
Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook



Abstract
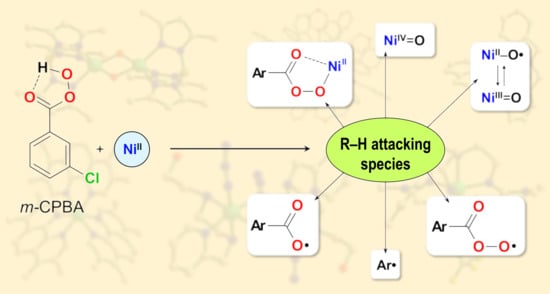
:
1. Introduction
2. Discussion

| Cat. | Adamantane a | MeCyH b | Proposed C–H Attacking Species | Proposed O–O Splitting Type | Reaction Media | Ref. |
|---|---|---|---|---|---|---|
| Ni1 | 13 | – c | NiO+ | – | CH2Cl2/CH3CN d | [35] |
| Ni1 | 36 e/14 f | – | ArC(O)O• | homo | CH2Cl2/CH3CN d | [23] |
| Ni1 | 13 | – | ArC(O)O•/NiII–O• | homo | CH2Cl2/CH3CN d | [62] |
| Ni2 | – | – | NiII–O• | homo | CH2Cl2/CH3CN d | [27] |
| Ni3 | 13–17 | – | NiII–O• | homo | CH2Cl2/CH3CN d | [37] |
| Ni4 | 11–12 | – | NiII–O• | homo | CH2Cl2/CH3CN d | [38] |
| Ni5 | – | – | NiIII–O– | homo | CH2Cl2 | [39] |
| Ni6 | – | 5:1 | NiII–O•/NiIII=O | homo/concerted | CH2Cl2 | [42] |
| Ni7 | – | 8:2:1 | Ni7(m-CPBA) complex | – | CF3C6H5, CH3C6H5, or C6H6 | [48] |
| Ni8 | 18 | – | NiIII–O• | hetero | CH2Cl2 | [49] |
| Ni9 | – | – | Ni(III) or Ni(IV) | homo/hetero | CH2Cl2/CH3CN g | [50] |
| Ni10 | – | – | Ni(III) | homo/hetero | CH2Cl2/CH3CN g | [51] |
| Ni11 | – | – | – | homo/hetero | CH3C6H5, CH2Cl2, CH3CN, or CH3CN/H2O h | [52] |
| Ni12 | 19 | 9:1 | – | homo | CH2Cl2/CH3CN d | [56] |
| Ni13 | – | – | – | – | DCE i or DCE/TCB j,k | [58] |
| Ni14 | – | 8:1 | NiII–O•/ArC(O)O•/Ar• | homo | CH2Cl2/CH3CN g | [59] |
| Ni15 | – | – | – | – | CH3CN | [60] |
| Ni16–Ni22 | 30–44 e/13–21 f | – | ArC(O)O• | homo | CH2Cl2/CH3CN d | [23] |
| Ni23 | – | – | ArC(O)O•/NiII–O• | homo | CH2Cl2/CH3CN d | [62,63] |
| Ni24 | – | – | ArC(O)O•/NiII–O• | homo | CH2Cl2/CH3CN d | [62,63] |
| NiCl2 | 35 e/17 f | – | – | – | CH2Cl2/CH3CN d | [23] |
| Ni(OAc)2 | – | 8:1 | – | – | CH2Cl2/CH3CN g | [59] |
| cat. free l | 44 a | 9:1 | – | – | CH2Cl2/CH3CN d | [23,59] |
| other cat. m | 2 | 6:3:1–26:7:1 n | HO• | – | CH3CN | [64,65] |
| other cat. m | 10 | 60:10:1–150:15:1 n | tBuO• | – | CH3CN | [64,66,67] |
| cat. free. o | >6 | – | Ar2C(O)O• p | – | ClC6H5 | [68] |
| cat. free q | 33 e | – | PhC(O)OO• | – | DCE | [23] |
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hou, H.L.; Zeng, X.K.; Zhang, X.W. Production of Hydrogen Peroxide by Photocatalytic Processes. Angew. Chem. Int. Edit. 2020, 59, 17356–17376. [Google Scholar] [CrossRef]
- Yi, H.; Zhang, G.T.; Wang, H.M.; Huang, Z.Y.; Wang, J.; Singh, A.K.; Lei, A.W. Recent Advances in Radical C-H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, H.; O’Reilly, K.; Gupta, M.K.; Horgan, C.; O’Leary, E.M.; O’Sullivan, T.P. Advances in the synthesis of acyclic peroxides. RSC Adv. 2017, 7, 19506–19556. [Google Scholar] [CrossRef] [Green Version]
- Shilov, A.E.; Shul’pin, G.B. Activation of C-H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, D.S.; Nesterova, O.V.; Pombeiro, A.J.L. Homo- and heterometallic polynuclear transition metal catalysts for alkane C-H bonds oxidative functionalization: Recent advances. Coord. Chem. Rev. 2018, 355, 199–222. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Maji, S.; Chen, P.R.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.; Kohler, V.; Flitsch, S.L.; Turner, N.J. Cytochromes P450 as useful biocatalysts: Addressing the limitations. Chem. Commun. 2011, 47, 2490–2501. [Google Scholar] [CrossRef]
- Targhan, H.; Evans, P.; Bahrami, K. A Review of the Role of Hydrogen Peroxide in Organic Transformations. J. Ind. Eng. Chem. 2021. [Google Scholar] [CrossRef]
- Ghosh, P.; Ganguly, B.; Das, S. NaI/KI/NH4I and TBHP as powerful oxidation systems: Use in the formation of various chemical bonds. Org. Biomol. Chem. 2021, 19, 2146–2167. [Google Scholar] [CrossRef]
- Wu, X.F.; Gong, J.L.; Qi, X.X. A powerful combination: Recent achievements on using TBAI and TBHP as oxidation system. Org. Biomol. Chem. 2014, 12, 5807–5817. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Fokin, A.A.; Schreiner, P.R. Selective alkane transformations via radicals and radical cations: Insights into the activation step from experiment and theory. Chem. Rev. 2002, 102, 1551–1593. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Bjorsvik, H.R.; Fontana, F.; Minisci, F.; Serri, A. Radical versus “oxenoid” oxygen insertion mechanism in the oxidation of alkanes and alcohols by aromatic peracids. New synthetic developments. J. Org. Chem. 1996, 61, 9409–9416. [Google Scholar] [CrossRef]
- Hussain, H.; Al-Harrasi, A.; Green, I.R.; Ahmed, I.; Abbas, G.; Rehman, N.U. meta-Chloroperbenzoic acid (mCPBA): A versatile reagent in organic synthesis. RSC Adv. 2014, 4, 12882–12917. [Google Scholar] [CrossRef]
- Chen, M.M.; Coelho, P.S.; Arnold, F.H. Utilizing Terminal Oxidants to Achieve P450-Catalyzed Oxidation of Methane. Adv. Synth. Catal. 2012, 354, 964–968. [Google Scholar] [CrossRef] [Green Version]
- Kudrik, E.V.; Afanasiev, P.; Alvarez, L.X.; Dubourdeaux, P.; Clemancey, M.; Latour, J.M.; Blondin, G.; Bouchu, D.; Albrieux, F.; Nefedov, S.E.; et al. An N-bridged high-valent diiron-oxo species on a porphyrin platform that can oxidize methane. Nat. Chem. 2012, 4, 1024–1029. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. Non-heme oxoiron(V) intermediates in chemo-, regio- and stereoselective oxidation of organic substrates. Coord. Chem. Rev. 2019, 384, 126–139. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Hypervalent Iodine-Catalyzed Oxidative Functionalizations Including Stereoselective Reactions. Chem. Asian J. 2014, 9, 950–971. [Google Scholar] [CrossRef]
- Clavier, H.; Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2012, 354, 3347–3403. [Google Scholar] [CrossRef]
- Ray, K.; Pfaff, F.F.; Wang, B.; Nam, W. Status of Reactive Non-Heme Metal-Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 13942–13958. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, M.; Balamurugan, M.; Palaniandavar, M. Alkane and alkene oxidation reactions catalyzed by nickel(II) complexes: Effect of ligand factors. Coord. Chem. Rev. 2020, 403, 14. [Google Scholar] [CrossRef]
- Qiu, Y.H.; Hartwig, J.F. Mechanism of Ni-Catalyzed Oxidations of Unactivated C(sp3)-H Bonds. J. Am. Chem. Soc. 2020, 142, 19239–19248. [Google Scholar] [CrossRef]
- Pirovano, P.; Twamley, B.; McDonald, A.R. Modulation of Nickel Pyridinedicarboxamidate Complexes to Explore the Properties of High-valent Oxidants. Chem. Eur. J. 2018, 24, 5238–5245. [Google Scholar] [CrossRef] [Green Version]
- Corona, T.; Draksharapu, A.; Padamati, S.K.; Gamba, I.; Martin-Diaconesou, V.; Acuna-Pares, F.; Browne, W.R.; Company, A. Rapid Hydrogen and Oxygen Atom Transfer by a High-Valent Nickel-Oxygen Species. J. Am. Chem. Soc. 2016, 138, 12987–12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirovano, P.; Farquhar, E.R.; Swart, M.; McDonald, A.R. Tuning the Reactivity of Terminal Nickel(III)-Oxygen Adducts for C-H Bond Activation. J. Am. Chem. Soc. 2016, 138, 14362–14370. [Google Scholar] [CrossRef] [Green Version]
- Nagataki, T.; Ishii, K.; Tachi, Y.; Itoh, S. Ligand effects on NiII-catalysed alkane-hydroxylation with m-CPBA. Dalton Trans. 2007, 1120–1128. [Google Scholar] [CrossRef]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon-Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Olivo, G.; Cusso, O.; Borrell, M.; Costas, M. Oxidation of alkane and alkene moieties with biologically inspired nonheme iron catalysts and hydrogen peroxide: From free radicals to stereoselective transformations. J. Biol. Inorg. Chem. 2017, 22, 425–452. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. Stereoselective oxidation of alkanes with m-CPBA as an oxidant and cobalt complex with isoindole-based ligands as catalysts. RSC Adv. 2016, 6, 93756–93767. [Google Scholar] [CrossRef]
- Olivo, G.; Lanzalunga, O.; Di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; Garcia-Espana, E.; Basallote, M.G.; Munck, E.; Que, L.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C-H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Kasyanova, K.V.; Makhankova, V.G.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Stereospecific sp3 C-H oxidation with m-CPBA: A CoIII Schiff base complex as pre-catalyst vs. its CoIIICdII heterometallic derivative. Appl. Catal. A 2018, 560, 171–184. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Kasyanova, K.V.; Buvaylo, E.A.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterov, D.S.; Pombeiro, A.J.L. Heterometallic CoIIIZnII Schiff Base Catalyst for Mild Hydroxylation of C(sp3)-H Bonds of Unactivated Alkanes: Evidence for Dual Mechanism Controlled by the Promoter. Catalysts 2019, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Nagataki, T.; Tachi, Y.; Itoh, S. NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA. Chem. Commun. 2006, 4016–4018. [Google Scholar] [CrossRef] [PubMed]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective C-H oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar] [CrossRef]
- Balamurugan, M.; Mayilmurugan, R.; Suresh, E.; Palaniandavar, M. Nickel(II) complexes of tripodal 4N ligands as catalysts for alkane oxidation using m-CPBA as oxidant: Ligand stereoelectronic effects on catalysis. Dalton Trans. 2011, 40, 9413–9424. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, M.; Balamurugan, M.; Palaniandavar, M.; Vadivelu, P.; Suresh, C.H. Nickel(II) Complexes of Pentadentate N5 Ligands as Catalysts for Alkane Hydroxylation by Using m-CPBA as Oxidant: A Combined Experimental and Computational Study. Chem. Eur. J. 2014, 20, 11346–11361. [Google Scholar] [CrossRef]
- Pfaff, F.F.; Heims, F.; Kundu, S.; Mebs, S.; Ray, K. Spectroscopic capture and reactivity of S=1/2 nickel(III)-oxygen intermediates in the reaction of a NiII-salt with mCPBA. Chem. Commun. 2012, 48, 3730–3732. [Google Scholar] [CrossRef]
- Krzystek, J.; Ozarowski, A.; Telser, J. Multi-frequency, high-field EPR as a powerful tool to accurately determine zero-field splitting in high-spin transition metal coordination complexes. Coord. Chem. Rev. 2006, 250, 2308–2324. [Google Scholar] [CrossRef]
- Krzystek, J.; Park, J.H.; Meisel, M.W.; Hitchman, M.A.; Stratemeier, H.; Brunel, L.C.; Telser, J. EPR spectra from "EPR-silent" species: High-frequency and high-field EPR spectroscopy of pseudotetrahedral complexes of nickel(II). Inorg. Chem. 2002, 41, 4478–4487. [Google Scholar] [CrossRef]
- Hikichi, S.; Hanaue, K.; Fujimura, T.; Okuda, H.; Nakazawa, J.; Ohzu, Y.; Kobayashi, C.; Akita, M. Characterization of nickel(II)-acylperoxo species relevant to catalytic alkane hydroxylation by nickel complex with mCPBA. Dalton Trans. 2013, 42, 3346–3356. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, S.; Kobayashi, C.; Yoshizawa, M.; Akita, M. Tuning the Stability and Reactivity of Metal-bound Alkylperoxide by Remote Site Substitution of the Ligand. Chem. Asian J. 2010, 5, 2086–2092. [Google Scholar] [CrossRef]
- Astakhov, G.S.; Bilyachenko, A.N.; Korlyukov, A.A.; Levitsky, M.M.; Shul’pina, L.S.; Bantreil, X.; Lamaty, F.; Vologzhanina, A.V.; Shubina, E.S.; Dorovatovskii, P.V.; et al. High-Cluster (Cu9) Cage Silsesquioxanes: Synthesis, Structure, and Catalytic Activity. Inorg. Chem. 2018, 57, 11524–11529. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Levitsky, M.M.; Yalymov, A.I.; Korlyukov, A.A.; Vologzhanina, A.V.; Kozlov, Y.N.; Shul’pina, L.S.; Nesterov, D.S.; Pombeiro, A.J.L.; Lamaty, F.; et al. A heterometallic (Fe6Na8) cage-like silsesquioxane: Synthesis, structure, spin glass behavior and high catalytic activity. RSC Adv. 2016, 6, 48165–48180. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599–1606. [Google Scholar] [CrossRef]
- Talsi, E.P.; Ottenbacher, R.V.; Bryliakov, K.P. Bioinspired oxidations of aliphatic C-H groups with H2O2 in the presence of manganese complexes. J. Organomet. Chem. 2015, 793, 102–107. [Google Scholar] [CrossRef]
- Nakazawa, J.; Terada, S.; Yamada, M.; Hikichi, S. Structural Characterization and Oxidation Reactivity of a Nickel(II) Acylperoxo Complex. J. Am. Chem. Soc. 2013, 135, 6010–6013. [Google Scholar] [CrossRef] [PubMed]
- Corona, T.; Pfaff, F.F.; Acuna-Pares, F.; Draksharapu, A.; Whiteoak, C.J.; Martin-Diaconescu, V.; Lloret-Fillol, J.; Browne, W.R.; Ray, K.; Company, A. Reactivity of a Nickel(II) Bis(amidate) Complex with meta-Chloroperbenzoic Acid: Formation of a Potent Oxidizing Species. Chem. Eur. J. 2015, 21, 15029–15038. [Google Scholar] [CrossRef]
- Bok, K.H.; Lee, M.M.; You, G.R.; Ahn, H.M.; Ryu, K.Y.; Kim, S.-J.; Kim, Y.; Kim, C. Synthesis, Characterization, and Catalytic Activities of a Nickel(II) Monoamido-Tetradentate Complex: Evidence for NiIII-Oxo and NiIV-Oxo Species. Chem. Eur. J. 2017, 23, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Jeong, A.R.; Shin, J.W.; Jeong, J.H.; Bok, K.H.; Kim, C.; Jeong, D.; Cho, J.; Hayami, S.; Min, K.S. Dinuclear Iron(III) and Nickel(II) Complexes Containing N-(2-Pyridylmethyl)-N′-(2-hydroxyethyl)ethylenediamine: Catalytic Oxidation and Magnetic Properties. Chem. Eur. J. 2017, 23, 3023–3033. [Google Scholar] [CrossRef]
- Kim, C.; Ahn, H.; Bae, J.; Kim, M.; Bok, K.; Jeong, H.; Lee, S. Synthesis, characterization, and efficient catalytic activities of a nickel(II) porphyrin: Remarkable solvent and substrate effects on participation of multiple active oxidants. Chem. Eur. J. 2017, 23, 11969–11976. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V.; Kopylovich, M.N.; Pombeiro, A.J.L. Pronounced retention of stereoconfiguration upon sp3 C-H bonds hydroxylation of dimethylcyclohexanes and decahydronaphthalenes with m-CPBA oxidant and a Co-phthalocyanine catalyst. Mol. Catal. 2018, 459, 8–15. [Google Scholar] [CrossRef]
- Guo, M.; Lee, Y.M.; Fukuzumi, S.; Nam, W. Biomimetic metal-oxidant adducts as active oxidants in oxidation reactions. Coord. Chem. Rev. 2021, 435, 23. [Google Scholar] [CrossRef]
- Larson, V.A.; Battistella, B.; Ray, K.; Lehnert, N.; Nam, W. Iron and manganese oxo complexes, oxo wall and beyond. Nat. Rev. Chem. 2020, 4, 404–419. [Google Scholar] [CrossRef]
- Terao, I.; Horii, S.; Nakazawa, J.; Okamura, M.; Hikichi, S. Efficient alkane hydroxylation catalysis of nickel(II) complexes with oxazoline donor containing tripodal tetradentate ligands. Dalton Trans. 2020, 49, 6108–6118. [Google Scholar] [CrossRef]
- Nesterova, O.V.; Nesterov, D.S.; Jezierska, J.; Pombeiro, A.J.L.; Ozarowski, A. Copper(II) Complexes with Bulky N-Substituted Diethanolamines: High-Field Electron Paramagnetic Resonance, Magnetic, and Catalytic Studies in Oxidative Cyclohexane Amidation. Inorg. Chem. 2018, 57, 12384–12397. [Google Scholar] [CrossRef] [PubMed]
- Bunescu, A.; Lee, S.W.; Li, Q.; Hartwig, J.F. Catalytic Hydroxylation of Polyethylenes. Acs. Cent. Sci. 2017, 3, 895–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, T.; Matsuba, N.; Nakazawa, J.; Hikichi, S. Aliphatic C–H hydroxylation activity and durability of a nickel complex catalyst according to the molecular structure of the bis(oxazoline) ligands. Mol. Catal. 2021, 511, 111718. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Yalymov, A.I.; Shul’pina, L.S.; Mandelli, D.; Korlyukov, A.A.; Vologzhanina, A.V.; Es’kova, M.A.; Shubina, E.S.; Levitsky, M.M.; Shul’pin, G.B. Novel Cage-Like Hexanuclear Nickel(II) Silsesquioxane. Synthesis, Structure, and Catalytic Activity in Oxidations with Peroxides. Molecules 2016, 21, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shul’pin, G.B.; Kozlov, Y.N.; Nizova, G.V.; Suss-Frank, G.; Stanislas, S.; Kitaygorodskiy, A.; Kulikova, V.S. Oxidations by the reagent O2-H2O2-vanadium derivative-pyrazine-2-carboxylic acid. Part 12. Main features, kinetics and mechanism of alkane hydroperoxidation. J. Chem. Soc. Perkin Trans. 2 2001, 1351–1371. [Google Scholar] [CrossRef]
- Shinke, T.; Itoh, M.; Wada, T.; Morimoto, Y.; Yanagisawa, S.; Sugimoto, H.; Kubo, M.; Itoh, S. Revisiting Alkane Hydroxylation with m-CPBA (m-Chloroperbenzoic Acid Catalyzed by Nickel(II) Complexes. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Nagataki, T.; Itoh, S. Catalytic alkane hydroxylation reaction with nickel(II) complexes supported by di- and triphenol ligands. Chem. Lett. 2007, 36, 748–749. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Kozlov, Y.N.; Bilyachenko, A.N.; Nesterov, D.S.; Shul’pina, L.S.; Zubavichus, Y.V.; Pombeiro, A.J.L.; Levitsky, M.M.; Yalymov, A.I.; Shul’pin, G.B. Alkane oxidation with peroxides catalyzed by cage-like copper(II) silsesquioxanes. New J. Chem. 2015, 39, 187–199. [Google Scholar] [CrossRef]
- Buvaylo, E.A.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Nesterova, O.V.; Pombeiro, A.J.L. Copper(II) complex of the 2-pyridinecarbaldehyde aminoguanidine Schiff base: Crystal structure and catalytic behaviour in mild oxidation of alkanes. Inorg. Chem. Commun. 2017, 78, 85–90. [Google Scholar] [CrossRef]
- Biswas, A.N.; Pariyar, A.; Bose, S.; Das, P.; Bandyopadhyay, P. Mild oxidation of hydrocarbons catalyzed by iron corrole with tert-butylhydroperoxide. Catal. Commun. 2010, 11, 1008–1011. [Google Scholar] [CrossRef]
- Costas, M.; Chen, K.; Que, L. Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 200, 517–544. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Krasutsky, A.P.; Kuehl, C.J.; Simonsen, A.J.; Woodward, J.K.; Mismash, B.; Bolz, J.T. Preparation, X-ray crystal structure, and chemistry of stable azidoiodinanes-derivatives of benziodoxole. J. Am. Chem. Soc. 1996, 118, 5192–5197. [Google Scholar] [CrossRef]
- Schneider, H.J.; Muller, W. Selective Functionalization of Hydrocarbons. 6. Mechanistic and Preparative Studies on the Regioselective and Stereoselective Paraffin Hydroxylation with Peracids. J. Org. Chem. 1985, 50, 4609–4615. [Google Scholar] [CrossRef]
- Cho, K.B.; Wu, X.; Lee, Y.M.; Kwon, Y.H.; Shaik, S.; Nam, W. Evidence for an Alternative to the Oxygen Rebound Mechanism in C-H Bond Activation by Non-Heme FeIVO Complexes. J. Am. Chem. Soc. 2012, 134, 20222–20225. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Fontana, F.; Minisci, F.; Serri, A. ‘Oxenoid’ oxygen insertion vs. a radical mechanism in the oxidation of alkanes and alcohols: The case of aromatic peracids. J. Chem. Soc. Chem. Commun. 1996, 1843–1844. [Google Scholar] [CrossRef]
- Oloo, W.N.; Que, L. Bioinspired Nonheme Iron Catalysts for C-H and C=C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res. 2015, 48, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B.; Loginov, D.A.; Shul’pina, L.S.; Ikonnikov, N.S.; Idrisov, V.O.; Vinogradov, M.M.; Osipov, S.N.; Nelyubina, Y.V.; Tyubaeva, P.M. Stereoselective Alkane Oxidation with meta-Chloroperoxybenzoic Acid (MCPBA) Catalyzed by Organometallic Cobalt Complexes. Molecules 2016, 21, 1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandeepan, P.; Muller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, P.; Rose, J.; Braunstein, P. Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]





































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterov, D.S.; Nesterova, O.V. Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook. Catalysts 2021, 11, 1148. https://doi.org/10.3390/catal11101148
Nesterov DS, Nesterova OV. Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook. Catalysts. 2021; 11(10):1148. https://doi.org/10.3390/catal11101148
Chicago/Turabian StyleNesterov, Dmytro S., and Oksana V. Nesterova. 2021. "Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook" Catalysts 11, no. 10: 1148. https://doi.org/10.3390/catal11101148
APA StyleNesterov, D. S., & Nesterova, O. V. (2021). Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook. Catalysts, 11(10), 1148. https://doi.org/10.3390/catal11101148