules on transition metal carbides: Surface science insight into potential applications in catalysis and electrocatalysis. Surf. Sci. Rep. 2012, 67, 201–232. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Liu, P.; Dvorak, J.; Jirsak, T.; Gomes, J.; Takahashi, Y.; Nakamura, K. Adsorption of sulfur on TiC(001): Photoemission and first-principles studies. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 69, 1–10. [Google Scholar] [CrossRef]
- Darujati, A.R.S.; LaMont, D.C.; Thomson, W.J. Oxidation stability of Mo2C catalysts under fuel reforming conditions. Appl. Catal. A Gen. 2003, 253, 397–407. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Muckerman, J.T. Sulfur adsorption and sulfidation of transition metal carbides as hydrotreating catalysts. J. Mol. Catal. A Chem. 2005, 239, 116–124. [Google Scholar] [CrossRef]
- Toth, E.L. Transition Metal Carbides and Nitrides; Academic Press: New York, NY, USA, 1971; ISBN 9780323157223. [Google Scholar]
- Rodriguez, J.A.; Evans, J.; Feria, L.; Vidal, A.B.; Liu, P.; Nakamura, K.; Illas, F. CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: Production of CO, methanol, and methane. J. Catal. 2013, 307, 162–169. [Google Scholar] [CrossRef]
- Oyama, S.T. Introduction to the chemistry of transition metal carbides and nitrides. In The Chemistry of Transition Metal Carbides and Nitrides; Oyama, S.T., Ed.; Online; Springer: Dordrecht, The Netherlands, 1996; ISBN 978-94-009-1565-7. [Google Scholar] [CrossRef]
- Xiao, Y.; Hwang, J.Y.; Sun, Y.K. Transition metal carbide-based materials: Synthesis and applications in electrochemical energy storage. J. Mater. Chem. A 2016, 4, 10379–10393. [Google Scholar] [CrossRef]
- Zheng, W.; Cotter, T.P.; Kaghazchi, P.; Jacob, T.; Frank, B.; Schlichte, K.; Zhang, W.; Su, D.S.; Schüth, F.; Schlögl, R. Experimental and theoretical investigation of molybdenum carbide and nitride as catalysts for ammonia decomposition. J. Am. Chem. Soc. 2013, 135, 3458–3464. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, H.; Zhu, Y.; Tian, D.; Wang, C.; Lu, X. Mo/Mo2C encapsulated in nitrogen-doped carbon nanofibers as efficiently integrated heterojunction electrocatalysts for hydrogen evolution reaction in wide pH range. Appl. Surf. Sci. 2019, 496, 143672. [Google Scholar] [CrossRef]
- Wang, P.; Chen, W.; Chiang, F.K.; Dugulan, A.I.; Song, Y.; Pestman, R.; Zhang, K.; Yao, J.; Feng, B.; Miao, P.; et al. Synthesis of stable and low-CO2 selective ϵ-iron carbide Fischer-Tropsch catalysts. Sci. Adv. 2018, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Qin, Y.; Xue, X.; Wang, X.; Yao, M.; Huang, H. Intrinsic Properties Affecting the Catalytic Activity of 3d Transition-Metal Carbides in Li-O2 Battery. J. Phys. Chem. C 2018, 122, 17812–17819. [Google Scholar] [CrossRef]
- Zhu, K.; Zhang, H.; Ye, K.; Zhao, W.; Yan, J.; Cheng, K.; Wang, G.; Yang, B.; Cao, D. Two-Dimensional Titanium Carbide MXene as a Capacitor-Type Electrode for Rechargeable Aqueous Li-Ion and Na-Ion Capacitor Batteries. ChemElectroChem 2017, 4, 3018–3025. [Google Scholar] [CrossRef]
- Tackett, B.M.; Sheng, W.; Chen, J.G. Opportunities and Challenges in Utilizing Metal-Modified Transition Metal Carbides as Low-Cost Electrocatalysts. Joule 2017, 1, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Feria, L.; Rodriguez, J.A.; Jirsak, T.; Illas, F. Interaction of SO2 with Cu/TiC(0 0 1) and Au/TiC(0 0 1): Toward a new family of DeSOx catalysts. J. Catal. 2011, 279, 352–360. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. Heterogeneous catalytic CO2 conversion to value-added hydrocarbons. Energy Environ. Sci. 2010, 3, 884–890. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Baldwin, J.W.; Peng, X.; Mpourmpakis, G.; Willauer, H.D. Potassium-promoted molybdenum carbide as a highly active and selective catalyst for CO2 conversion to CO. ChemSusChem 2017, 10, 2408–2415. [Google Scholar] [CrossRef]
- Zhang, Q.; Pastor-Pérez, L.; Jin, W.; Gu, S.; Reina, T.R. Understanding the promoter effect of Cu and Cs over highly effective β-Mo2C catalysts for the reverse water-gas shift reaction. Appl. Catal. B Environ. 2019, 244, 889–898. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Water-gas-shift reaction on molybdenum carbide surfaces: Essential role of the oxycarbide. J. Phys. Chem. B 2006, 110, 19418–19425. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.S.; Asensio, J.M.; Estrader, M.; Werner, M.; Bordet, A.; Yi, D.; Chaudret, B. Iron carbide or iron carbide/cobalt nanoparticles for magnetically-induced CO2 hydrogenation over Ni/SiRAlOx catalysts. Catal. Sci. Technol. 2019, 9, 2601–2607. [Google Scholar] [CrossRef]
- Brungs, A.J.; York, A.P.E.; Green, M.L.H. Comparison of the group V and VI transition metal carbides for methane dry reforming and thermodynamic prediction of their relative stabilities. Catal. Lett. 1999, 57, 65–69. [Google Scholar] [CrossRef]
- Yan, Q.; Lu, Y.; To, F.; Li, Y.; Yu, F. Synthesis of tungsten carbide nanoparticles in biochar matrix as a catalyst for dry reforming of methane to syngas. Catal. Sci. Technol. 2015, 5, 3270–3280. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Evans, J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly Active Au/δ-MoC and Cu/δ-MoC Catalysts for the Conversion of CO2: The Metal/C Ratio as a Key Factor Defining Activity, Selectivity, and Stability. J. Am. Chem. Soc. 2016, 138, 8269–8278. [Google Scholar] [CrossRef] [Green Version]
- Ono, L.K.; Sudfeld, D.; Cuenya, B.R. In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation. Surf. Sci. 2006, 600, 5041–5050. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Illas, F. Activation of noble metals on metal-carbide surfaces: Novel catalysts for CO oxidation, desulfurization and hydrogenation reactions. Phys. Chem. Chem. Phys. 2012, 14, 427–438. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Kattel, S.; Li, W.H.; Liu, P.; Chen, J.G. Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides. Chem. Commun. 2015, 51, 6988–6991. [Google Scholar] [CrossRef]
- Kunkel, C.; Viñes, F.; Illas, F. Transition metal carbides as novel materials for CO2 capture, storage, and activation. Energy Environ. Sci. 2016, 9, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Regmi, Y.N.; Leonard, B.M. Multiple phases of molybdenum carbide as electrocatalysts for the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2014, 53, 6407–6410. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ramirez, P.J.; Stacchiola, D.; Rodriguez, J.A. Synthesis of α-MoC1-x and β-MoCy catalysts for CO2 hydrogenation by thermal carburization of Mo-oxide in hydrocarbon and hydrogen mixtures. Catal. Lett. 2014, 144, 1418–1424. [Google Scholar] [CrossRef]
- Liu, X.; Kunkel, C.; de la Piscina, P.R.; Homs, N.; Viñes, F.; Illas, F. Effective and Highly Selective CO Generation from CO2 Using a Polycrystalline α-Mo2C Catalyst. ACS Catal. 2017, 7, 4323–4335. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Ma, D. Highly Dispersed Copper over β-Mo2C as an Efficient and Stable Catalyst for the Reverse Water Gas Shift (rWGS) Reaction. ACS Catal. 2017, 7, 912–918. [Google Scholar] [CrossRef]
- Xu, W.; Ramírez, P.J.; Stacchiola, D.; Brito, J.L.; Rodriguez, J.A. The Carburization of Transition Metal Molybdates (MxMoO4, M = Cu, Ni or Co) and the Generation of Highly Active Metal/Carbide Catalysts for CO2 Hydrogenation. Catal. Lett. 2015, 145, 1365–1373. [Google Scholar] [CrossRef]
- Prasad, P.S.S.; Bae, J.W.; Jun, K.W.; Lee, K.W. Fischer-Tropsch synthesis by carbon dioxide hydrogenation on Fe-based catalysts. Catal. Surv. Asia 2008, 12, 170–183. [Google Scholar] [CrossRef]
- Yao, L.; Wang, Y.; Galvez, M.E.; Hu, C.; da Costa, P. γ-Alumina-Supported Ni-Mo Carbides as Promising Catalysts for CO2 Methanation. Mod. Res. Catal. 2017, 06, 135–145. [Google Scholar] [CrossRef] [Green Version]
- SPosada-Pérez, S.; Viñes, F.; Ramirez, P.J.; Vidal, A.B.; Rodriguez, J.A.; Illas, F. The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces. Phys. Chem. Chem. Phys. 2014, 16, 14912–14921. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Gao, H.; Yao, Z.; Jia, R.; Shi, Y.; Sun, Y.; Fan, Q.; Wang, H. Simple synthesis of ultrasmall β-Mo2C and α-MoC1−x nanoparticles and new insights into their catalytic mechanisms for dry reforming of methane. Catal. Sci. Technol. 2017, 7, 3312–3324. [Google Scholar] [CrossRef]
- York, A.P.E.; Claridge, J.B.; Brungs, A.J.; Tsang, S.C.; Green, M.L.H. Molybdenum and tungsten carbides as catalysts for the conversion of methane to synthesis gas using stoichiometric feedstocks. Chem. Commun. 1997, 1, 39–40. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, A.; Li, X.; Zhang, S.; Zhu, A.; Ma, Y.; Au, C. Ni-modified Mo2C catalysts for methane dry reforming. Appl. Catal. A Gen. 2012, 431, 164–170. [Google Scholar] [CrossRef]
- Yao, Z.; Jiang, J.; Zhao, Y.; Luan, F.; Zhu, J.; Shi, Y.; Gao, H.; Wang, H. Insights into the deactivation mechanism of metal carbide catalysts for dry reforming of methane via comparison of nickel-modified molybdenum and tungsten carbides. RSC Adv. 2016, 6, 19944–19951. [Google Scholar] [CrossRef]
- Mounfield, W.P.; Harale, A.; Román-Leshkov, Y. Impact of morphological effects on the activity and stability of tungsten carbide catalysts for dry methane reforming. Energy Fuels 2019, 33, 5544–5550. [Google Scholar] [CrossRef]
- Shao, H.; Kugler, E.L.; Ma, W.; Dadyburjor, D.B. Effect of temperature on structure and performance of in-house cobalt-tungsten carbide catalyst for dry reforming of methane. Ind. Eng. Chem. Res. 2005, 44, 4914–4921. [Google Scholar] [CrossRef]
- Morse, J.R.; Juneau, M.; Baldwin, J.W.; Porosoff, M.D.; Willauer, H.D. Alkali promoted tungsten carbide as a selective catalyst for the reverse water gas shift reaction. J. CO2 Util. 2020, 35, 38–46. [Google Scholar] [CrossRef]
- Bukur, D.B.; Okabe, K.; Rosynek, M.P.; Li, C.P.; Wang, D.J.; Rao, K.R.P.M.; Huffman, G.P. Activation Studies with a Precipitated Iron Catalyst for Fischer-Tropsch Synthesis. I. Characterization Studies. J. Catal. 1995, 155, 353–365. [Google Scholar] [CrossRef]
- Shroff, M.D.; Datye, A.K. The importance of passivation in the study of iron Fischer-Tropsch catalysts. Catal. Lett. 1996, 37, 101–106. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, H.; Hou, Y.; Ma, D. Fe5C2 nanoparticles: A facile bromide-induced synthesis and as an active phase for Fischer-Tropsch synthesis. J. Am. Chem. Soc. 2012, 134, 15814–15821. [Google Scholar] [CrossRef]
- Chen, B.; Wang, D.; Duan, X.; Liu, W.; Li, Y.; Qian, G.; Yuan, W.; Holmen, A.; Zhou, X.; Chen, D. Charge-Tuned CO Activation over a χ-Fe5C2 Fischer-Tropsch Catalyst. ACS Catal. 2018, 8, 2709–2714. [Google Scholar] [CrossRef]
- Herranz, T.; Rojas, S.; Pérez-Alonso, F.J.; Ojeda, M.; Terreros, P.; Fierro, J.L.G. Genesis of iron carbides and their role in the synthesis of hydrocarbons from synthesis gas. J. Catal. 2006, 243, 199–211. [Google Scholar] [CrossRef]
- Liu, X.; Cao, C.; Tian, P.; Zhu, M.; Zhang, Y.; Xu, J.; Tian, Y.; Han, Y.F. Resolving CO2 activation and hydrogenation pathways over iron carbides from DFT investigation. J. CO2 Util. 2020, 38, 10–15. [Google Scholar] [CrossRef]
- Wang, W.; Gong, J. Methanation of carbon dioxide: An overview. Front. Chem. Eng. China 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Abenojar, E.C.; Wickramasinghe, S.; Bas-Concepcion, J.; Samia, A.C.S. Structural effects on the magnetic hyperthermia properties of iron oxide nanoparticles. Prog. Nat. Sci. Mater. Int. 2016, 26, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Bordet, A.; Asensio, J.M.; Soulantica, K.; Chaudret, B. Enhancement of Carbon Oxides Hydrogenation on Iron-Based Nanoparticles by In-Situ Water Removal. ChemCatChem 2018, 10, 4047–4051. [Google Scholar] [CrossRef]
- López, M.; Broderick, L.; Carey, J.J.; Viñes, F.; Nolan, M.; Illas, F. Tuning transition metal carbide activity by surface metal alloying: A case study on CO2 capture and activation. Phys. Chem. Chem. Phys. 2018, 20, 22179–22186. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Viñes, F.; Nolan, M.; Illas, F. Predicting the Effect of Dopants on CO2 Adsorption in Transition Metal Carbides: Case Study on TiC (001). J. Phys. Chem. C 2020, 124, 15969–15976. [Google Scholar] [CrossRef]
- Vidal, A.B.; Feria, L.; Evans, J.; Takahashi, Y.; Liu, P.; Nakamura, K.; Rodriguez, J.A. CO2 activation and methanol synthesis on novel Au/TiC and Cu/TiC catalysts. J. Phys. Chem. Lett. 2012, 3, 2275–2280. [Google Scholar] [CrossRef]
- Asara, G.G.; Ricart, J.M.; Rodriguez, J.A.; Illas, F. Exploring the activity of a novel Au/TiC(001) model catalyst towards CO and CO2 hydrogenation. Surf. Sci. 2015, 640, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Dubois, J.L.; Sayama, K.; Arakawa, H. CO2 Hydrogenation over Carbide Catalysts. Chem. Lett. 1992, 21, 5–8. [Google Scholar] [CrossRef]
- Kim, K.Y.; Lee, H.; Noh, W.Y.; Shin, J.; Han, S.J.; Kim, S.K.; An, K.; Lee, J.S. Cobalt Ferrite Nanoparticles to Form a Catalytic Co–Fe Alloy Carbide Phase for Selective CO2 Hydrogenation to Light Olefins. ACS Catal. 2020, 10, 8660–8671. [Google Scholar] [CrossRef]
- Pajares, A.; Prats, H.; Romero, A.; Viñes, F.; de la Piscina, P.R.; Sayós, R.; Homs, N.; Illas, F. Critical effect of carbon vacancies on the reverse water gas shift reaction over vanadium carbide catalysts. Appl. Catal. B Environ. 2020, 267, 118719. [Google Scholar] [CrossRef]
- Mehdad, A.; Jentoft, R.E.; Jentoft, F.C. Passivation agents and conditions for Mo2C and W2C: Effect on catalytic activity for toluene hydrogenation. J. Catal. 2017, 347, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Hanif, A.; Xiao, T.; York, A.P.E.; Sloan, J.; Green, M.L.H. Study on the structure and formation mechanism of molybdenum carbides. Chem. Mater. 2002, 14, 1009–1015. [Google Scholar] [CrossRef]
- Rocha, A.S.; Rocha, A.B.; da Silva, V.T. Benzene adsorption on Mo2C: A theoretical and experimental study. Appl. Catal. A Gen. 2010, 379, 54–60. [Google Scholar] [CrossRef]
- Nagai, M.; Kurakami, T.; Omi, S. Activity of carbided molybdena—Alumina for CO2 hydrogenation. Catal. Today 1998, 45, 235–239. [Google Scholar] [CrossRef]
- Shou, H.; Ferrari, D.; Barton, D.G.; Jones, C.W.; Davis, R.J. Influence of Passivation on the Reactivity of Unpromoted and Rb-Promoted Mo2C Nanoparticles for CO Hydrogenation. ACS Catal. 2012, 2, 1408–1416. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Structures | Stacking Sequence | Crystal View |
|---|---|---|---|
| α-MoC1-X (NaCl type) | Cubic | ABCABC |  |
| β-Mo2C (Fe2N type) | Hexagonal | ABAB |  |
| γ-MoC (WC type) | Hexagonal | AAAA |  |
| η-MoC (MoC type) | Hexagonal | ABCABC |  |
| Catalyst | CO2 Conversion (%) | TOF (min−1) | CO:CH4 Ratio |
|---|---|---|---|
| PtCo/CeO2 | 6.6 | 14.6 | 4.5 |
| PdNi/CeO2 | 2.5 | 5.6 | 0.6 |
| Mo2C | 8.7 | 25.7 | 14.5 |
| Co-Mo2C | 9.5 | 16.1 | 51.3 |
| Supports | Active Metals | Promoters | Reactions | Comments |
|---|---|---|---|---|
| β-Mo2C | Co | rWGS (reverse water gas shift) | CoMoCyOz phase in Co-Mo2C is an active phase which can dissociate CH4 hence improve the catalytic performance (especially CO selectivity) of Mo2C [2]. | |
| Cu | The strong interaction between Cu and β-Mo2C can effectively promote the dispersion of supported copper and prevent the aggregation of Cu particles [67]. | |||
| Cu, Ni, Co | CO selectivity: Cu/Mo2C > β-Mo2C > Ni/Mo2C > Co/Mo2C. CO2 conversion: Cu/Mo2C < β-Mo2C < Ni/Mo2C < Co/Mo2C [68]. | |||
| Cu | Cs | The electropositive character of Cs facilitates the electronic transfer from Cs to Mo and leads to an electronically rich surface which favours the selectivity towards CO [54]. | ||
| α-Mo2C | CO2 dissociation toward CO can happen at ambient temperature at the surface of α-Mo2C and the presence of Mo2C (101)-Mo/C surface facet can be serve as a possible explanation of the observed reactivity [66]. | |||
| Al2O3 | Mo2C/Mo | Ni | CO2 methanation | The carburization process enhances the basicity of Ni-Mo2C/Al2O3 and thus CO2 absorption on their surface [70]. |
| β-Mo2C,δ-MoC and TiC | Methanol synthesis | TMC catalysts with a metal/carbon ratio of 1 (δ-MoC and TiC) were considered to be more selective than the catalysts with a metal/carbon ratio of 2 (β-Mo2C) [59]. | ||
| α-MoC1−x β-Mo2C | DRM (dry reforming of methane) | The phase of the molybdenum carbides obtained depended considerably on the precursor preparation method and flowing gas composition (Ar or H2). α-MoC1−x phase showed better stability than β-Mo2C phase in the DRM reaction [72]. | ||
| β-Mo2C | Testing pressure is one of the critical reasons for catalyst deactivation. Clear deactivation occurred at β-Mo2C after 8h test at ambient pressure. At same condition, high activity was maintained for 144 h with no bulk carbon deposition at 8 bar [73]. | |||
| Mo2C | Ni | DRM | The ratio of active metal sites (Ni) and Mo will influence the deactivation of Mo2C catalysts in DRM reaction. The Ni-Mo2C catalyst with Ni/Mo molar ratios of 1/2 is the most stable one [74]. | |
| SiRAlOx | Co, Ni | Fe2.2C | CO2 methanation | As a magnetically soft material, Fe2.2C NPs (nanoparticles) can be used as heating agent in a magnetic hyperthermia system for CO2 methanation [56]. |
| TiC | Cu, Au | Methanol synthesis | Charge polarization happened between Au, Cu particles and TiC (001) surface makes them very active for CO2 activation and the catalytic synthesis of methanol [90]. | |
| Cu, Au, Ni | CO2 hydrogenation | Main product Au-TiC and Cu-TiC: CO, methanolNi-TiC: CO, methanol, methane [42] | ||
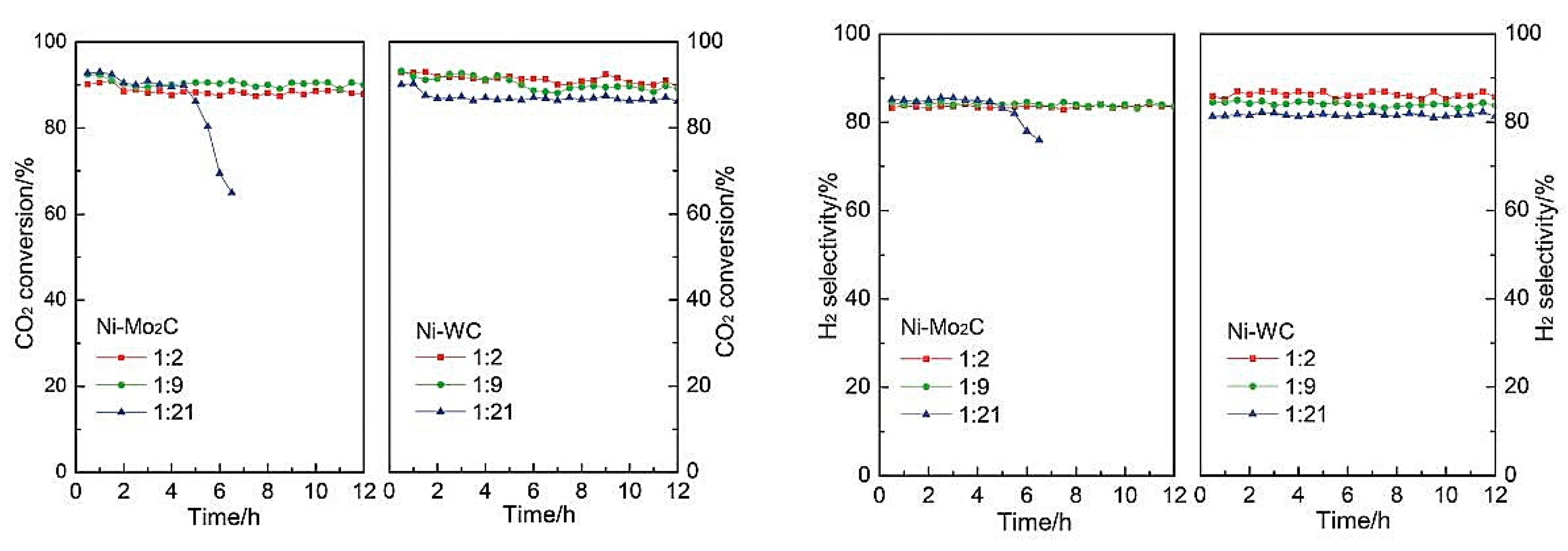
| WC Mo2C | Ni | DRM | Ni-WC is more stable than Ni-Mo2C in DRM reaction due to the fact that compared with Ni-Mo2C, the Ni-WC catalysts showed better ability to keep control of crystal structure and better resistance to sintering [75]. | |
| γ-Al2O3 | α-WC β-W2C | β-W2C nanoparticles exhibited higher stability than α-WC nanorods due to the inherent disorder and presence of carbon vacancies in the β-W2C phase nanoparticles which can facilitate the reaction and prevent from coking [76]. | ||
| biochar | WC | Uniform particle distribution with little coke formation after 500 h on stream, indicating that the small particle size contributed directly to the stability of the catalyst [58]. | ||
| Co6W6C | Co6W6C is ineffective for DRM below 850 °C. It is active after 30 h DRM inlet gas treatment at 850 °C, because Co, WC were formed after the treatment. Co, WC are the actual active phase [77]. | |||
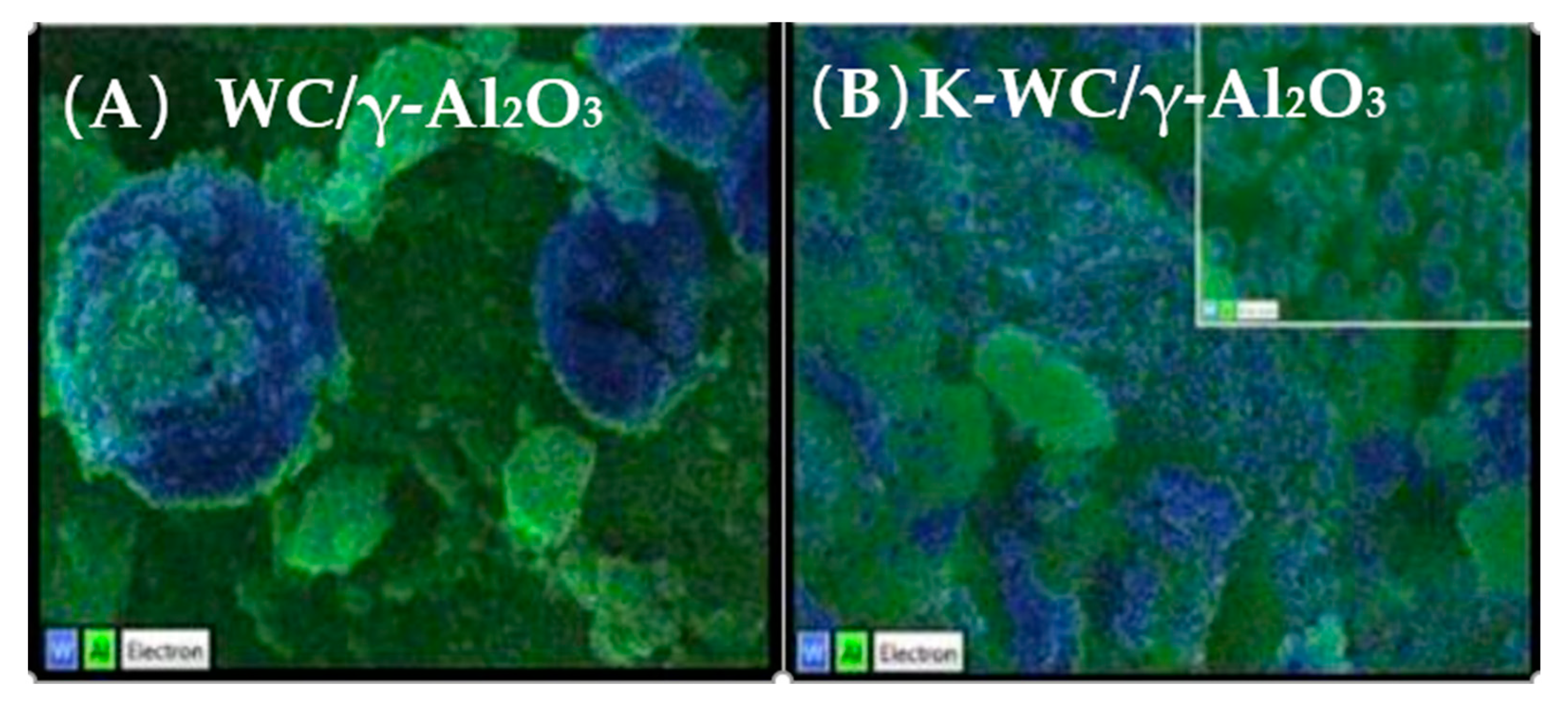
| γ-Al2O3 | WC | K | rWGS | K acted as a structural and electronic promoter in K-WC/γ-Al2O3, attenuating surface chemistry and catalyst dispersion hence improve the CO selectivity in rWGS [78]. |
| CNT | CoFe2O4 | Na | CO2 hydrogenation | (Fe1–xCox)5C2 with a Hägg carbide structure is the active site in the catalyst for enhanced activity and preferential chain growth hence improve the catalytic performance [93]. |
| VC/V8C7 | rWGS | V8C7 shows a higher CO2 conversion and CO selectivity and C vacancies in V8C7 are responsible for the better catalytic behavior [94]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Pastor-Pérez, L.; Gu, S.; Ramirez Reina, T. Transition Metal Carbides (TMCs) Catalysts for Gas Phase CO2 Upgrading Reactions: A Comprehensive Overview. Catalysts 2020, 10, 955. https://doi.org/10.3390/catal10090955
Zhang Q, Pastor-Pérez L, Gu S, Ramirez Reina T. Transition Metal Carbides (TMCs) Catalysts for Gas Phase CO2 Upgrading Reactions: A Comprehensive Overview. Catalysts. 2020; 10(9):955. https://doi.org/10.3390/catal10090955
Chicago/Turabian StyleZhang, Qi, Laura Pastor-Pérez, Sai Gu, and Tomas Ramirez Reina. 2020. "Transition Metal Carbides (TMCs) Catalysts for Gas Phase CO2 Upgrading Reactions: A Comprehensive Overview" Catalysts 10, no. 9: 955. https://doi.org/10.3390/catal10090955
APA StyleZhang, Q., Pastor-Pérez, L., Gu, S., & Ramirez Reina, T. (2020). Transition Metal Carbides (TMCs) Catalysts for Gas Phase CO2 Upgrading Reactions: A Comprehensive Overview. Catalysts, 10(9), 955. https://doi.org/10.3390/catal10090955