
Boosting the H2 Production Efficiency via Photocatalytic Organic Reforming: The Role of Additional Hole Scavenging System





Abstract
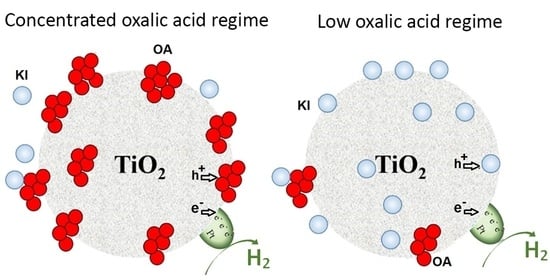
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. The Fate of Oxalic Acid during the Photocatalytic Reforming
2.2. Stoichiometric and Mechanistic Investigations
2.3. Isotopic Studies
2.4. The Role of Intermediates
2.5. Maximizing the Total Yields by the Addition of KI
3. Experimental Section
3.1. Materials
3.2. Photocatalytic Experiments
3.3. Electron Paramagnetic Resonance (EPR) Experiments
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeon, T.H.; Koo, M.S.; Kim, H.; Choi, W. Dual-Functional Photocatalytic and Photoelectrocatalytic Systems for Energy- and Resource-Recovering Water Treatment. ACS Catal. 2018, 8, 11542–11563. [Google Scholar] [CrossRef]
- Niaz, S.; Manzoor, T.; Pandith, A.H. Hydrogen storage: Materials, methods and perspectives. Renew. Sustain. Energy Rev. 2015, 50, 457–469. [Google Scholar] [CrossRef]
- Hakki, A.; AlSalka, Y.; Mendive, C.B.; Ubogui, J.; dos Santos Claro, P.C.; Bahnemann, D. Hydrogen Production by Heterogeneous Photocatalysis. In Encyclopedia of Interfacial Chemistry; Wandelt, K., Ed.; Elsevier: Oxford, UK, 2018; pp. 413–419. [Google Scholar] [CrossRef]
- Kennedy, J.; Bahruji, H.; Bowker, M.; Davies, P.R.; Bouleghlimat, E.; Issarapanacheewin, S. Hydrogen generation by photocatalytic reforming of potential biofuels: Polyols, cyclic alcohols, and saccharides. J. Photochem. Photobiol. A Chem. 2018, 356, 451–456. [Google Scholar] [CrossRef]
- AlSalka, Y. Photocatalytic Water Splitting for Solar Hydrogen Production and Simultaneous Decontamination of Organic Pollutants. Ph.D. Thesis, Gottfried Wilhelm Leibniz Universität, Hannover, Germany, 8 July 2020. [Google Scholar]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Pellegrin, Y.; Odobel, F. Sacrificial electron donor reagents for solar fuel production. C. R. Chim. 2017, 20, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Al-Madanat, O.; AlSalka, Y.; Ramadan, W.; Bahnemann, D.W. TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels. Catalysts 2021, 11, 317. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Coord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Ramadan, W.; Dillert, R.; Koch, J.; Tegenkamp, C.; Bahnemann, D.W. Changes in the solid-state properties of bismuth iron oxide during the photocatalytic reformation of formic acid. Catal. Today 2019, 326, 22–29. [Google Scholar] [CrossRef]
- Friehs, E.; AlSalka, Y.; Jonczyk, R.; Lavrentieva, A.; Jochums, A.; Walter, J.-G.; Stahl, F.; Scheper, T.; Bahnemann, D. Toxicity, phototoxicity and biocidal activity of nanoparticles employed in photocatalysis. J. Photochem. Photobiol. C Photochem. Rev. 2016, 29, 1–28. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Schneider, J.; Bahnemann, D.W. Co-catalyst-free photocatalytic hydrogen evolution on TiO2: Synthesis of optimized photocatalyst through statistical material science. Appl. Catal. B Environ. 2018, 238, 422–433. [Google Scholar] [CrossRef]
- Ramadan, W.; Feldhoff, A.; Bahnemann, D. Assessing the photocatalytic oxygen evolution reaction of BiFeO3 loaded with IrO2 nanoparticles as cocatalyst. Sol. Energy Mater. Sol. Cells 2021, 232, 111349. [Google Scholar] [CrossRef]
- AlSalka, Y.; Granone, L.I.; Ramadan, W.; Hakki, A.; Dillert, R.; Bahnemann, D.W. Iron-based photocatalytic and photoelectrocatalytic nano-structures: Facts, perspectives, and expectations. Appl. Catal. B Environ. 2019, 244, 1065–1095. [Google Scholar] [CrossRef]
- AlSalka, Y.; Al-Madanat, O.; Curti, M.; Hakki, A.; Bahnemann, D.W. Photocatalytic H2 Evolution from Oxalic Acid: Effect of Cocatalysts and Carbon Dioxide Radical Anion on the Surface Charge Transfer Mechanisms. ACS Appl. Energy Mater. 2020, 3, 6678–6691. [Google Scholar] [CrossRef]
- Li, Y.; Lu, G.; Li, S. Photocatalytic hydrogen generation and decomposition of oxalic acid over platinized TiO2. Appl. Catal. A Gen. 2001, 214, 179–185. [Google Scholar] [CrossRef]
- Franch, M.I.; Ayllón, J.A.; Peral, J.; Domènech, X. Photocatalytic degradation of short-chain organic diacids. Catal. Today 2002, 76, 221–233. [Google Scholar] [CrossRef]
- Bowker, M.; Bahruji, H.; Kennedy, J.; Jones, W.; Hartley, G.; Morton, C. The Photocatalytic Window: Photo-Reforming of Organics and Water Splitting for Sustainable Hydrogen Production. Catal. Lett. 2015, 145, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Miyahigashi, T.; Ohkubo, K.; Fukuzumi, S. Photocatalytic hydrogen evolution from carbon-neutral oxalate with 2-phenyl-4-(1-naphthyl)quinolinium ion and metal nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 10564–10571. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Fleisch, M.; Bahnemann, D.W. Understanding the degradation pathways of oxalic acid in different photocatalytic systems: Towards simultaneous photocatalytic hydrogen evolution. J. Photochem. Photobiol. A Chem. 2018, 366, 81–90. [Google Scholar] [CrossRef]
- Kmetykó, Á.; Mogyorósi, K.; Gerse, V.; Kónya, Z.; Pusztai, P.; Dombi, A.; Hernádi, K. Photocatalytic H2 production using Pt-TiO2 in the presence of oxalic acid: Influence of the noble metal size and the carrier gas flow rate. Materials 2014, 7, 7022–7038. [Google Scholar] [CrossRef] [Green Version]
- Kandiel, T.A.; Ivanova, I.; Bahnemann, D.W. Long-term investigation of the photocatalytic hydrogen production on platinized TiO2: An isotopic study. Energy Environ. Sci. 2014, 7, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Belhadj, H.; Hamid, S.; Robertson, P.K.J.; Bahnemann, D.W. Mechanisms of Simultaneous Hydrogen Production and Formaldehyde Oxidation in H2O and D2O over Platinized TiO2. ACS Catal. 2017, 7, 4753–4758. [Google Scholar] [CrossRef] [Green Version]
- Al-Madanat, O.; AlSalka, Y.; Curti, M.; Dillert, R.; Bahnemann, D.W. Mechanistic Insights into Hydrogen Evolution by Photocatalytic Reforming of Naphthalene. ACS Catal. 2020, 10, 7398–7412. [Google Scholar] [CrossRef]
- Yuzawa, H.; Aoki, M.; Itoh, H.; Yoshida, H. Adsorption and Photoadsorption States of Benzene Derivatives on Titanium Oxide Studied by NMR. J. Phys. Chem. Lett. 2011, 2, 1868–1873. [Google Scholar] [CrossRef]
- Yates, J.T., Jr.; McKee, D.W. Kinetic isotope effect in the heterogeneous reaction of graphite with H2O (D2O). J. Chem. Phys. 1981, 75, 2711–2714. [Google Scholar] [CrossRef]
- Buettner, G.R. Spin Trapping: ESR parameters of spin adducts 1474 1528V. Free Radic. Biol. Med. 1987, 3, 259–303. [Google Scholar] [CrossRef]
- Mendive, C.B.; Bredow, T.; Schneider, J.; Blesa, M.; Bahnemann, D. Oxalic acid at the TiO2/water interface under UV (A) illumination: Surface reaction mechanisms. J. Catal. 2015, 322, 60–72. [Google Scholar] [CrossRef]
- Draganic, Z.D.; Kosanic, M.; Nenadovic, M. Competition studies of the hydroxyl radical reactions in some gamma-ray irradiated aqueous solutions at different pH values. J. Phys. Chem. 1967, 71, 2390–2395. [Google Scholar] [CrossRef]
- Doudrick, K.; Monzón, O.; Mangonon, A.; Hristovski, K.; Westerhoff, P. Nitrate reduction in water using commercial titanium dioxide photocatalysts (P25, P90, and Hombikat UV100). J. Environ. Eng. 2011, 138, 852–861. [Google Scholar] [CrossRef]
- Mora-Sero, I.; Villarreal, T.L.; Bisquert, J.; Pitarch, Á.; Gomez, R.; Salvador, P. Photoelectrochemical behavior of nanostructured TiO2 thin-film electrodes in contact with aqueous electrolytes containing dissolved pollutants: A model for distinguishing between direct and indirect interfacial hole transfer from photocurrent measurements. J. Phys. Chem. B 2005, 109, 3371–3380. [Google Scholar] [CrossRef]
- Mulazzani, Q.G.; D’Angelantonio, M.; Venturi, M.; Hoffman, M.Z.; Rodgers, M.A. Interaction of formate and oxalate ions with radiation-generated radicals in aqueous solution. Methylviologen as a mechanistic probe. J. Phys. Chem. 1986, 90, 5347–5352. [Google Scholar] [CrossRef]
- Yamada, Y.; Nomura, A.; Tadokoro, H.; Fukuzumi, S. A composite photocatalyst of an organic electron donor–acceptor dyad and a Pt catalyst supported on semiconductor nanosheets for efficient hydrogen evolution from oxalic acid. Catal. Sci. Technol. 2015, 5, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chang, W.; Ji, H.; Chen, C.; Ma, W.; Zhao, J. An Unexpected Fluctuating Reactivity for Odd and Even Carbon Numbers in the TiO2-Based Photocatalytic Decarboxylation of C2-C6 Dicarboxylic Acids. Chem. A Eur. J. 2014, 20, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Hykaway, N.; Sears, W.M.; Morisaki, H.; Morrison, S.R. Current-doubling reactions on titanium dioxide photoanodes. J. Phys. Chem. 1986, 90, 6663–6667. [Google Scholar] [CrossRef]
- Nogami, G.; Kennedy, J.H. Investigation of “Current Doubling” Mechanism of Organic Compounds by the Rotating Ring Disk Electrode Technique. J. Electrochem. Soc. 1989, 136, 2583. [Google Scholar] [CrossRef]
- Francàs, L.; Burns, E.; Steier, L.; Cha, H.; Solà-Hernández, L.; Li, X.; Shakya Tuladhar, P.; Bofill, R.; García-Antón, J.; Sala, X.; et al. Rational design of a neutral pH functional and stable organic photocathode. Chem. Commun. 2018, 54, 5732–5735. [Google Scholar] [CrossRef] [Green Version]
- Puga, A.V.; Barka, N.; Imizcoz, M. Simultaneous H2 Production and Bleaching via Solar Photoreforming of Model Dye-polluted Wastewaters on Metal/Titania. ChemCatChem 2021, 13, 1513–1529. [Google Scholar] [CrossRef]
- Bagotzky, V.S.; Osetrova, N.V. Investigations of hydrogen ionization on platinum with the help of micro-electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1973, 43, 233–249. [Google Scholar] [CrossRef]
- Gossenberger, F.; Roman, T.; Groß, A. Hydrogen and halide co-adsorption on Pt(111) in an electrochemical environment: A computational perspective. Electrochim. Acta 2016, 216, 152–159. [Google Scholar] [CrossRef]
- Palominos, R.; Freer, J.; Mondaca, M.A.; Mansilla, H.D. Evidence for hole participation during the photocatalytic oxidation of the antibiotic flumequine. J. Photochem. Photobiol. A Chem. 2008, 193, 139–145. [Google Scholar] [CrossRef]
- Schneider, J.T.; Firak, D.S.; Ribeiro, R.R.; Peralta-Zamora, P. Use of scavenger agents in heterogeneous photocatalysis: Truths, half-truths, and misinterpretations. Phys. Chem. Chem. Phys. 2020, 22, 15723–15733. [Google Scholar] [CrossRef]
- Rowley, J.; Meyer, G.J. Reduction of I2/I3− by Titanium Dioxide. J. Phys. Chem. C 2009, 113, 18444–18447. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Ivanova, I.; Schneider, J.; Gutzmann, H.; Kliemann, J.-O.; Gärtner, F.; Klassen, T.; Bahnemann, D.; Mendive, C.B. Photocatalytic degradation of oxalic and dichloroacetic acid on TiO2 coated metal substrates. Catal. Today 2013, 209, 84–90. [Google Scholar] [CrossRef]
- Yamamoto, S.; Back, R.A. The gas-phase photochemistry of oxalic acid. J. Phys. Chem. 1985, 89, 622–625. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlSalka, Y.; Al-Madanat, O.; Hakki, A.; Bahnemann, D.W. Boosting the H2 Production Efficiency via Photocatalytic Organic Reforming: The Role of Additional Hole Scavenging System. Catalysts 2021, 11, 1423. https://doi.org/10.3390/catal11121423
AlSalka Y, Al-Madanat O, Hakki A, Bahnemann DW. Boosting the H2 Production Efficiency via Photocatalytic Organic Reforming: The Role of Additional Hole Scavenging System. Catalysts. 2021; 11(12):1423. https://doi.org/10.3390/catal11121423
Chicago/Turabian StyleAlSalka, Yamen, Osama Al-Madanat, Amer Hakki, and Detlef W. Bahnemann. 2021. "Boosting the H2 Production Efficiency via Photocatalytic Organic Reforming: The Role of Additional Hole Scavenging System" Catalysts 11, no. 12: 1423. https://doi.org/10.3390/catal11121423
APA StyleAlSalka, Y., Al-Madanat, O., Hakki, A., & Bahnemann, D. W. (2021). Boosting the H2 Production Efficiency via Photocatalytic Organic Reforming: The Role of Additional Hole Scavenging System. Catalysts, 11(12), 1423. https://doi.org/10.3390/catal11121423