
In Situ Growth of Exsolved Nanoparticles under Varying rWGS Reaction Conditions—A Catalysis and Near Ambient Pressure-XPS Study

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
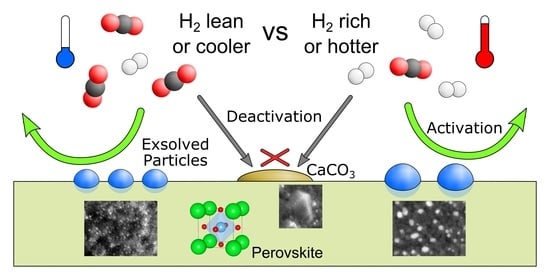
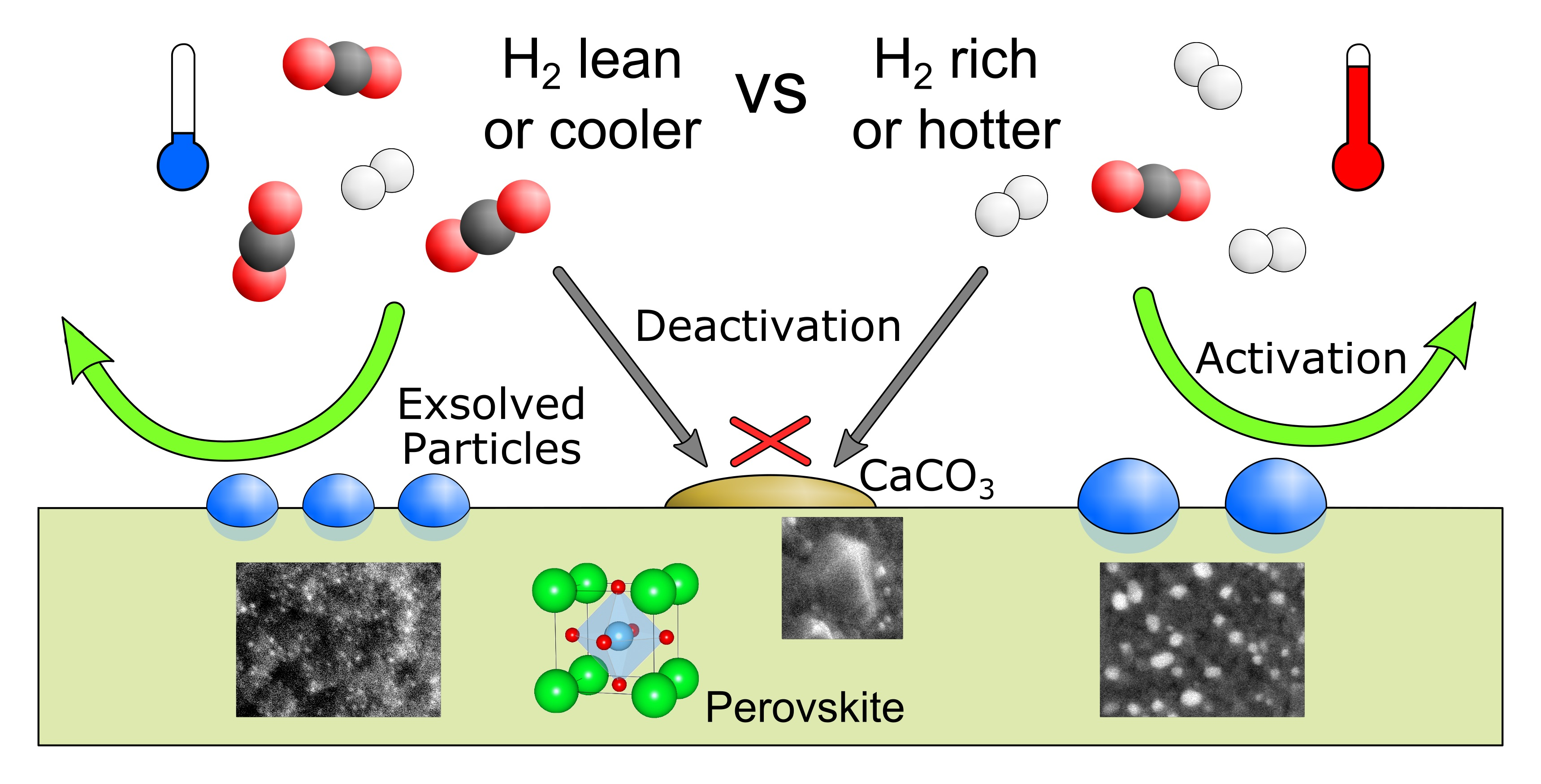
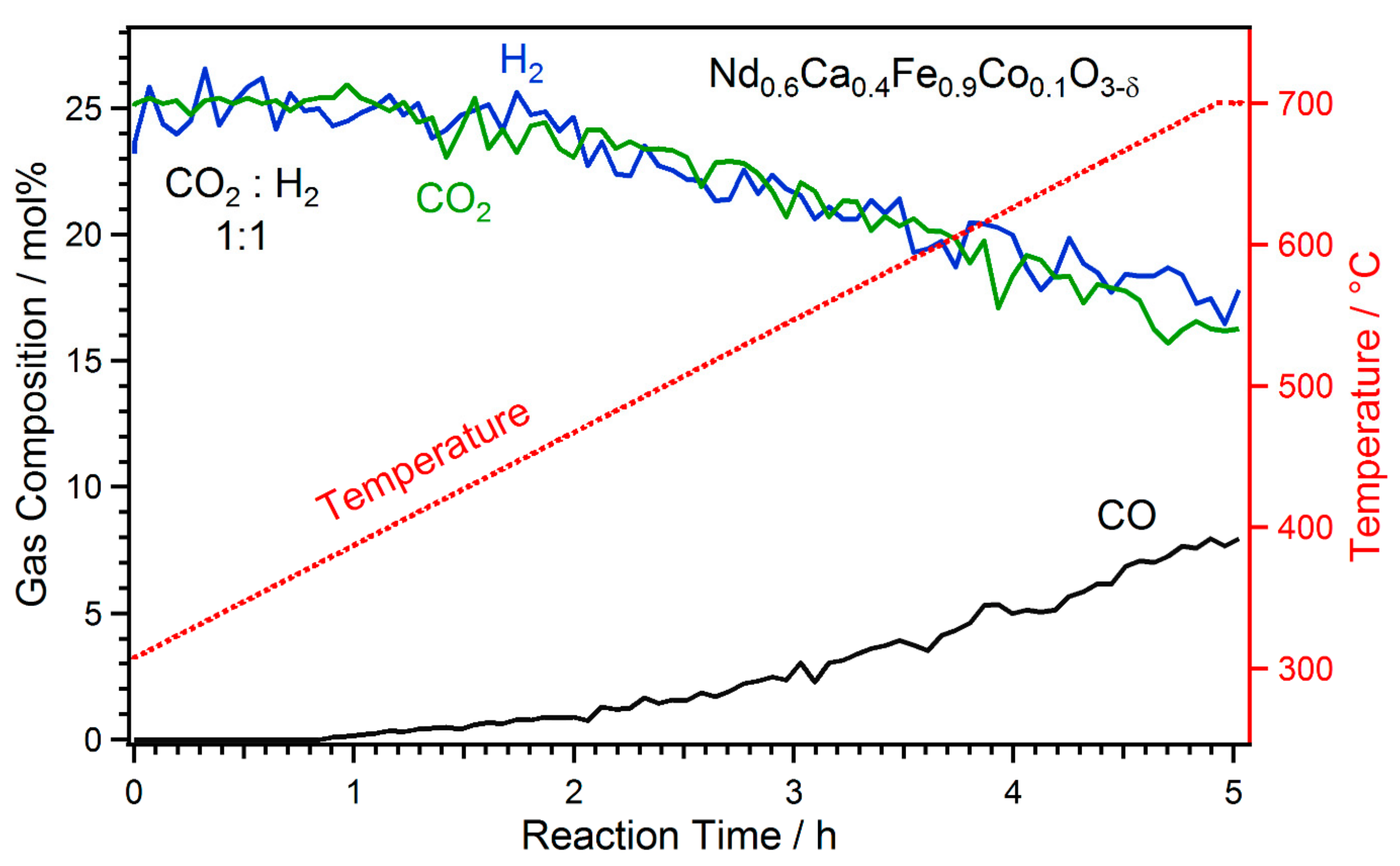
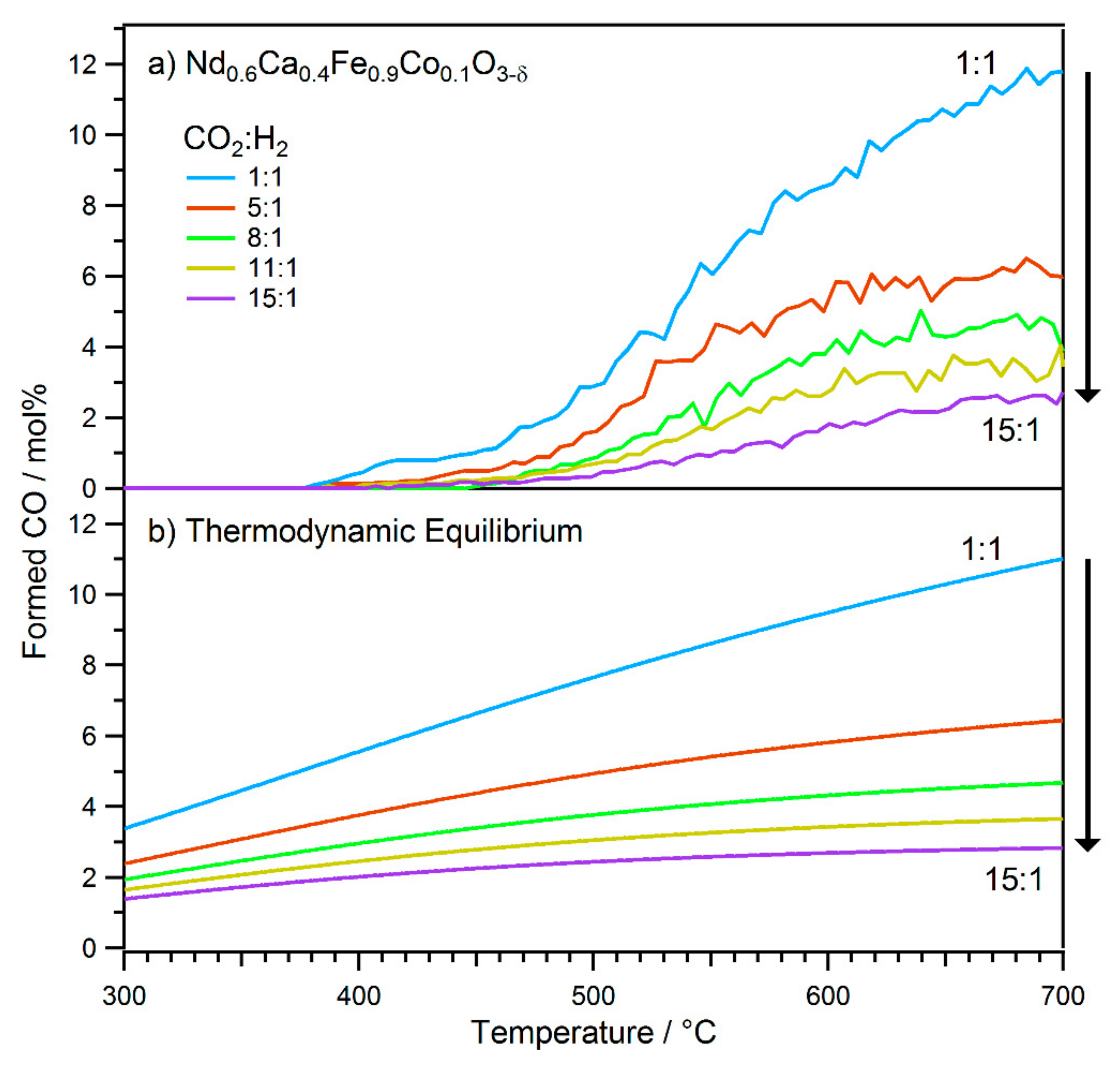
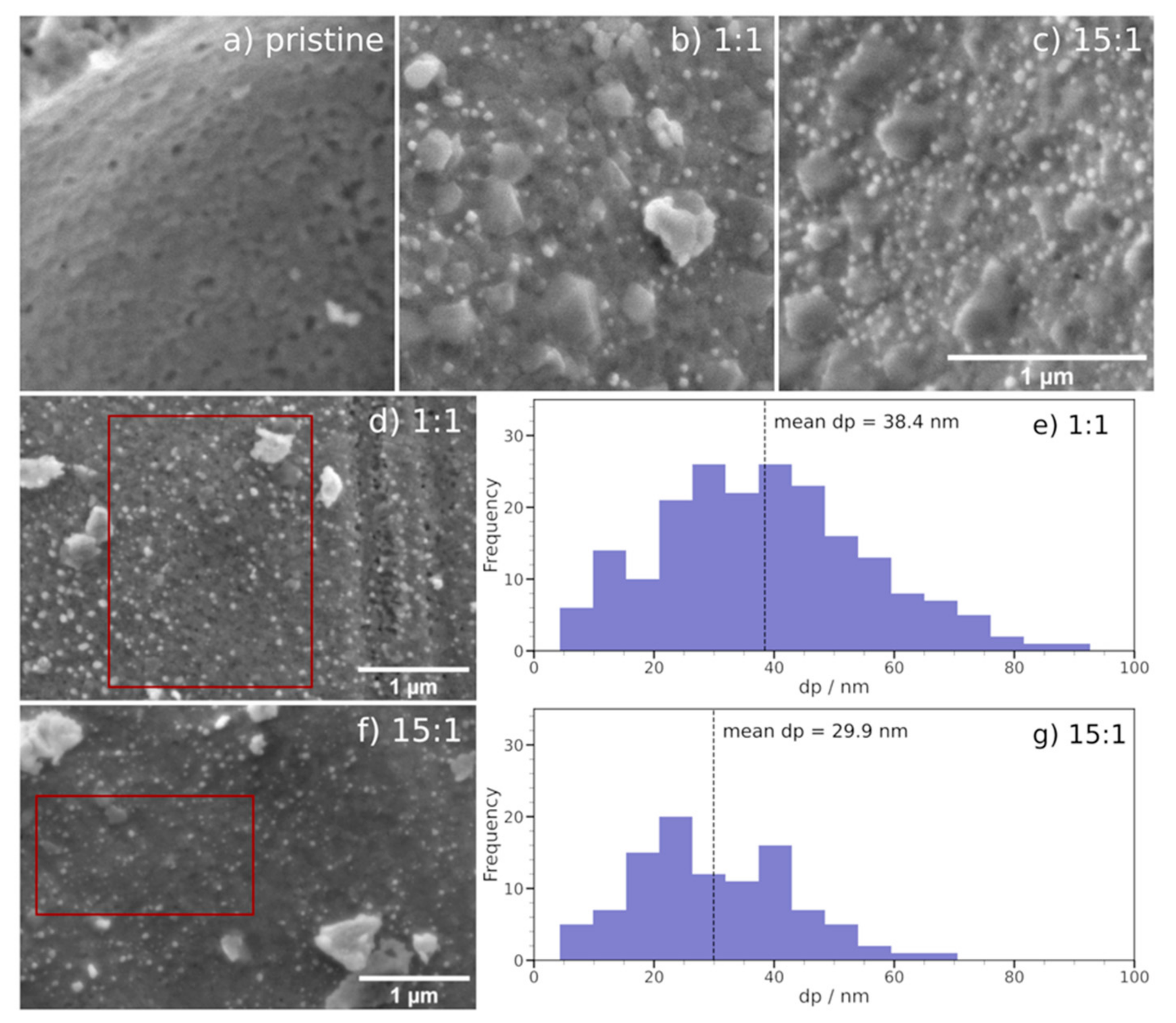
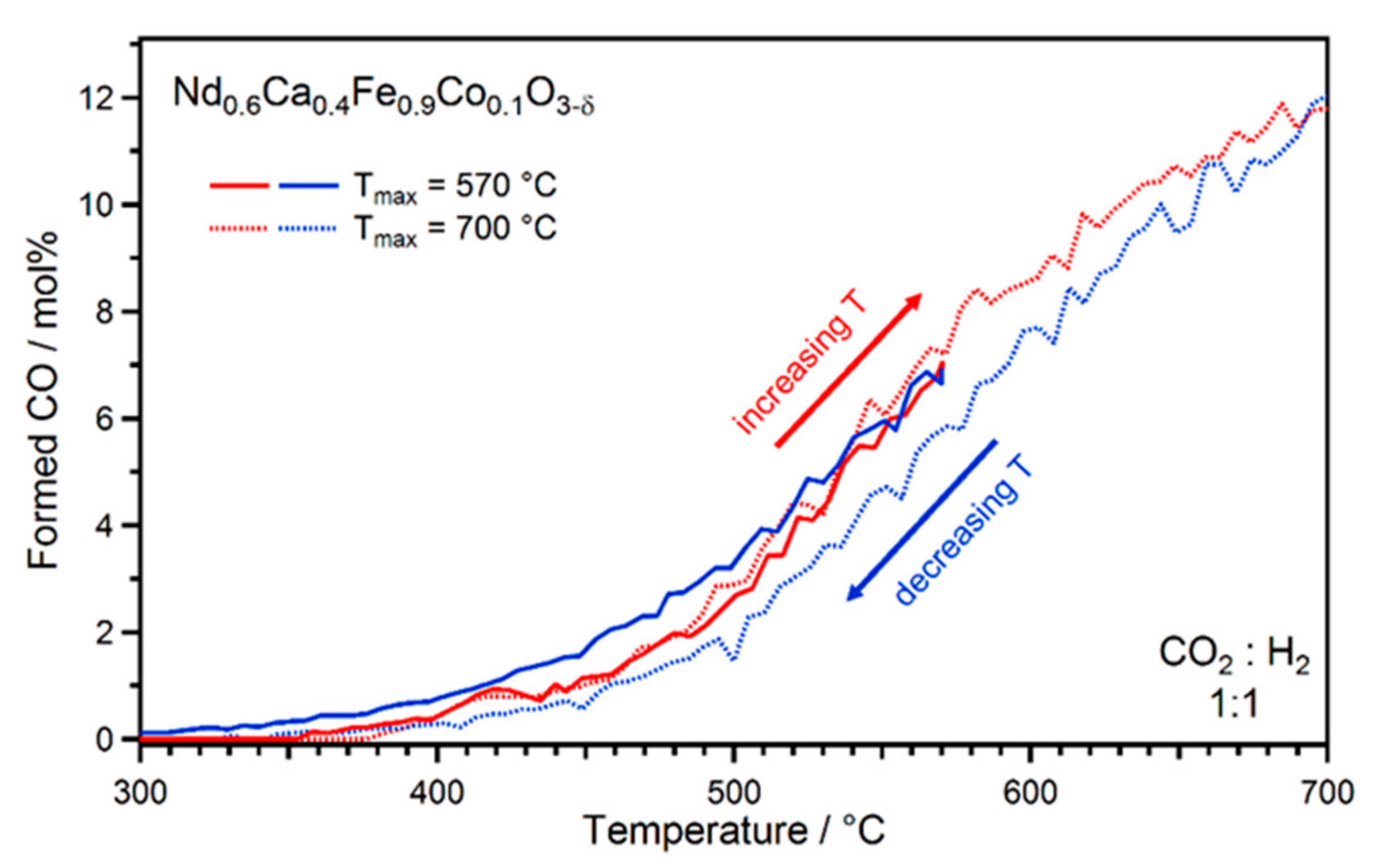
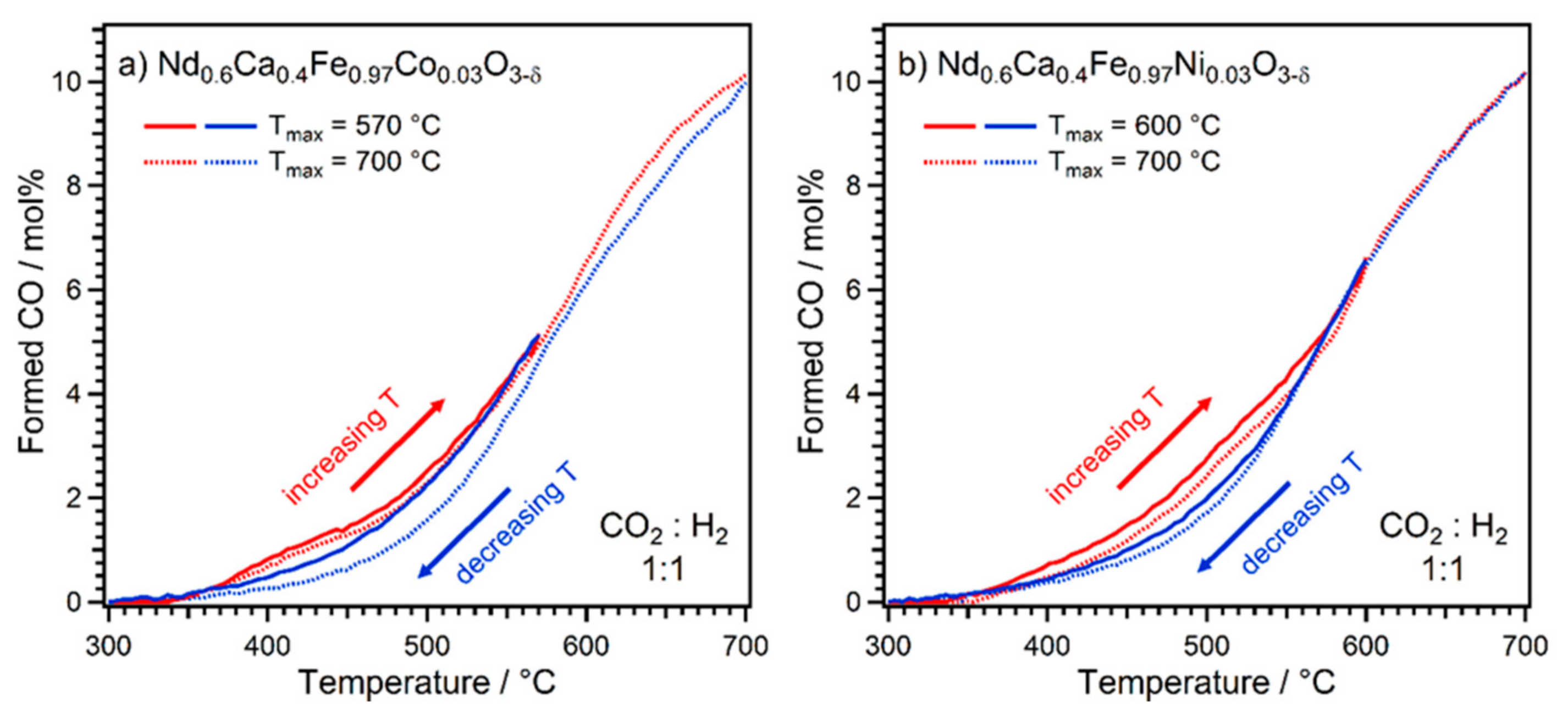
2.1. Exsolution Induced by rWGS
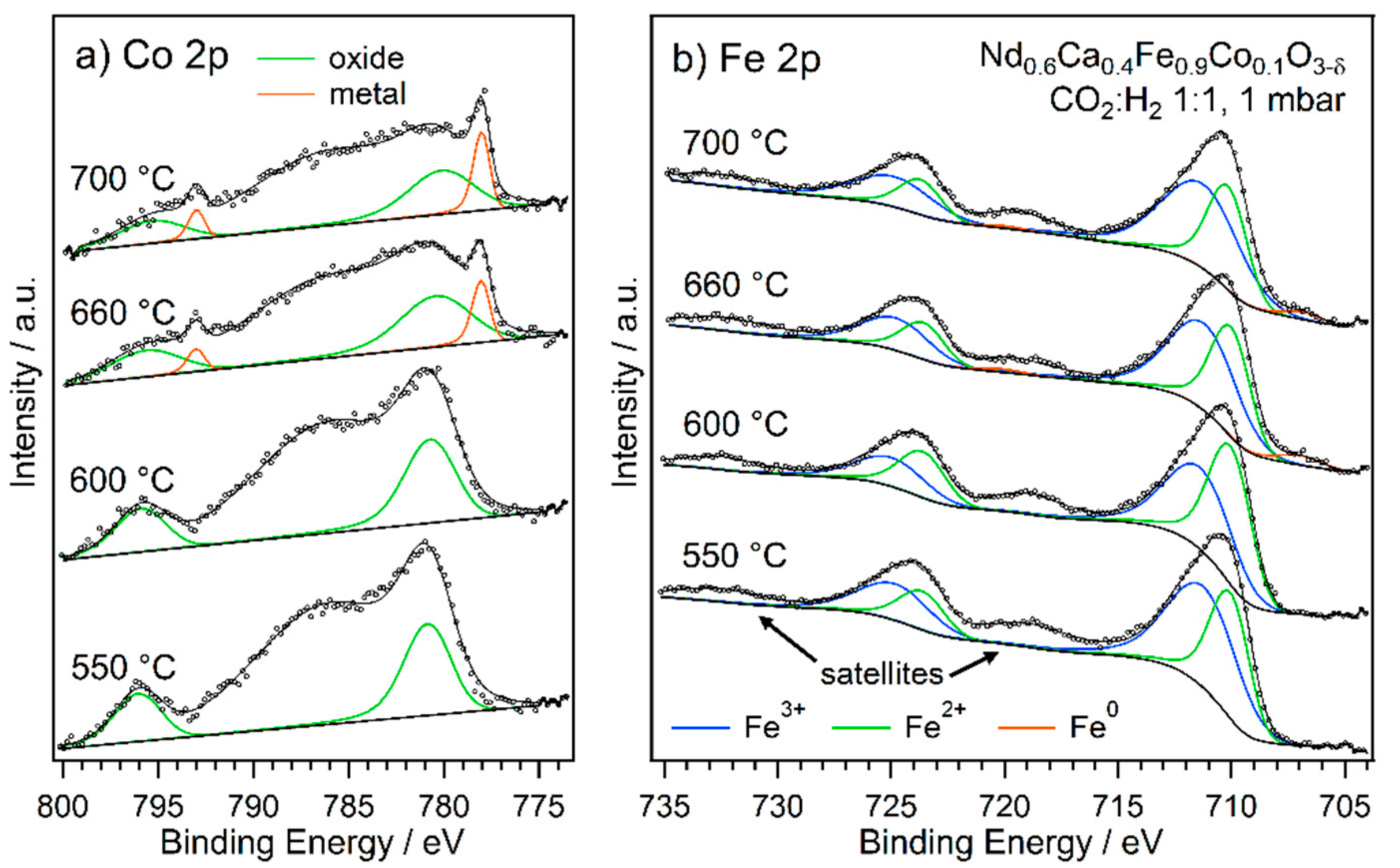
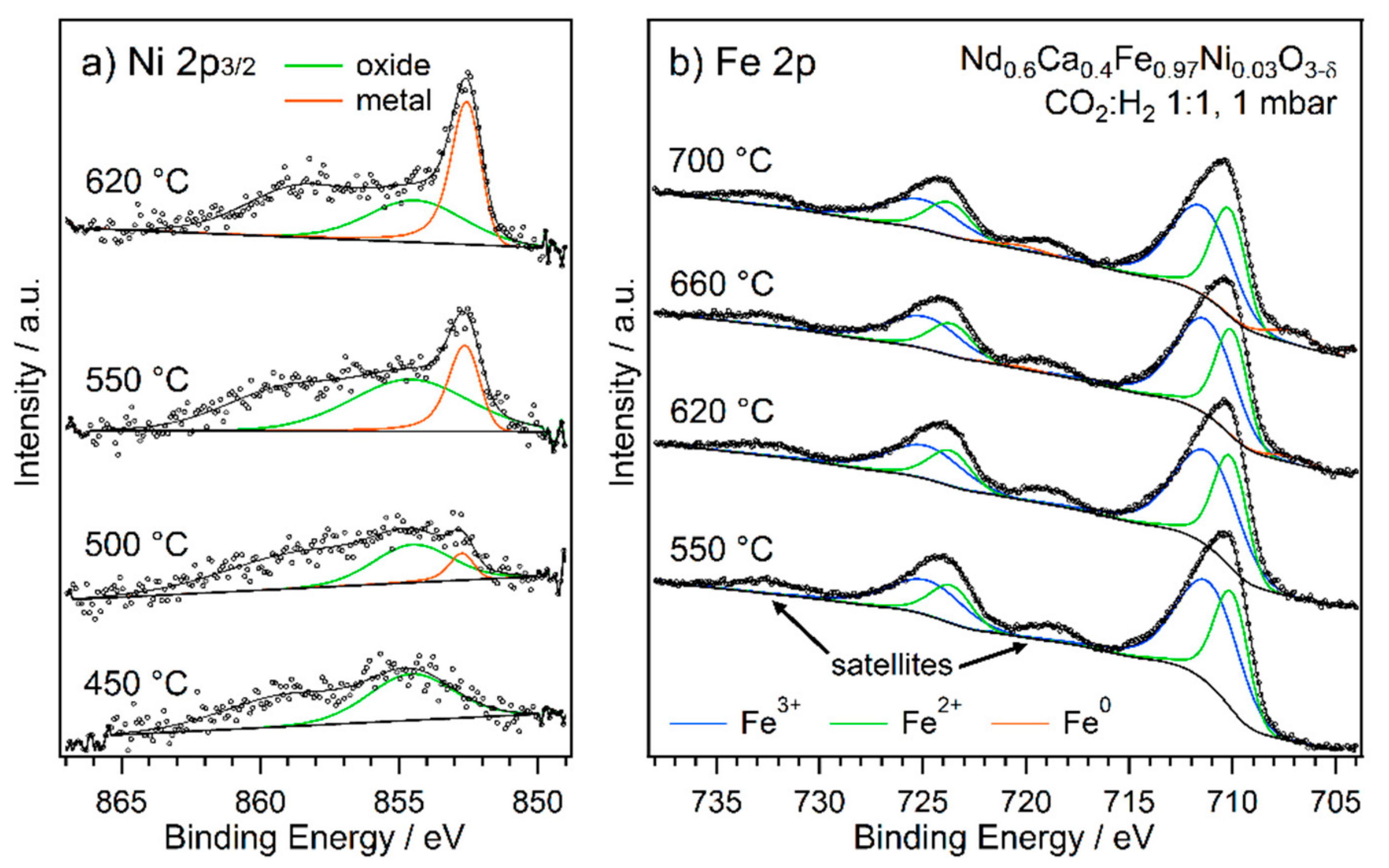
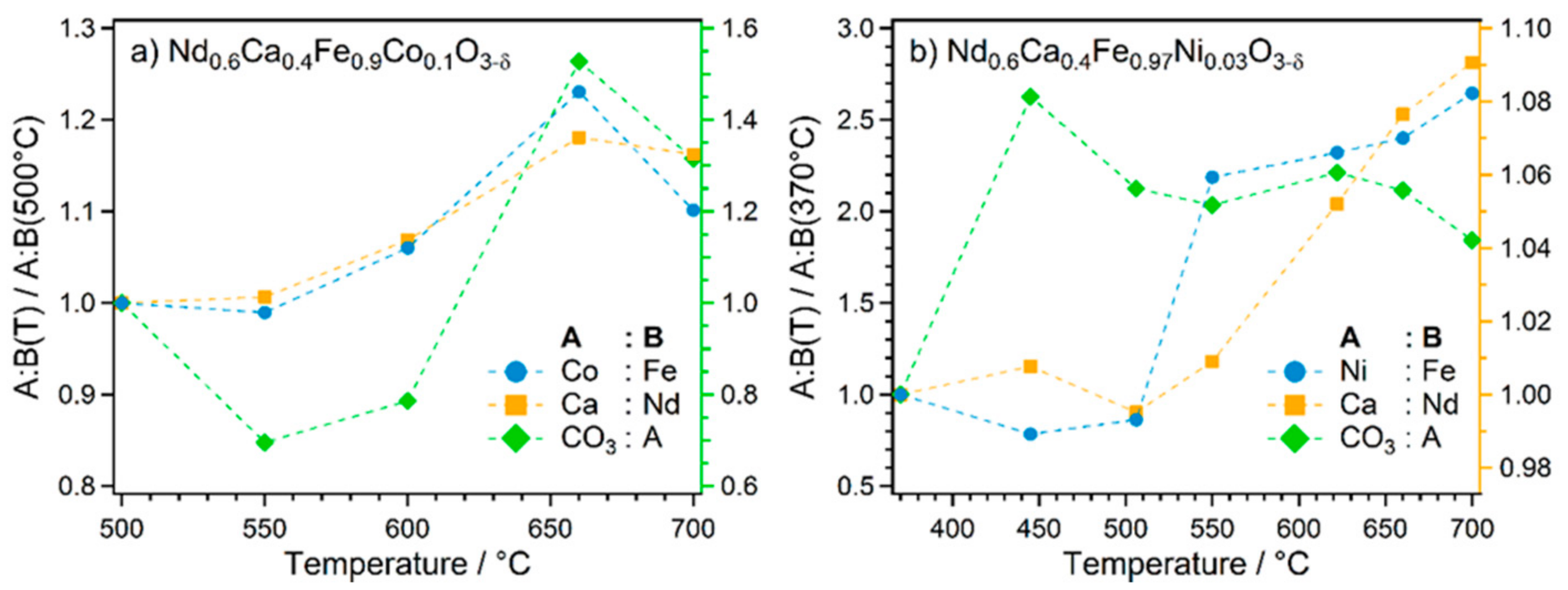
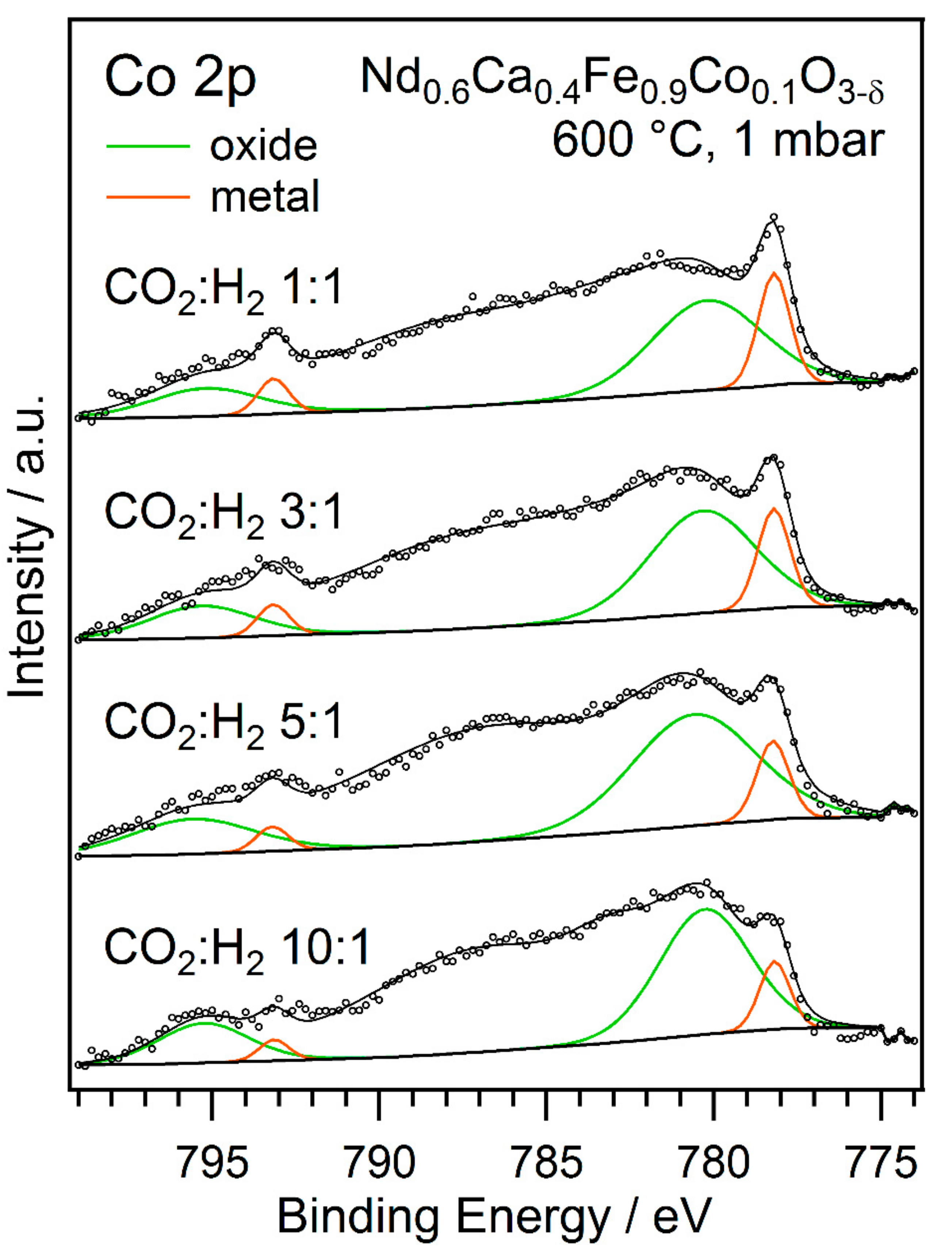
2.2. Exsolution Followed by NAP-XPS
- (i)
- The first type was performed with Nd0.6Ca0.4Fe0.9Co0.1O3-δ and Nd0.6Ca0.4Fe0.97Ni0.03O3-δ, mimicking the catalytic test runs in an rWGS atmosphere with a CO2 to H2 ratio of 1:1. Thus, the respective sample was first oxidised in O2 (1 mbar) at 600 °C. After cooling down and switching to the reaction atmosphere (CO2:H2 = 1:1, 1 mbar), the temperature was increased in steps. At each temperature step, a set of XPS spectra was obtained.
- (ii)
- For the second type of experiment, the temperature was held constant while different gas ratios were tested. Here, for each measurement block, a fresh thin film sample of Nd0.6Ca0.4Fe0.9Co0.1O3-δ was prepared and oxidised in O2 (1 mbar) at 600 °C to ensure the same starting conditions. Then, the respective gas mixtures (1 mbar total pressure) were applied to the measurement chamber at room temperature, and the sample was heated to 600 °C (due to laser heating this took only 2–3 min). A set of XPS spectra was recorded at these conditions. A total of four different gas ratios for CO2 to H2 were tested this way (10:1, 5:1, 3:1, and 1:1).
2.3. Summary and Discussion
- (i)
- The exsolution of nanoparticles that at least predominantly consisted of the catalytically active dopant element. The particle composition, the high surface area of the small particles, and their good anchoring within the perovskite support led to an activation effect on catalytic conversion.
- (ii)
- The formation of CaCO3 covering the catalyst surface and blocking active sites. This caused deactivation.
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent Advances in CO2 Capture and Utilization. Chemsuschem 2008, 1, 893–899. [Google Scholar] [CrossRef]
- Falkowski, P.; Scholes, R.J.; Boyle, E.; Canadell, J.; Canfield, D.; Elser, J.; Gruber, N.; Hibbard, K.; Hogberg, P.; Linder, S.; et al. The global carbon cycle: A test of our knowledge of earth as a system. Science 2000, 290, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 36, 2975–2992. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Mayr, L.; Shi, X.R.; Kopfle, N.; Milligan, C.A.; Zemlyanov, D.Y.; Knop-Gericke, A.; Havecker, M.; Klotzer, B.; Penner, S. Chemical vapor deposition-prepared sub-nanometer Zr clusters on Pd surfaces: Promotion of methane dry reforming. Phys. Chem. Chem. Phys. 2016, 18, 31586–31599. [Google Scholar] [CrossRef] [Green Version]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Rameshan, C.; Li, H.; Anic, K.; Roiaz, M.; Pramhaas, V.; Rameshan, R.; Blume, R.; Haevecker, M.; Knudsen, J.; Knop-Gericke, A.; et al. In situ NAP-XPS spectroscopy during methane dry reforming on ZrO2/Pt(111) inverse model catalyst. J. Phys. Condens. Matter 2018, 30, 264007. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M. Promoting the Synthesis of Methanol: Understanding the Requirements for an Industrial Catalyst for the Conversion of CO2. Angew. Chem. -Int. Ed. 2016, 55, 14906–14908. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Claeys, M.; Schulz, H.; Schaub, G.; Nam, S.S.; Jun, K.W.; Choi, M.J.; Kishan, G.; Lee, K.W. Comparative study of Fischer-Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts. Appl. Catal. A Gen. 1999, 186, 201–213. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic aspects of the selective methanation of CO over Ru/TiO2 catalyst. Catal. Today 2012, 181, 138–147. [Google Scholar] [CrossRef]
- Goguet, A.; Meunier, F.C.; Tibiletti, D.; Breen, J.P.; Burch, R. Spectrokinetic investigation of reverse water-gas-shift reaction intermediates over a Pt/CeO2 catalyst. J. Phys. Chem. B 2004, 108, 20240–20246. [Google Scholar] [CrossRef] [Green Version]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef]
- Galvan, C.A.; Schumann, J.; Behrens, M.; Fierro, J.L.G.; Schlogl, R.; Frei, E. Reverse water-gas shift reaction at the Cu/ZnO interface: Influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B Environ. 2016, 195, 104–111. [Google Scholar] [CrossRef]
- Wang, L.H.; Liu, H.; Chen, Y.; Yang, S.Q. Reverse water-gas shift reaction over co-precipitated Co-CeO2 catalysts: Effect of Co content on selectivity and carbon formation. Int. J. Hydrog. Energy 2017, 42, 3682–3689. [Google Scholar] [CrossRef]
- Wang, L.H.; Liu, H.; Liu, Y.; Chen, Y.; Yang, S.Q. Effect of precipitants on Ni-CeO2 catalysts prepared by a co-precipitation method for the reverse water-gas shift reaction. J. Rare Earths 2013, 31, 969–974. [Google Scholar] [CrossRef]
- Collins, S.E.; Delgado, J.J.; Mira, C.; Calvino, J.J.; Bernal, S.; Chiavassa, D.L.; Baltanas, M.A.; Bonivardi, A.L. The role of Pd-Ga bimetallic particles in the bifunctional mechanism of selective methanol synthesis via CO2 hydrogenation on a Pd/Ga2O3 catalyst. J. Catal. 2012, 292, 90–98. [Google Scholar] [CrossRef]
- Liu, L.N.; Das, S.; Chen, T.J.; Dewangan, N.; Ashok, J.; Xi, S.B.; Borgna, A.; Li, Z.W.; Kawi, S. Low temperature catalytic reverse water-gas shift reaction over perovskite catalysts in DBD plasma. Appl. Catal. B Environ. 2020, 265, 118573. [Google Scholar] [CrossRef]
- Lorenz, H.; Zhao, Q.; Turner, S.; Lebedev, O.I.; van Tendeloo, G.; Kloetzer, B.; Rameshan, C.; Penner, S. Preparation and structural characterization of SnO2 and GeO2 methanol steam reforming thin film model catalysts by (HR)TEM. Mater. Chem. Phys. 2010, 122, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Perez, L.; Baibars, F.; Le Sache, E.; Arellano-Garcia, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Daza, Y.A.; Maiti, D.; Kent, R.A.; Bhethanabotla, V.R.; Kuhn, J.N. Isothermal reverse water gas shift chemical looping on La0.75Sr0.25Co1-YFeYO3 perovskite-type oxides. Catal. Today 2015, 258, 691–698. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, J.L.; Park, E.J.; Kim, Y.D.; Uhm, S. Dopant Effect of Barium Zirconate-Based Perovskite-Type Catalysts for the Intermediate-Temperature Reverse Water Gas Shift Reaction. ACS Catal. 2014, 4, 3117–3122. [Google Scholar] [CrossRef]
- Bligaard, T.; Norskov, J.K.; Dahl, S.; Matthiesen, J.; Christensen, C.H.; Sehested, J. The Bronsted-Evans-Polanyi relation and the volcano curve in heterogeneous catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Pastor-Perez, L.; Shah, M.; le Sache, E.; Reina, T.R. Improving Fe/Al2O3 Catalysts for the Reverse Water-Gas Shift Reaction: On the Effect of Cs as Activity/Selectivity Promoter. Catalysts 2018, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Jurkovic, D.L.; Pohar, A.; Dasireddy, V.; Likozar, B. Effect of Copper-based Catalyst Support on Reverse Water-Gas Shift Reaction (RWGS) Activity for CO2 Reduction. Chem. Eng. Technol. 2017, 40, 973–980. [Google Scholar] [CrossRef]
- Lindenthal, L.; Rameshan, R.; Summerer, H.; Ruh, T.; Popovic, J.; Nenning, A.; Löffler, S.; Opitz, A.K.; Blaha, P.; Rameshan, C. Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution. Catalysts 2020, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Shin, T.H.; Myung, J.H.; Verbraeken, M.; Kim, G.; Irvine, J.T.S. Oxygen deficient layered double perovskite as an active cathode for CO2 electrolysis using a solid oxide conductor. Faraday Discuss. 2015, 182, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Stöger, B.; Hieckel, M.; Mittendorfer, F.; Wang, Z.M.; Fobes, D.; Peng, J.; Mao, Z.Q.; Schmid, M.; Redinger, J.; Diebold, U. High Chemical Activity of a Perovskite Surface: Reaction of CO with Sr3Ru2O7. Phys. Rev. Lett. 2014, 113, 116101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, M.B.; Zhang, S.Y.; Duan, Y.W.; Wang, H.J.; Fang, M.H.; Zhang, K.; Li, B.H.; Graham, G.W.; Pan, X.Q. Reversible precipitation/dissolution of precious-metal clusters in perovskite-based catalyst materials: Bulk versus surface re-dispersion. J. Catal. 2012, 293, 145–148. [Google Scholar] [CrossRef]
- Thalinger, R.; Opitz, A.K.; Kogler, S.; Heggen, M.; Stroppa, D.; Schmidmair, D.; Tappert, R.; Fleig, J.; Klotzer, B.; Penner, S. Water-Gas Shift and Methane Reactivity on Reducible Perovskite-Type Oxides. J. Phys. Chem. C 2015, 119, 11739–11753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, D.N.; Machala, M.L.; Bluhm, H.; Chueh, W.C. Redox activity of surface oxygen anions in oxygen-deficient perovskite oxides during electrochemical reactions. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neagu, D.; Tsekouras, G.; Miller, D.N.; Menard, H.; Irvine, J.T.S. In situ growth of nanoparticles through control of non-stoichiometry. Nat. Chem. 2013, 5, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.F.; Li, J.H.; Zeng, Y.M.; Amirkhiz, B.S.; Wang, M.N.; Behnamian, Y.; Luo, J.L. A-site deficient perovskite: The parent for in situ exsolution of highly, regenerable nano-particles as SOFC anodes. J. Mater. Chem. A 2015, 3, 11048–11056. [Google Scholar] [CrossRef]
- Papargyriou, D.; Irvine, J.T.S. Nickel nanocatalyst exsolution from (La,Sr) (Cr,M,Ni)O3 (M=Mn,Fe) perovskites for the fuel oxidation layer of Oxygen Transport Membranes. Solid State Ion. 2016, 288, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Neagu, D.; Oh, T.S.; Miller, D.N.; Menard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenthal, L.; Ruh, T.; Rameshan, R.; Summerer, H.; Nenning, A.; Herzig, C.; Loffler, S.; Limbeck, A.; Opitz, A.K.; Blaha, P.; et al. Ca-doped rare earth perovskite materials for tailored exsolution of metal nanoparticles. Acta Crystallogr. Sect. B 2020, 76, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.S.; Rahani, E.K.; Neagu, D.; Irvine, J.T.S.; Shenoy, V.B.; Gorte, R.J.; Vohs, J.M. Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites during Exsolution. J. Phys. Chem. Lett. 2015, 6, 5106–5110. [Google Scholar] [CrossRef] [Green Version]
- Haag, J.M.; Barnett, S.A.; Richardson, J.W.; Poeppelmeier, K.R. Structural and Chemical Evolution of the SOFC Anode La0.30Sr0.70Fe0.70Cr0.30O3-δ upon Reduction and Oxidation: An in Situ Neutron Diffraction Study. Chem. Mater. 2010, 22, 3283–3289. [Google Scholar] [CrossRef]
- Gotsch, T.; Schlicker, L.; Bekheet, M.F.; Doran, A.; Grunbacher, M.; Praty, C.; Tada, M.; Matsui, H.; Ishiguro, N.; Gurlo, A.; et al. Structural investigations of La0.6Sr0.4FeO3-δ under reducing conditions: Kinetic and thermodynamic limitations for phase transformations and iron exsolution phenomena. RSC Adv. 2018, 8, 3120–3131. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.B.; Graham, G.W.; Duan, Y.W.; Liu, H.; Adamo, C.; Schlom, D.G.; Pan, X.Q. Self-Regeneration of Pd-LaFeO3 Catalysts: New Insight from Atomic-Resolution Electron Microscopy. J. Am. Chem. Soc. 2011, 133, 18090–18093. [Google Scholar] [CrossRef]
- Lv, H.F.; Lin, L.; Zhang, X.M.; Song, Y.F.; Matsumoto, H.; Zeng, C.B.; Ta, N.; Liu, W.; Gao, D.F.; Wang, G.X.; et al. In Situ Investigation of Reversible Exsolution/Dissolution of CoFe Alloy Nanoparticles in a Co-Doped Sr2Fe1.5Mo0.5O6-δ Cathode for CO2 Electrolysis. Adv. Mater. 2020, 32, 1906193. [Google Scholar] [CrossRef]
- Neagu, D.; Kyriakou, V.; Roiban, I.L.; Aouine, M.; Tang, C.Y.; Caravaca, A.; Kousi, K.; Schreur-Piet, I.; Metcalfe, I.S.; Vernoux, P.; et al. In Situ Observation of Nanoparticle Exsolution from Perovskite Oxides: From Atomic Scale Mechanistic Insight to Nanostructure Tailoring. ACS Nano 2019, 13, 12996–13005. [Google Scholar] [CrossRef]
- Jo, Y.R.; Koo, B.; Seo, M.J.; Kim, J.K.; Lee, S.; Kim, K.; Han, J.W.; Jung, W.; Kim, B.J. Growth Kinetics of Individual Co Particles Ex-solved on SrTi0.75Co0.25O3-δ Polycrystalline Perovskite Thin Films. J. Am. Chem. Soc. 2019, 141, 6690–6697. [Google Scholar] [CrossRef]
- Lu, J.H.; Zhu, C.L.; Pan, C.C.; Lin, W.L.; Lemmon, J.P.; Chen, F.L.; Li, C.S.; Xie, K. Highly efficient electrochemical reforming of CH4/CO2 in a solid oxide electrolyser. Sci. Adv. 2018, 4, eaar5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gines, M.J.L.; Marchi, A.J.; Apesteguia, C.R. Kinetic study of the reverse water-gas shift reaction over CuO/ZnO/Al2O3 catalysts. Appl. Catal. A Gen. 1997, 154, 155–171. [Google Scholar] [CrossRef]
- Opitz, A.K.; Nenning, A.; Rameshan, C.; Kubicek, M.; Goetsch, T.; Blume, R.; Haevecker, M.; Knop-Gericke, A.; Rupprechter, G.; Kloetzer, B.; et al. Surface Chemistry of Perovskite-Type Electrodes During High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy. ACS Appl. Mater. Interfaces 2017, 9, 35847–35860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Gao, M.R.; Luo, J.L. In Situ Exsolved Metal Nanoparticles: A Smart Approach for Optimization of Catalysts. Chem. Mater. 2020, 32, 5424–5441. [Google Scholar] [CrossRef]
- Rameshan, R.; Nenning, A.; Raschhofer, J.; Lindenthal, L.; Ruh, T.; Summerer, H.; Opitz, A.K.; Huber, T.M.; Rameshan, C. Novel Sample-Stage for Combined Near Ambient Pressure X-ray Photoelectron Spectroscopy, Catalytic Characterization and Electrochemical Impedance Spectroscopy. Crystals 2020, 10, 947. [Google Scholar] [CrossRef]
- Zeng, D.W.; Qiu, Y.; Peng, S.; Chen, C.; Zeng, J.M.; Zhang, S.; Xiao, R. Enhanced hydrogen production performance through controllable redox exsolution within CoFeAlOx spinel oxygen carrier materials. J. Mater. Chem. A 2018, 6, 11306–11316. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Lindenthal, L.; Popovic, J.; Rameshan, R.; Huber, J.; Schrenk, F.; Ruh, T.; Nenning, A.; Löffler, S.; Opitz, A.K.; Rameshan, C. Novel perovskite catalysts for CO2 utilization—Exsolution enhanced reverse water-gas shift activity. Appl. Catal. B Environ. 2021, 292, 120183. [Google Scholar] [CrossRef]
- Sambalova, O.; Billeter, E.; Mann, J.; Miyayama, T.; Burnat, D.; Heel, A.; Bleiner, D.; Borgschulte, A. Hard and soft X-ray photoelectron spectroscopy for selective probing of surface and bulk chemical compositions in a perovskite-type Ni catalyst. Surf. Interface Anal. 2020, 52, 811–817. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, D.; Saccoccio, M.; Lu, Z.; Ciucci, F. From material design to mechanism study: Nanoscale Ni exsolution on a highly active A-site deficient anode material for solid oxide fuel cells. Nano Energy 2016, 27, 499–508. [Google Scholar] [CrossRef]
- Kwon, O.; Joo, S.; Choi, S.; Sengodan, S.; Kim, G. Review on exsolution and its driving forces in perovskites. J. Phys. Energy 2020, 2, 032001. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to Form a Capacitor. U.S. Patent 3,330,697, 11 July 1967. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromey, D.W. Avoiding Twisted Pixels: Ethical Guidelines for the Appropriate Use and Manipulation of Scientific Digital Images. Sci. Eng. Ethics 2010, 16, 639–667. [Google Scholar] [CrossRef] [PubMed]
- Price-Whelan, A.M.; Sipocz, B.M.; Gunther, H.M.; Lim, P.L.; Crawford, S.M.; Conseil, S.; Shupe, D.L.; Craig, M.W.; Dencheva, N.; Ginsburg, A.; et al. The Astropy Project: Building an Open-science Project and Status of the v2.0 Core Package. Astron. J. 2018, 156, 123. [Google Scholar] [CrossRef]
- Robitaille, T.P.; Tollerud, E.J.; Greenfield, P.; Droettboom, M.; Bray, E.; Aldcroft, T.; Davis, M.; Ginsburg, A.; Price-Whelan, A.M.; Kerzendorf, W.E.; et al. Astropy: A community Python package for astronomy. Astron. Astrophys. 2013, 558, A33. [Google Scholar] [CrossRef]
- Burges, D.R., Jr. Thermochemical Data. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2018. [Google Scholar] [CrossRef]
- Lindenthal, L.; Buchinger, R.; Drexler, H.; Schrenk, F.; Ruh, T.; Rameshan, C. Exsolution Catalysts—Increasing Metal Efficiency. Encyclopedia 2021, 1, 249–260. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, X.; Chen, J.; Deng, J.; Yu, R.; Xing, X. Shape controllable synthesis of NdFeO3 micro single crystals by a hydrothermal route. CrystEngComm 2014, 16, 858–862. [Google Scholar] [CrossRef]
- Lerou, J.J.; Froment, G.F. Velocity, Temperature and Conversion Profiles in Fixed-Bed Catalytic Reactors. Chem. Eng. Sci. 1977, 32, 853–861. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO2 | H2 | CO | H2O | |
|---|---|---|---|---|
| Initial | 0 | 0 | ||
| Reacted |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindenthal, L.; Huber, J.; Drexler, H.; Ruh, T.; Rameshan, R.; Schrenk, F.; Löffler, S.; Rameshan, C. In Situ Growth of Exsolved Nanoparticles under Varying rWGS Reaction Conditions—A Catalysis and Near Ambient Pressure-XPS Study. Catalysts 2021, 11, 1484. https://doi.org/10.3390/catal11121484
Lindenthal L, Huber J, Drexler H, Ruh T, Rameshan R, Schrenk F, Löffler S, Rameshan C. In Situ Growth of Exsolved Nanoparticles under Varying rWGS Reaction Conditions—A Catalysis and Near Ambient Pressure-XPS Study. Catalysts. 2021; 11(12):1484. https://doi.org/10.3390/catal11121484
Chicago/Turabian StyleLindenthal, Lorenz, Joel Huber, Hedda Drexler, Thomas Ruh, Raffael Rameshan, Florian Schrenk, Stefan Löffler, and Christoph Rameshan. 2021. "In Situ Growth of Exsolved Nanoparticles under Varying rWGS Reaction Conditions—A Catalysis and Near Ambient Pressure-XPS Study" Catalysts 11, no. 12: 1484. https://doi.org/10.3390/catal11121484
APA StyleLindenthal, L., Huber, J., Drexler, H., Ruh, T., Rameshan, R., Schrenk, F., Löffler, S., & Rameshan, C. (2021). In Situ Growth of Exsolved Nanoparticles under Varying rWGS Reaction Conditions—A Catalysis and Near Ambient Pressure-XPS Study. Catalysts, 11(12), 1484. https://doi.org/10.3390/catal11121484