
Fine Crystalline Mg-Al Hydrotalcites as Catalysts for Baeyer-Villiger Oxidation of Cyclohexanone with H2O2

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
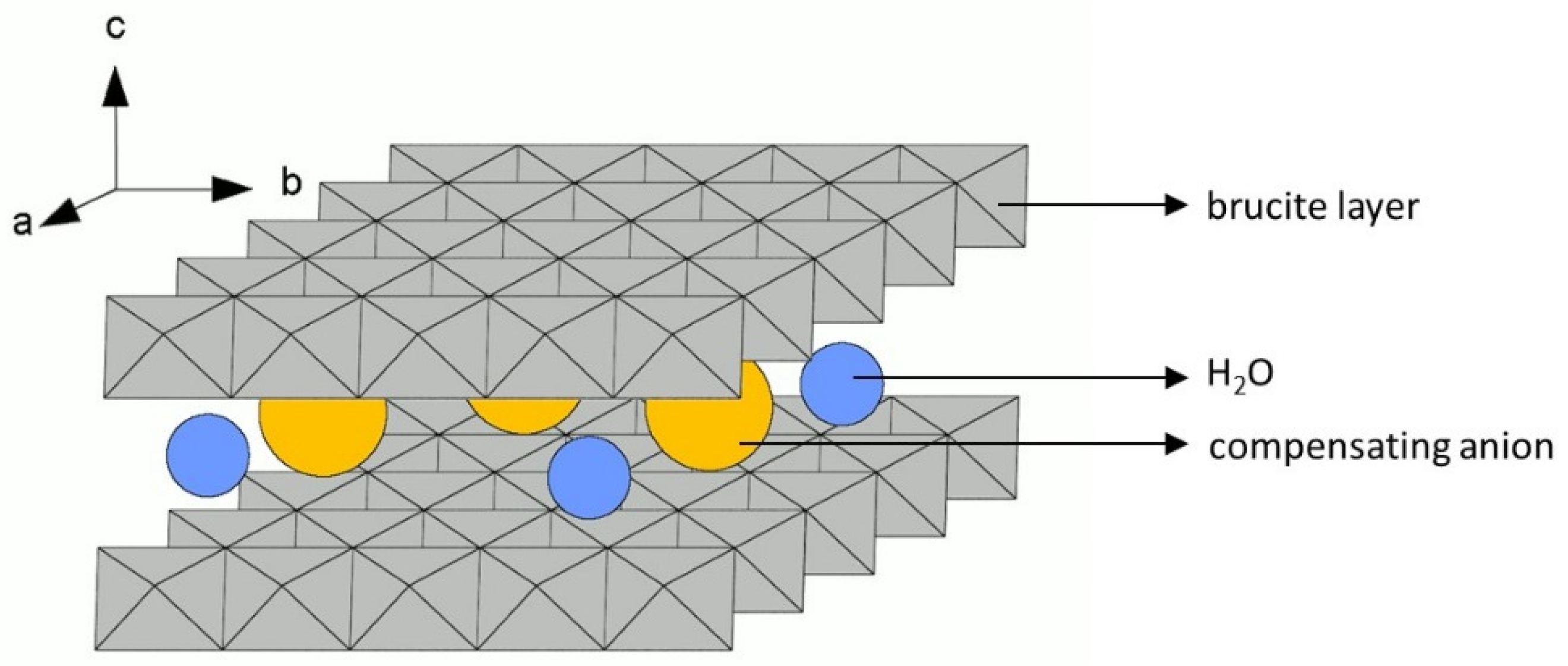
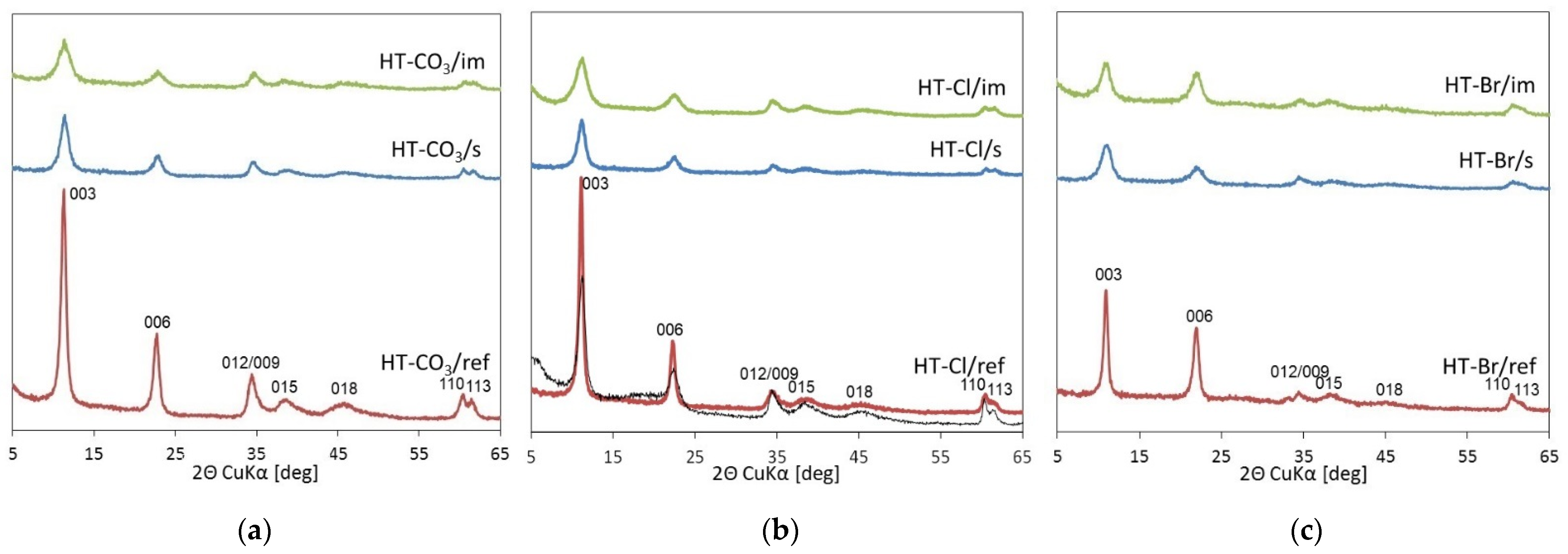
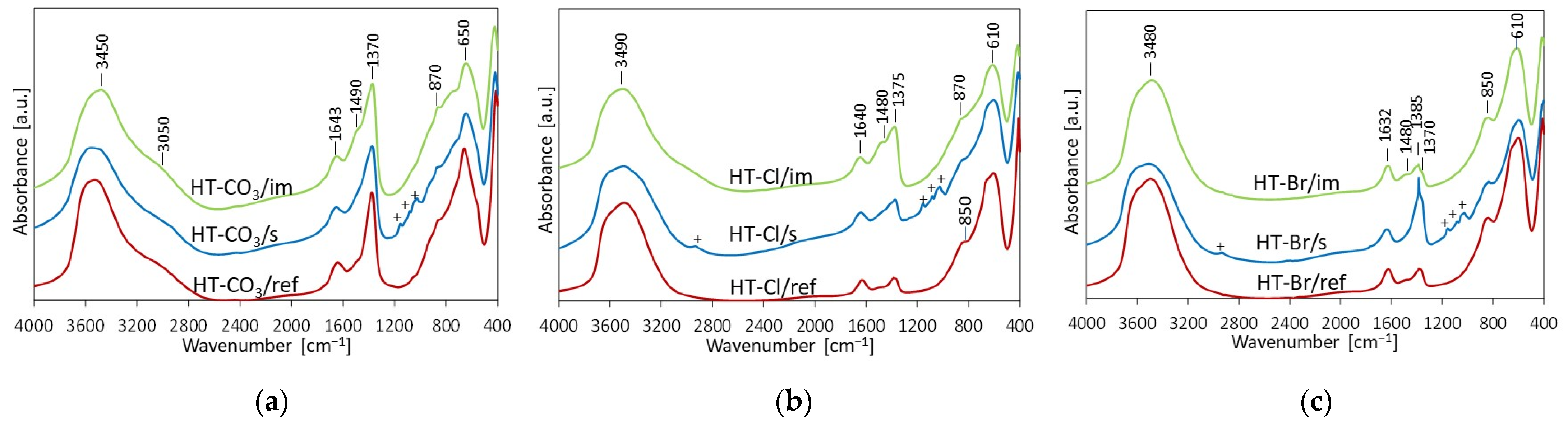
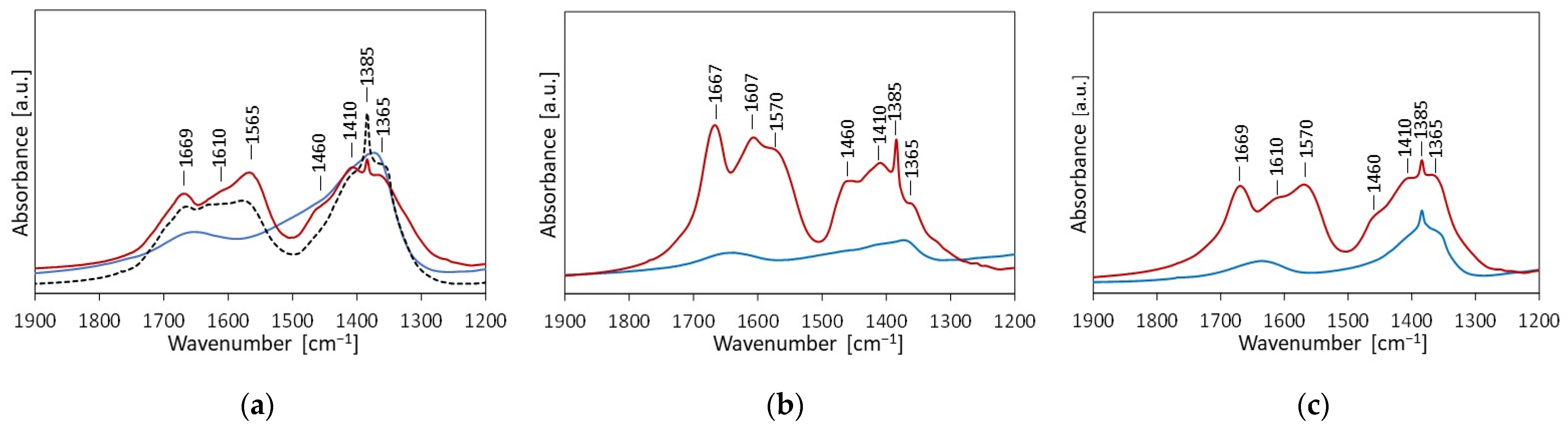
2.1. Physicochemical Characterization
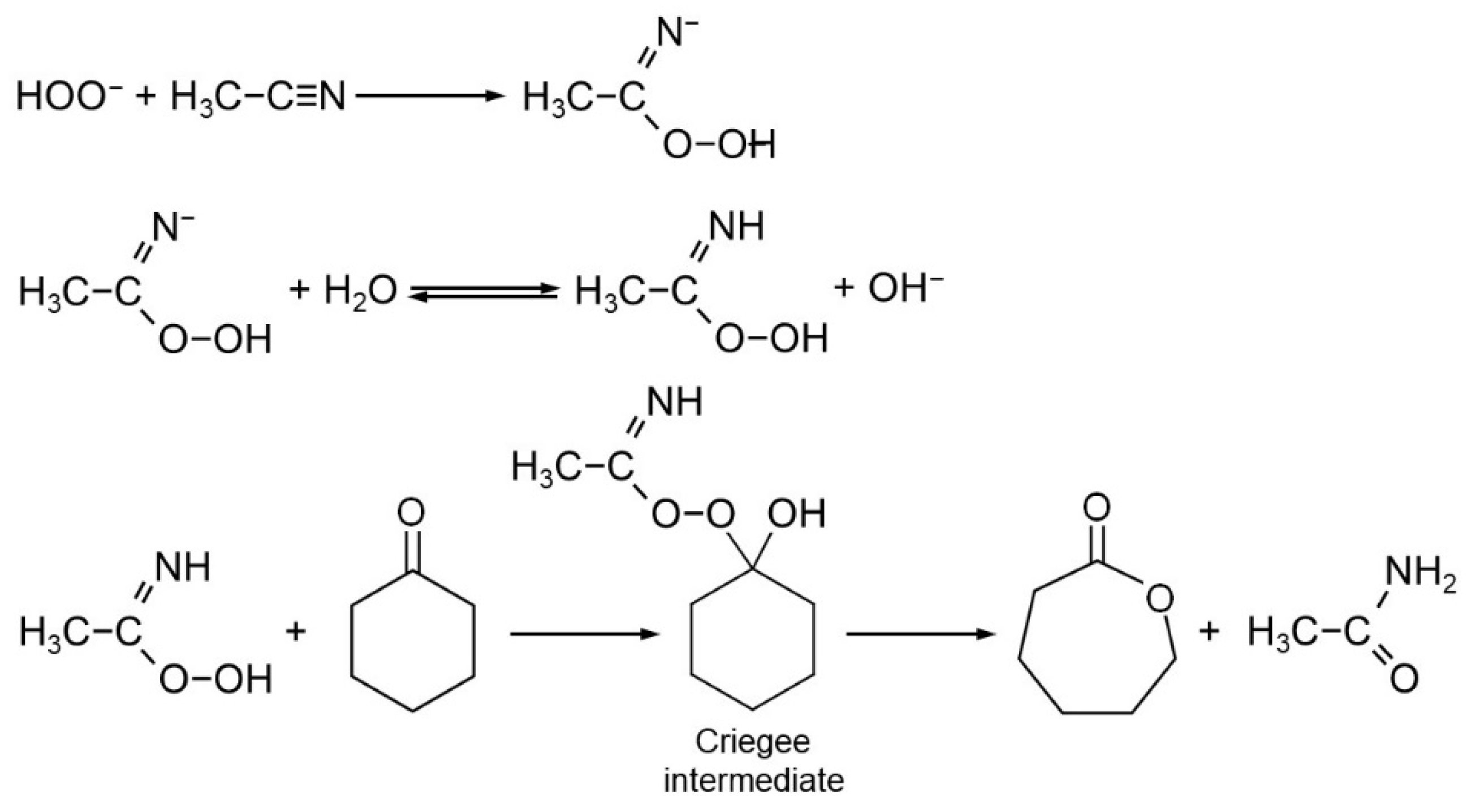
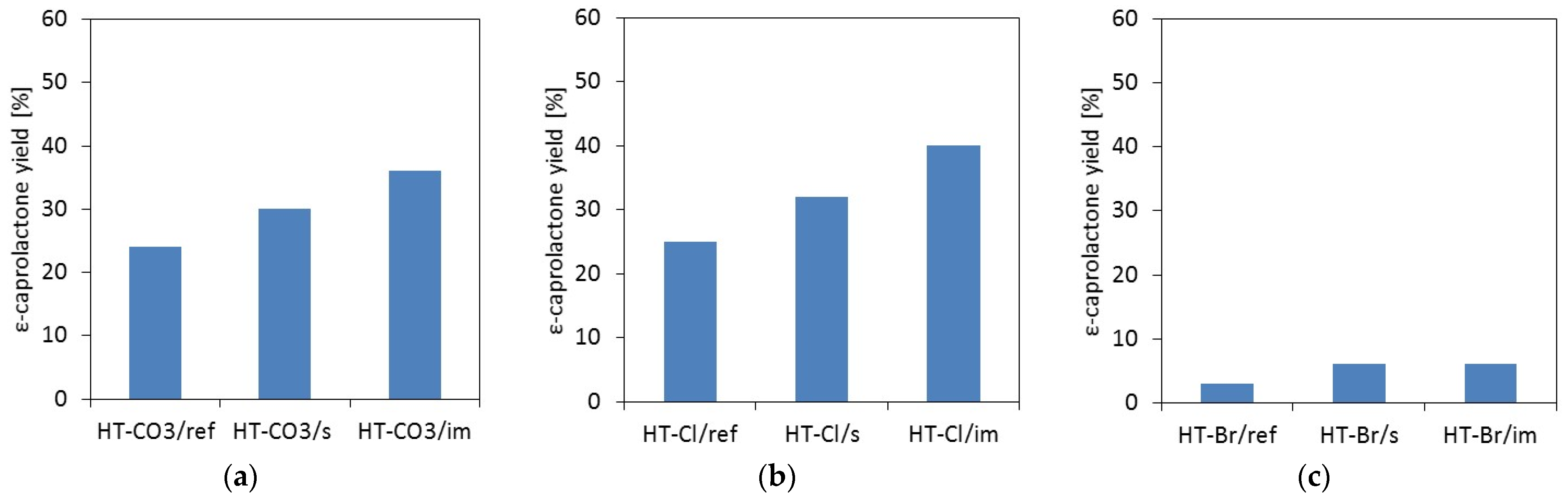
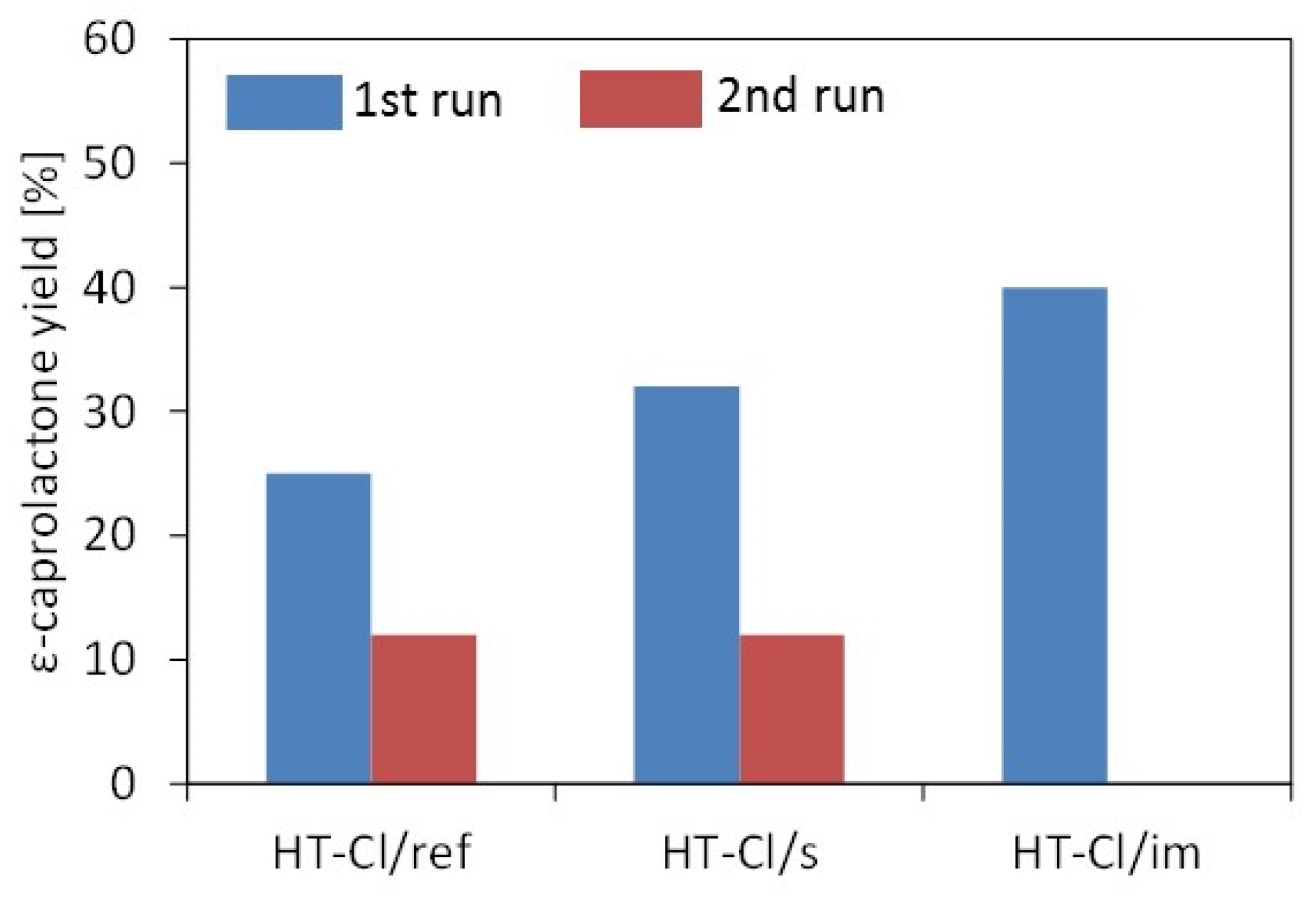
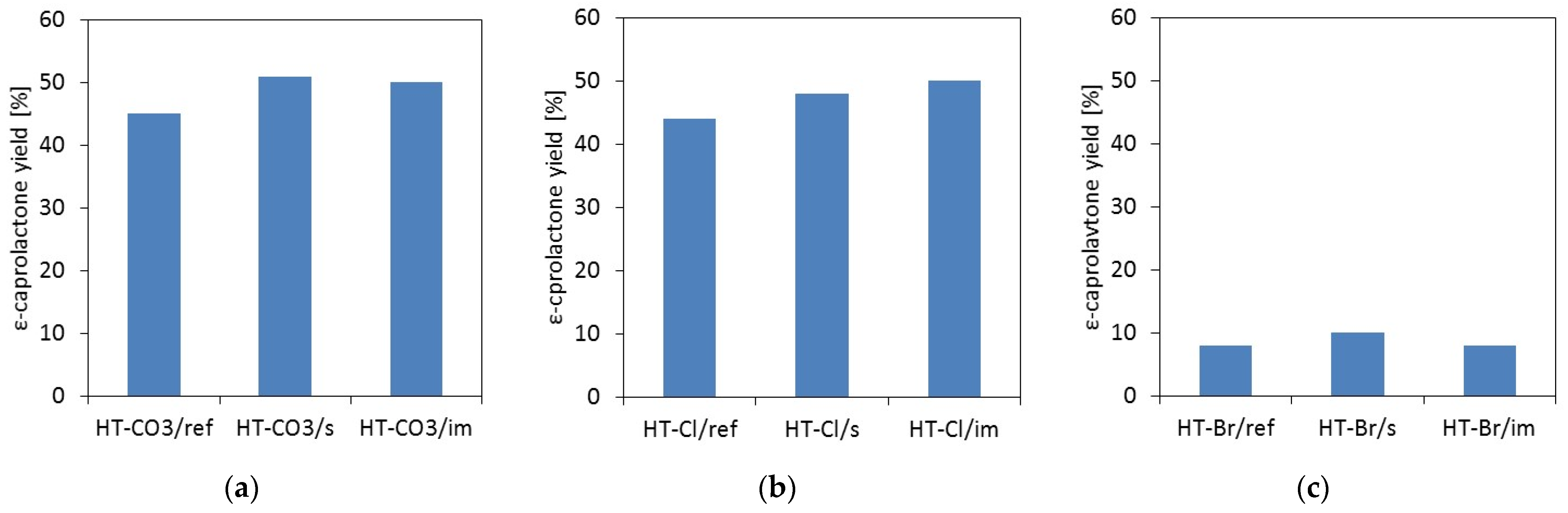
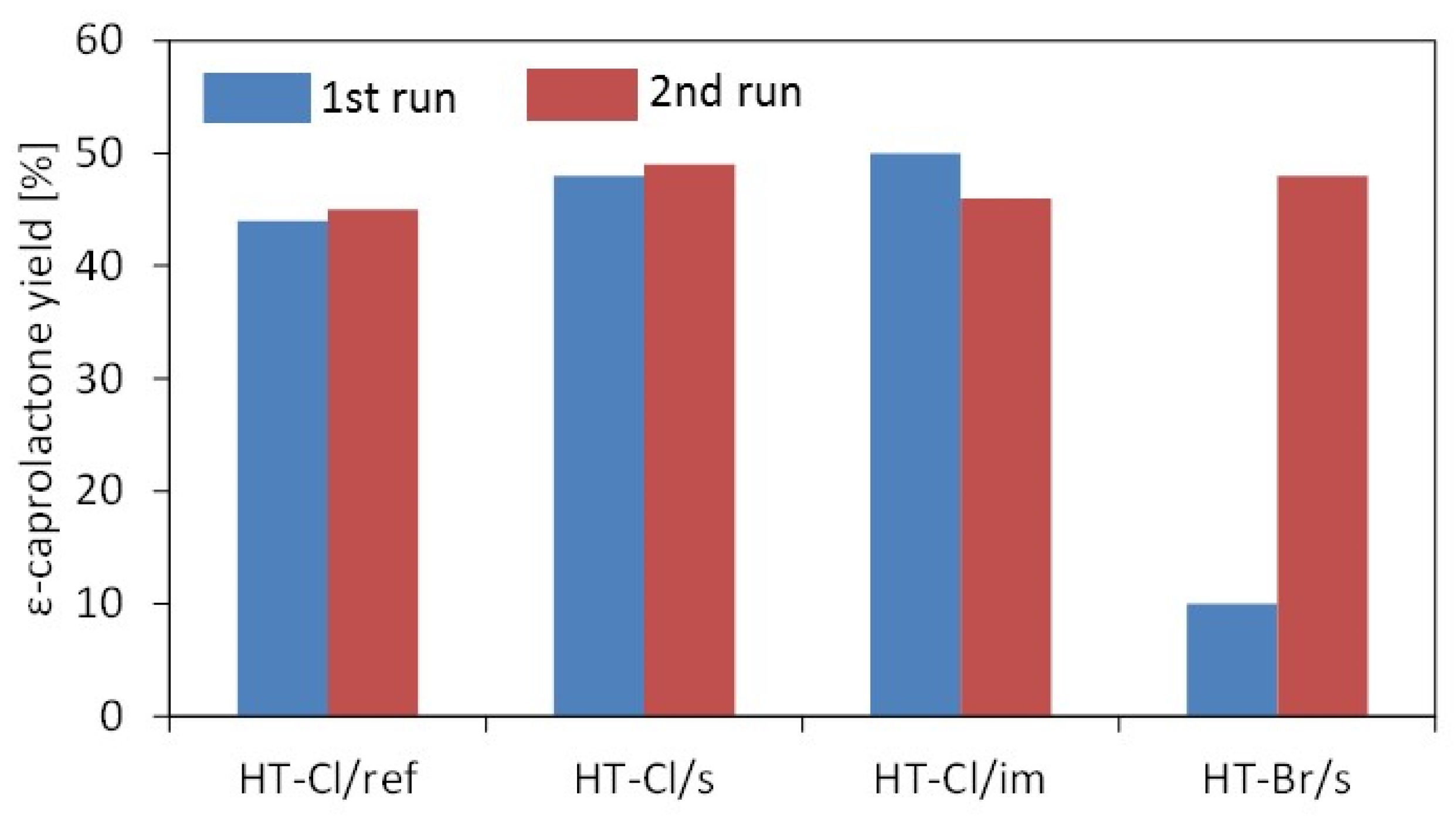
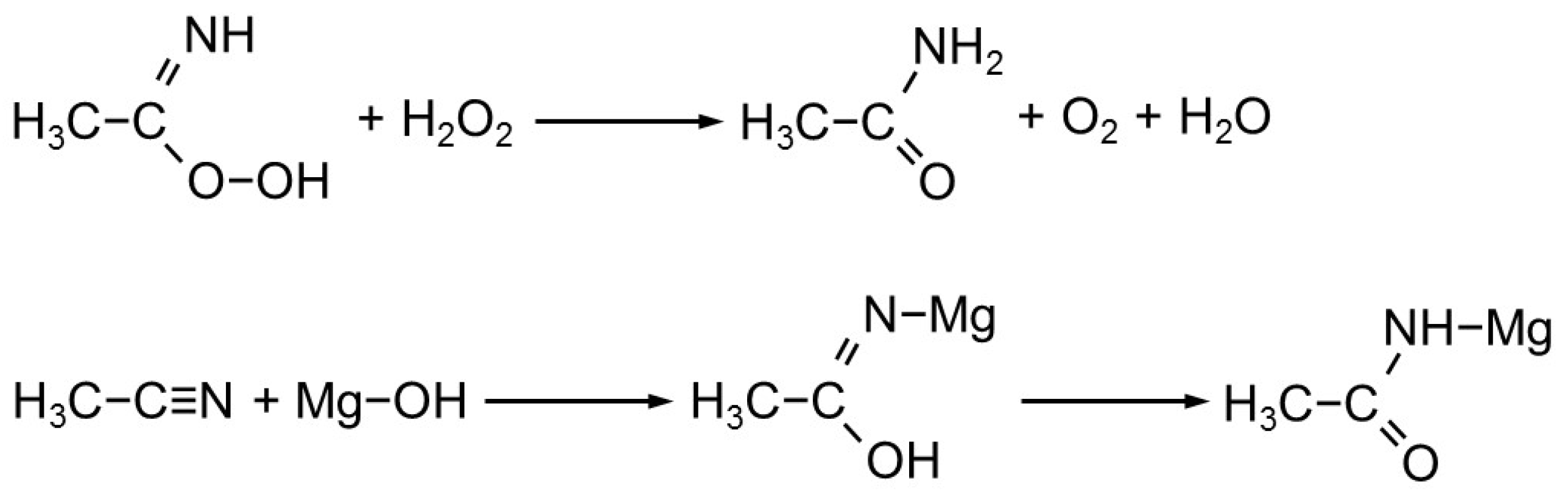
2.2. Catalytic Testing
3. Experimental Section
3.1. Materials
3.2. Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhitova, E.S.; Krivovichev, S.V.; Pekov, I.V.; Greenwell, H.C. Crystal chemistry of natural layered double hydroxides. 5. Single-crystal structure refinement of hydrotalcite, [Mg6Al2(OH)16](CO3)(H2O)4. Miner. Mag. 2019, 83, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Cat. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Catalysis by layered materials: A review. Micropor. Mesopor. Mater. 2008, 107, 3–15. [Google Scholar] [CrossRef]
- Othman, M.R.; Helwani, Z.; Martunus Fernando, W.J.N. Synthetic hydrotalcites from different routes and their application as catalysts and gas adsorbents: A review. Appl. Organomet. Chem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Roelofs, J.C.A.A.; Lensveld, D.J.; van Dillen, A.J.; de Jong, K.P. On the Structure of Activated Hydrotalcites as Solid Base Catalysts for Liquid-Phase Aldol Condensation. J. Catal. 2001, 203, 184–191. [Google Scholar] [CrossRef]
- Climent, M.; Corma, A.; Iborra, S.; Epping, K.; Velty, A. Increasing the basicity and catalytic activity of hydrotalcites by different synthesis procedures. J. Catal. 2004, 225, 316–326. [Google Scholar] [CrossRef]
- Abelló, S.; Medina, F.; Tichit, D.; Pérez-Ramírez, J.; Groen, J.C.; Sueiras, J.E.; Salagre, P.; Cesteros, Y. Aldol Condensations Over Reconstructed Mg-Al Hydrotalcites: Structure-Activity Relationships Related to the Rehydration Method. Chem. Eur. J. 2004, 11, 728–739. [Google Scholar] [CrossRef]
- Winter, F.; van Dillen, A.J.; de Jong, K.P. Supported hydrotalcites as highly active solid base catalysts. Chem. Commun. 2005, 3977–3979. [Google Scholar] [CrossRef]
- Kuśtrowski, P.; Sułkowska, D.; Chmielarz, L.; Dziembaj, R. Aldol condensation of citral and acetone over mesoporous catalysts obtained by thermal and chemical activation of magnesium–aluminum hydrotalcite-like precursors. Appl. Catal. A Gen. 2006, 302, 317–324. [Google Scholar] [CrossRef]
- Helwani, Z.; Othman, M.R.; Aziz, N.; Kim, J.; Fernando, W.J.N. Solid heterogeneous catalysts for transesterification of triglycerides with methanol: A review. Appl. Catal. A Gen. 2009, 363, 1–10. [Google Scholar] [CrossRef]
- Jothiramalingam, R.; Wang, M.K. Review of Recent Developments in Solid Acid, Base, and Enzyme Catalysts (Heterogeneous) for Biodiesel Production via Transesterification. Ind. Eng. Chem. Res. 2009, 48, 6162–6172. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, Tuning, and Exploiting the Basicity of Hydrotalcites for Applications in Heterogeneous Catalysis. Chem. Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef]
- Lee, G.; Jeong, Y.; Takagaki, A.; Jung, J.C. Sonication assisted rehydration of hydrotalcite catalyst for isomerization of glucose to fructose. J. Mol. Catal. A-Chem. 2014, 393, 289–295. [Google Scholar] [CrossRef]
- Delidovich, I.; Palkovits, R. Structure–performance correlations of Mg-Al hydrotalcite catalysts for the isomerization of glucose into fructose. J. Catal. 2015, 327, 1–9. [Google Scholar] [CrossRef]
- Kikhtyanin, O.; Hora, L.; Kubička, D. Unprecedented selectivities in aldol condensation over Mg-Al hydrotalcite in a fixed bed reactor setup. Catal. Commun. 2015, 58, 89–92. [Google Scholar] [CrossRef]
- Wang, Y.T.; Fang, Z.; Zhang, F.; Xue, B.J. One-step production of biodiesel from oils with high acid value by activated Mg-Al hydrotalcite nanoparticles. Bioresour. Technol. 2015, 193, 84–89. [Google Scholar] [CrossRef]
- Landeros, J.M.; Juaristi, E. Mechanochemical Synthesis of Dipeptides Using Mg-Al Hydrotalcite as Activating Agent under Solvent-Free Reaction Conditions. Eur. J. Org. Chem. 2017, 2017, 687–694. [Google Scholar] [CrossRef]
- Hincapié, G.; López, D.; Moreno, A. Infrared analysis of methanol adsorption on mixed oxides derived from Mg/Al hydrotalcite catalysts for transesterification reactions. Catal. Today 2018, 302, 277–285. [Google Scholar] [CrossRef]
- Wang, H.; Liu, W.; Wang, Y.; Tao, N.; Cai, H.; Liu, J.; Lv, J. Mg-Al Mixed Oxide Derived from Hydrotalcites Prepared by Solvent-free Method: A Stable Acid-base Bifunctional Catalyst for Continuous-Flow Transesterification of Dimethyl carbonate and Ethanol. Ind. Eng. Chem. Res. 2020, 59, 5591–5600. [Google Scholar] [CrossRef]
- Hussain, S.; Velisoju, V.K.; Rajan, N.P.; Kumar, B.P.; Chary, K.V.R. Synthesis of γ-Valerolactone from Levulinic Acid and Formic Acid over Mg-Al Hydrotalcite Like Compound. Chem. Sel. 2018, 3, 6186–6194. [Google Scholar] [CrossRef]
- Yabushita, M.; Shibayama, N.; Nakajima, K.; Fukuoka, A. Selective Glucose-to-Fructose Isomerization in Ethanol Catalyzed by Hydrotalcites. ACS Catal. 2019, 3, 2101–2109. [Google Scholar] [CrossRef]
- An, S.; Kwon, D.; Cho, J.; Jung, J.C. Effect of the Solvent on the Basic Properties of Mg-Al Hydrotalcite Catalysts for Glucose Isomerization. Catalysts 2020, 10, 1236. [Google Scholar] [CrossRef]
- Kaneda, K.; Ueno, S. Development of Hydrotalcite Catalysts in Heterogeneous Baeyer-Villiger Oxidation. ACS Symp. Ser. 1996, 638, 300–318. [Google Scholar]
- Pillai, U. Sn-exchanged hydrotalcites as catalysts for clean and selective Baeyer–Villiger oxidation of ketones using hydrogen peroxide. J. Mol. Catal. A-Chem. 2003, 191, 93–100. [Google Scholar] [CrossRef]
- Kawabata, T.; Fujisaki, N.; Shishido, T.; Nomura, K.; Sano, T.; Takehira, K. Improved Fe/Mg-Al hydrotalcite catalyst for Baeyer–Villiger oxidation of ketones with molecular oxygen and benzaldehyde. J. Mol. Catal. A-Chem. 2006, 253, 279–289. [Google Scholar] [CrossRef]
- Llamas, R.; Jiménez-Sanchidrián, C.; Ruiz, J.R. Heterogeneous Baeyer–Villiger oxidation of ketones with H2O2/nitrile, using Mg/Al hydrotalcite as catalyst. Tetrahedron 2007, 63, 1435–1439. [Google Scholar] [CrossRef]
- Jiménez-Sanchidrián, C.; Ruiz, J.R. The Baeyer–Villiger reaction on heterogeneous catalysts. Tetrahedron 2008, 64, 2011–2026. [Google Scholar] [CrossRef]
- Chen, C.; Peng, J.; Li, B.; Wang, L. The Catalytic Baeyer–Villiger Oxidation of Cyclohexanone to ε-Caprolactone over Stibium-containing Hydrotalcite. Catal. Lett. 2009, 131, 618–623. [Google Scholar] [CrossRef]
- Kaneda, K.; Mizugaki, T. Design of High-Performance Heterogeneous Catalysts Using Hydrotalcite for Selective Organic Transformations. Green Chem. 2019, 21, 1361–1389. [Google Scholar] [CrossRef]
- Olszówka, J.; Karcz, R.; Napruszewska, B.D.; Duraczyńska, D.; Gaweł, A.; Bahranowski, K.; Serwicka, E.M. Baeyer-Villiger oxidation of cyclohexanone with H2O2/acetonitrile over hydrotalcite-like catalysts: Effect of Mg/Al ratio on the ε-caprolactone yield. Catal. Commun. 2017, 100, 196–201. [Google Scholar] [CrossRef]
- Olszówka, J.E.; Karcz, R.; Napruszewska, B.D.; Michalik-Zym, A.; Duraczyńska, D.; Kryściak-Czerwenka, J.; Niecikowska, A.; Bahranowski, K.; Serwicka, E.M. Effect of Mg-Al hydrotalcite crystallinity on catalytic Baeyer-Villiger oxidation of cyclohexanone with H2O2/acetonitrile. Catal. Commun. 2018, 107, 48–52. [Google Scholar] [CrossRef]
- Olszówka, J.E.; Karcz, R.; Michalik-Zym, A.; Napruszewska, B.D.; Bielańska, E.; Kryściak-Czerwenka, J.; Socha, R.P.; Nattich-Rak, M.; Krzan, M.; Klimek, A.; et al. Effect of grinding on the physico-chemical properties of Mg-Al hydrotalcite and its performance as a catalyst for Baeyer-Villiger oxidation of cyclohexanone. Catal. Today 2019, 333, 147–153. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Progr. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Suwantong, O. Biomedical applications of electrospun polycaprolactone fiber mats. Polym. Adv. Technol. 2016, 27, 1264–1273. [Google Scholar] [CrossRef]
- Bellezza, F.; Cipiciani, A.; Costantino, U.; Nocchetti, M.; Posati, T. Hydrotalcite-Like Nanocrystals from Water-in-Oil Microemulsions. Eur. J. Inorg. Chem. 2009, 2603–2611. [Google Scholar] [CrossRef]
- Holgado, P.H.; Holgado, M.J.; San Román, M.S.; Rives, V. Effect of surfactants on the properties of hydrotalcites prepared by the reverse micelle method. Mater. Chem. Phys. 2015, 151, 140–148. [Google Scholar] [CrossRef]
- Napruszewska, B.D.; Michalik-Zym, A.; Dula, R.; Bielańska, E.; Rojek, W.; Machej, T.; Socha, R.P.; Lityńska-Dobrzyńska, L.; Bahranowski, K.; Serwicka, E.M. Composites derived from exfoliated Laponite and Mn-Al hydrotalcite prepared in inverse microemulsion: A new strategy for design of robust VOCs combustion catalysts. Appl. Catal. B-Environ. 2017, 211, 46–56. [Google Scholar] [CrossRef]
- Napruszewska, B.D.; Michalik-Zym, A.; Dula, R.; Duraczyńska, D.; Rojek, W.; Socha, R.P.; Lityńska-Dobrzyńska, L.; Bahranowski, K.; Serwicka, E.M. VOCs combustion catalysts based on composites of exfoliated organo-Laponite and multimetallic (Mn, Al, Zr, Ce) hydrotalcites prepared by inverse microemulsion. Catal. Today 2019, 333, 183–189. [Google Scholar] [CrossRef]
- Michalik, A.; Napruszewska, B.D.; Walczyk, A.; Kryściak-Czerwenka, J.; Duraczyńska, D.; Serwicka, E.M. Synthesis of Nanocrystalline Mg-Al Hydrotalcites in the Presence of Starch—the Effect on Structure and Composition. Materials 2020, 13, 602. [Google Scholar] [CrossRef] [Green Version]
- Miyata, S. Anion-Exchange Properties of Hydrotalcite-Like Compounds. Clays Clay Miner. 1983, 31, 305–311. [Google Scholar] [CrossRef]
- Solin, S.A.; Hines, D.; Yun, S.K.; Pinnavaia, T.J.; Thorpe, M.F. Layer rigidity in 2D disordered Ni-Al layer double hydroxides. J. Non-Cryst. Solids 1995, 182, 212–220. [Google Scholar] [CrossRef]
- Roobottom, H.K.; Jenkins, H.D.B.; Passmore, J.; Glasser, L. Thermochemical Radii of Complex Ions. J. Chem. Educ. 1999, 76, 1570–1573. [Google Scholar] [CrossRef]
- Olszówka, J.E.; Karcz, R.; Bielańska, E.; Kryściak-Czerwenka, J.; Napruszewska, B.D.; Sulikowski, B.; Socha, R.P.; Gaweł, A.; Bahranowski, K.; Olejniczak, Z.; et al. New insight into the preferred valency of interlayer anions in hydrotalcite-like compounds: The effect of Mg/Al ratio. Appl. Clay Sci. 2018, 155, 84–94. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared and Raman Spectroscopic Studies of Layered Double Hydroxides (LDHs). In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; Nova Science Publishers: New York, NY, USA, 2001; Chapter 5; pp. 139–192. [Google Scholar]
- López, T.; Bosch, P.; Asomoza, M.; Gómez, R.; Ramos, E. DTA-TGA and FTIR spectroscopies of sol-gel hydrotalcites: Aluminum source effect on physicochemical properties. Mater. Lett. 1997, 31, 311–316. [Google Scholar] [CrossRef]
- Kagunya, W.; Baddour-Hadjean, R.; Kooli, F.; Jones, W. Vibrational modes in layered double hydroxides and their calcined derivatives. Chem. Phys. 1998, 236, 225–234. [Google Scholar] [CrossRef]
- Payne, G.B.; Deming, P.H.; Williams, P.H. Reactions of hydrogen peroxide. VII. Alkali-catalyzed Epoxidation and oxidation using a nitrile as co-reactant. J. Organomet. Chem. 1961, 26, 659–663. [Google Scholar] [CrossRef]
- Prihod’ko, R.; Sychev, M.; Kolomitsyn, I.; Stobbelaar, P.J.; Hensen, E.J.M.; van Santen, R.A. Layered double hydroxides as catalysts for aromatic nitrile hydrolysis. Micropor. Mesopor. Mat. 2002, 56, 241–255. [Google Scholar] [CrossRef]
- Jiménez-Sanchidrián, C.; Hidalgo, J.M.; Llamas, R.; Ruiz, J.R. Baeyer–Villiger oxidation of cyclohexanone with hydrogen peroxide/benzonitrile over hydrotalcites as catalysts. Appl. Catal. A-Gen. 2006, 312, 86–94. [Google Scholar] [CrossRef]
- Bray, W.C.; Livingston, R.S. The catalytic decomposition of hydrogen peroxide in a bromine-bromide solution, and a study of the steady state. J. Am. Chem. Soc. 1923, 45, 1251–1271. [Google Scholar] [CrossRef]
- Karcz, R.; Olszówka, J.E.; Napruszewska, B.D.; Kryściak-Czerwenka, J.; Serwicka, E.M.; Klimek, A.; Bahranowski, K. Combined H2O2/nitrile/bicarbonate system for catalytic Baeyer-Villiger oxidation of cyclohexanone to ε-caprolactone over Mg Al hydrotalcite catalysts. Catal. Commun. 2019, 132, 105821. [Google Scholar] [CrossRef]
- Richardson, D.E.; Yao, H.; Frank, K.M.; Bennett, D.A. Equilibria, kinetics, and mechanism in the bicarbonate activation of hydrogen peroxide: Oxidation of sulfides by peroxymonocarbonate. J. Am. Chem. Soc. 2000, 122, 1729–1739. [Google Scholar] [CrossRef]
- Uno, T.; Machida, K.; Saito, Y. Out-of-plane vibrations of acetamide and partially N-deuterated acetamide. Spectrochim. Acta A-Mol. Spectrosc. 1971, 27, 833–844. [Google Scholar] [CrossRef]
- Koubowetz, F.; Latzel, J.; Noller, H. Adsorption of acetonitrile on magnesia, an IR and TPD study. J. Coll. Interface Sci. 1980, 74, 322–330. [Google Scholar] [CrossRef]
- Larrubia, M.A.; Ramis, G.; Busca, G. An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3-TiO2 SCR catalysts. Appl. Catal. B-Environ. 2001, 30, 101–110. [Google Scholar] [CrossRef]
- Lavalley, J.C. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Leitner, N.K.V.; Berger, P.; Legube, B. Oxidation of Amino Groups by Hydroxyl Radicals in Relation to the Oxidation Degree of the α-Carbon. Environ. Sci. Technol. 2002, 36, 3083–3089. [Google Scholar] [CrossRef]
- Olanrewaju, J.; Newalkar, B.L.; Mancino, C.; Komarneni, S. Simplified synthesis of nitrate form of layered double hydroxide. Mater. Lett. 2000, 45, 307–310. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mg/Al | Cl(Br)/Al | d003 [nm] | D003 [nm] | D110 [nm] |
|---|---|---|---|---|---|
| HT–CO3/im | 2.94 | - | 0.777 | 5.2 | 11.7 |
| HT–CO3/s | 3.08 | - | 0.775 | 7.1 | 13.5 |
| HT–CO3/ref | 2.86 | - | 0.774 | 13.9 | 14.4 |
| HT-Cl/im | 3.06 | 0.78 | 0.791 | 6.6 | 13.4 |
| HT-Cl/s | 3.00 | 0.84 (0.09) | 0.792 | 7.6 | 13.9 |
| HT-Cl/ref | 2.84 | 0.89 | 0.797 | 15.6 | 17.2 |
| HT–Br/im | 2.91 | 0.82 | 0.812 | 6.3 | 13.6 |
| HT–Br/s | 2.99 | 0.71 (0.03) | 0.809 | 7.0 | 13.1 |
| HT–Br/ref | 2.83 | 0.88 | 0.812 | 15.3 | 17.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karcz, R.; Napruszewska, B.D.; Michalik, A.; Kryściak-Czerwenka, J.; Duraczyńska, D.; Serwicka, E.M. Fine Crystalline Mg-Al Hydrotalcites as Catalysts for Baeyer-Villiger Oxidation of Cyclohexanone with H2O2. Catalysts 2021, 11, 1493. https://doi.org/10.3390/catal11121493
Karcz R, Napruszewska BD, Michalik A, Kryściak-Czerwenka J, Duraczyńska D, Serwicka EM. Fine Crystalline Mg-Al Hydrotalcites as Catalysts for Baeyer-Villiger Oxidation of Cyclohexanone with H2O2. Catalysts. 2021; 11(12):1493. https://doi.org/10.3390/catal11121493
Chicago/Turabian StyleKarcz, Robert, Bogna D. Napruszewska, Alicja Michalik, Joanna Kryściak-Czerwenka, Dorota Duraczyńska, and Ewa M. Serwicka. 2021. "Fine Crystalline Mg-Al Hydrotalcites as Catalysts for Baeyer-Villiger Oxidation of Cyclohexanone with H2O2" Catalysts 11, no. 12: 1493. https://doi.org/10.3390/catal11121493
APA StyleKarcz, R., Napruszewska, B. D., Michalik, A., Kryściak-Czerwenka, J., Duraczyńska, D., & Serwicka, E. M. (2021). Fine Crystalline Mg-Al Hydrotalcites as Catalysts for Baeyer-Villiger Oxidation of Cyclohexanone with H2O2. Catalysts, 11(12), 1493. https://doi.org/10.3390/catal11121493