
Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors




Abstract
:1. Introduction
2. Results
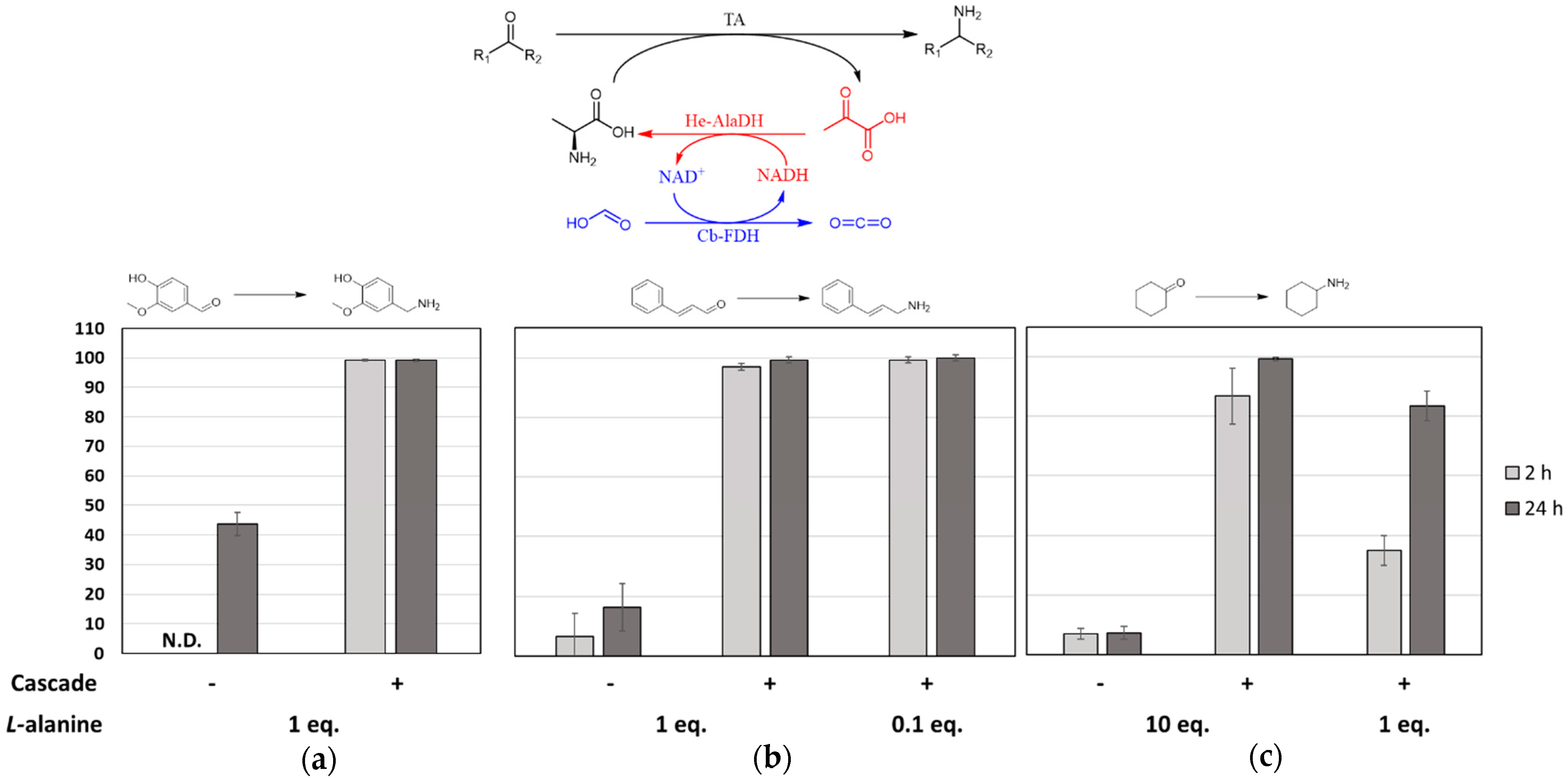
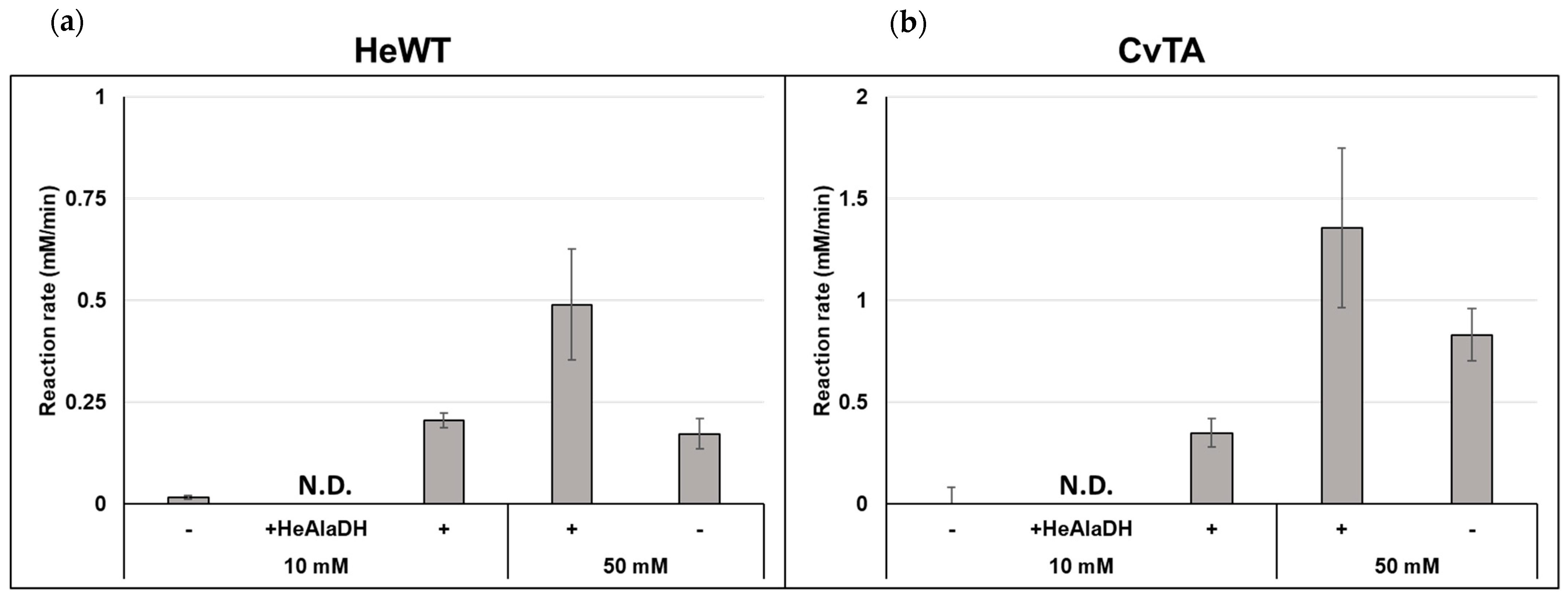
2.1. Batch Amination of Unfavourable Carbonyl Acceptors
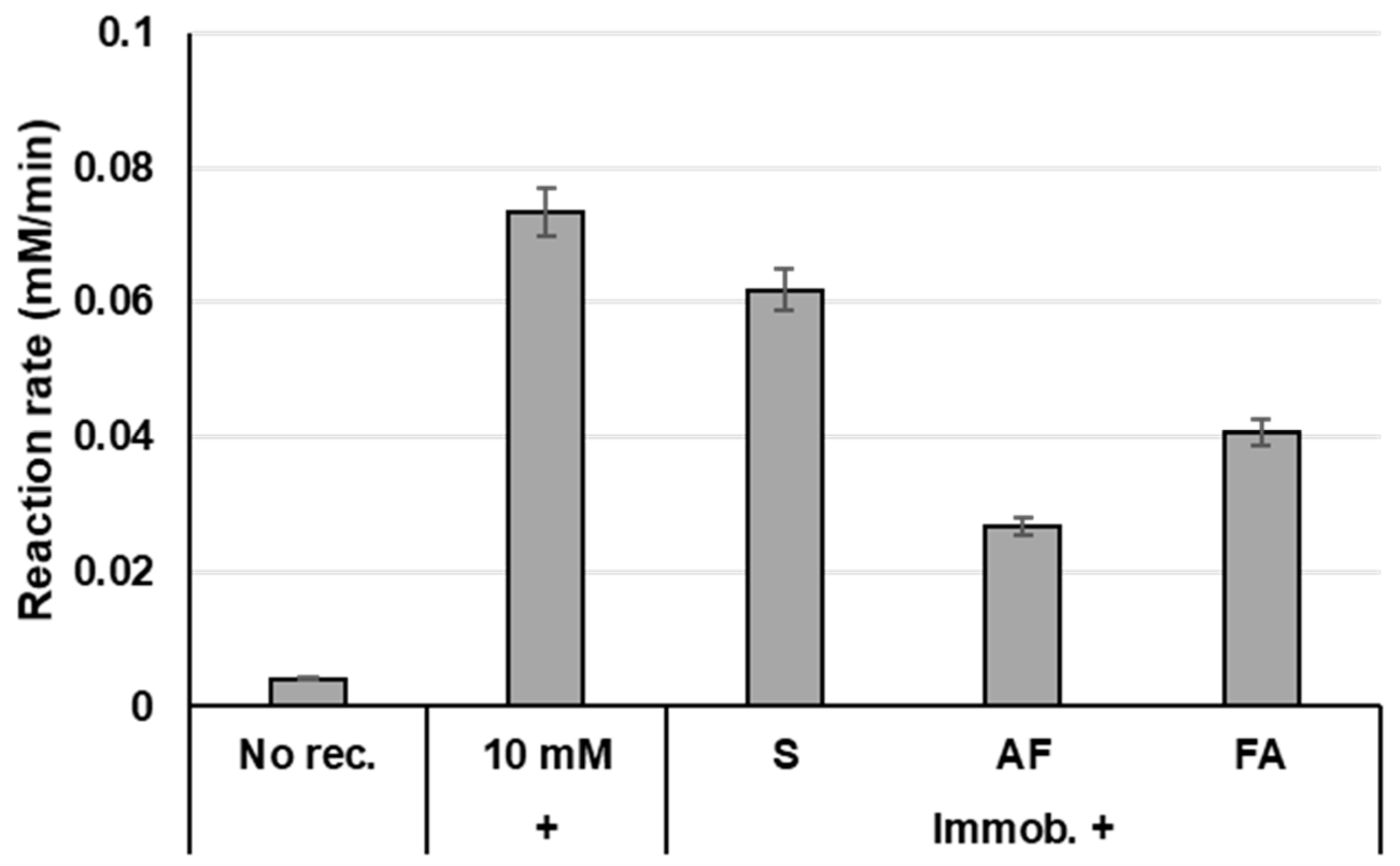
2.2. Enzyme Immobilisation
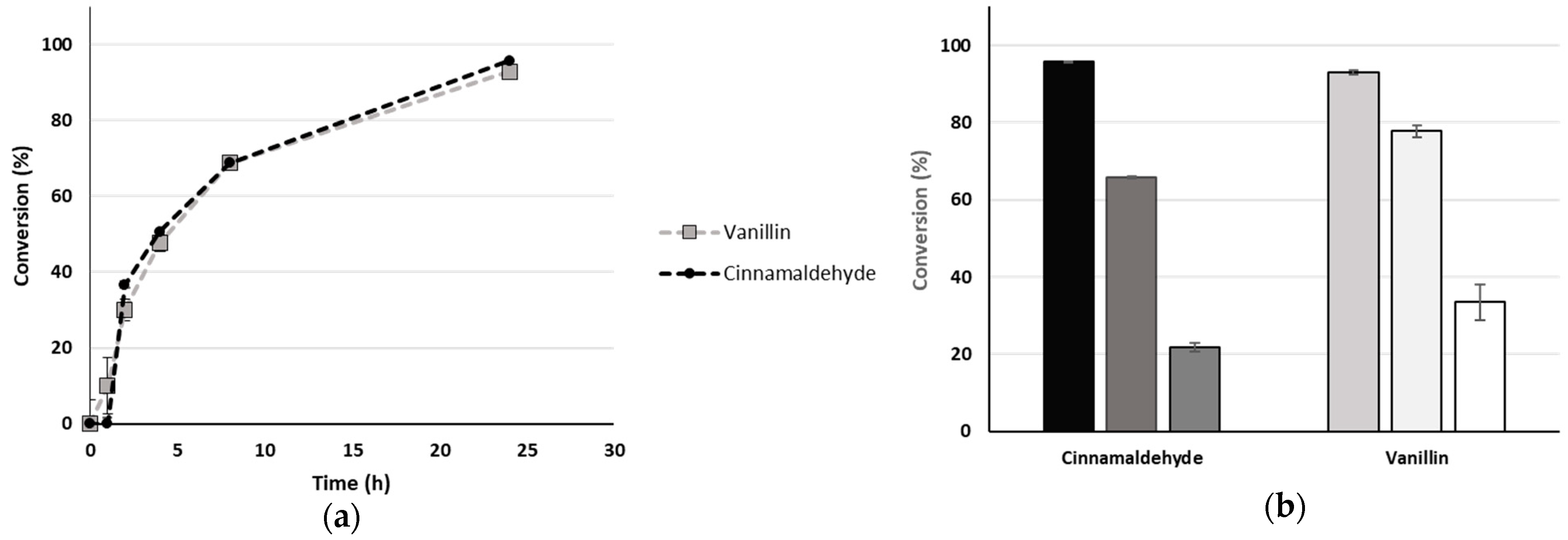
2.3. Scale Up: Continuous Flow and Reusability in Batch
3. Discussion
4. Materials and Methods
4.1. Materials, Strains, Vectors, and Culture Conditions
4.2. Protein Expression and Purification
4.3. Enzymatic Assay
4.4. SDS-PAGE
4.5. Batch Reactions
4.6. Immobilisation of HeAlaDH into Epoxy Functionalised Supports
4.7. Preparation of Dextran-Aldehyde
4.8. Post Immobilisation Coating of Immobilised HeAlaDH
4.9. Co-Immobilisation of HeAlaDH and CbFDH
4.10. Flow Reactions
4.11. Dialysis Assisted Reaction
4.12. HPLC Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gröger, H. Biocatalytic concepts for synthesizing amine bulk chemicals: Recent approaches towards linear and cyclic aliphatic primary amines and ω-substituted derivatives thereof. Appl. Microbiol. Biotechnol. 2019, 103, 83–95. [Google Scholar] [CrossRef]
- Pelckmans, M.; Renders, T.; Van De Vyver, S.; Sels, B.F. Bio-based amines through sustainable heterogeneous catalysis. Green Chem. 2017, 19, 5303–5331. [Google Scholar] [CrossRef]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Galman, J.L.; Slabu, I.; Weise, N.J.; Iglesias, C.; Parmeggiani, F.; Lloyd, R.C.; Turner, N.J. Biocatalytic transamination with near-stoichiometric inexpensive amine donors mediated by bifunctional mono- and di-amine transaminases. Green Chem. 2017, 19, 361–366. [Google Scholar] [CrossRef]
- Cassimjee, K.E.; Manta, B.; Himo, F. A quantum chemical study of the ω-transaminase reaction mechanism. Org. Biomol. Chem. 2015, 13, 8453–8464. [Google Scholar] [CrossRef] [Green Version]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.R.; Shonnard, D.; Pannuri, S.; Kamat, S. Characterization of free and immobilized (S)-aminotransferase for acetophenone production. Appl. Microbiol. Biotechnol. 2007, 76, 843–851. [Google Scholar] [CrossRef]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Börner, T.; Rämisch, S.; Bartsch, S.; Vogel, A.; Adlercreutz, P.; Grey, C. Three in One: Temperature, Solvent and Catalytic Stability by Engineering the Cofactor-Binding Element of Amine Transaminase. ChemBioChem 2017, 18, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Roura Padrosa, D.; Alaux, R.; Smith, P.; Dreveny, I.; López-Gallego, F.; Paradisi, F. Enhancing PLP-Binding Capacity of Class-III ω-Transaminase by Single Residue Substitution. Front. Bioeng. Biotechnol. 2019, 7, 282. [Google Scholar] [CrossRef] [Green Version]
- Börner, T.; Rämisch, S.; Reddem, E.R.; Bartsch, S.; Vogel, A.; Thunnissen, A.M.W.H.; Adlercreutz, P.; Grey, C. Explaining Operational Instability of Amine Transaminases: Substrate-Induced Inactivation Mechanism and Influence of Quaternary Structure on Enzyme-Cofactor Intermediate Stability. ACS Catal. 2017, 7, 1259–1269. [Google Scholar] [CrossRef]
- Kelefiotis-Stratidakis, P.; Tyrikos-Ergas, T.; Pavlidis, I.V. The challenge of using isopropylamine as an amine donor in transaminase catalysed reactions. Org. Biomol. Chem. 2019, 17, 1634–1642. [Google Scholar] [CrossRef]
- Huisman, G.W.; Collier, S.J. On the development of new biocatalytic processes for practical pharmaceutical synthesis. Curr. Opin. Chem. Biol. 2013, 17, 284–292. [Google Scholar] [CrossRef]
- Leipold, L.; Dobrijevic, D.; Jeffries, J.W.E.; Bawn, M.; Moody, T.S.; Ward, J.M.; Hailes, H.C. The identification and use of robust transaminases from a domestic drain metagenome. Green Chem. 2019, 21, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Green, A.P.; Turner, N.J.; Reilly, E.O. Chiral Amine Synthesis Using w -Transaminases: An Amine Donor that Displaces Equilibria and Enables High-Throughput Screening. Angewandte 2014, 10714–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, D.; Ladkau, N.; Moody, T.S.; Ward, J.M.; Hailes, H.C. A rapid, sensitive colorimetric assay for the high-throughput screening of transaminases in liquid or solid-phase. Chem. Commun. 2015, 51, 17225–17228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slabu, I.; Galman, J.L.; Weise, N.J.; Lloyd, R.C.; Turner, N.J. Putrescine Transaminases for the Synthesis of Saturated Nitrogen Heterocycles from Polyamines. ChemCatChem 2016, 8, 1038–1042. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. Stereoselective amination of racemic sec-alcohols through sequential application of laccases and transaminases. Green Chem. 2017, 19, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Guebitz, G.M.; Rozzell, D.; Kroutil, W. Formal asymmetric biocatalytic reductive amination. Angew. Chem. Int. Ed. 2008, 47, 9337–9340. [Google Scholar] [CrossRef] [PubMed]
- Richter, N.; Farnberger, J.E.; Pressnitz, D.; Lechner, H.; Zepeck, F.; Kroutil, W. A system for ω-transaminase mediated (R)-amination using l-alanine as an amine donor. Green Chem. 2015, 17, 2952–2958. [Google Scholar] [CrossRef] [Green Version]
- Klatte, S.; Wendisch, V.F. Role of L-alanine for redox self-sufficient amination of alcohols. Microb. Cell Fact. 2015, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω-transaminases. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar] [CrossRef]
- Contente, M.L.; Dall’Oglio, F.; Tamborini, L.; Molinari, F.; Paradisi, F. Highly Efficient Oxidation of Amines to Aldehydes with Flow-based Biocatalysis. ChemCatChem 2017, 9, 3843–3848. [Google Scholar] [CrossRef]
- Abaházi, E.; Sátorhelyi, P.; Erdélyi, B.; Vértessy, B.G.; Land, H.; Paizs, C.; Berglund, P.; Poppe, L. Covalently immobilized Trp60Cys mutant of Ω-transaminase from Chromobacterium violaceum for kinetic resolution of racemic amines in batch and continuous-flow modes. Biochem. Eng. J. 2018, 132, 270–278. [Google Scholar] [CrossRef]
- Planchestainer, M.; Contente, M.L.; Cassidy, J.; Molinari, F.; Tamborini, L.; Paradisi, F. Continuous flow biocatalysis: Production and in-line purification of amines by immobilised transaminase from Halomonas elongata. Green Chem. 2017, 19, 372–375. [Google Scholar] [CrossRef]
- Truppo, M.D.; Strotman, H.; Hughes, G. Development of an Immobilized Transaminase Capable of Operating in Organic Solvent. ChemCatChem 2012, 4, 1071–1074. [Google Scholar] [CrossRef]
- Agustian, J.; Kamaruddin, A.H.; Bhatia, S. Enzymatic membrane reactors: The determining factors in two separate phase operations. J. Chem. Technol. Biotechnol. 2011, 86, 1032–1048. [Google Scholar] [CrossRef]
- Possebom, G.; Nyari, N.L.D.; Zeni, J.; Steffens, J.; Rigo, E.; di Luccio, M. Esterification of fatty acids by Penicillium crustosum lipase in a membrane reactor. J. Sci. Food Agric. 2014, 94, 2905–2911. [Google Scholar] [CrossRef] [PubMed]
- Kaulmann, U.; Smithies, K.; Smith, M.E.B.; Hailes, H.C.; Ward, J.M. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzym. Microb. Technol. 2007, 41, 628–637. [Google Scholar] [CrossRef]
- Cerioli, L.; Planchestainer, M.; Cassidy, J.; Tessaro, D.; Paradisi, F. Characterization of a novel amine transaminase from Halomonas elongata. J. Mol. Catal. B Enzym. 2015, 120, 141–150. [Google Scholar] [CrossRef]
- Schütte, H.; Flossdorf, J.; Sahm, H.; Kula, M.-R. Purification and Properties of Formaldehyde Dehydrogenase and Formate Dehydrogenase from Candida boidinii. Eur. J. Biochem. 1976, 62, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Mateos, A.I.; Contente, M.L.; Velasco-Lozano, S.; Paradisi, F.; López-Gallego, F. Self-Sufficient Flow-Biocatalysis by Coimmobilization of Pyridoxal 5′-Phosphate and ω-Transaminases onto Porous Carriers. ACS Sustain. Chem. Eng. 2018, 6, 13151–13159. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Wilson, L.; Ferrarotti, S.A.; Fernandez-Lafuente, R.; Guisan, J.M.; Mateo, C. Evaluation of different immobilization strategies to prepare an industrial biocatalyst of formate dehydrogenase from Candida boidinii. Enzym. Microb. Technol. 2007, 40, 540–546. [Google Scholar] [CrossRef]
- Mateo, C.; Grazú, V.; Pessela, B.C.C.; Montes, T.; Palomo, J.M.; Torres, R.; López-Gallego, F.; Fernández-Lafuente, R.; Guisán, J.M. Advances in the design of new epoxy supports for enzyme immobilization–stabilization. Biochem. Soc. Trans. 2008, 35, 1593–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolivar, J.M.; Rocha-Martín, J.; Mateo, C.; Guisan, J.M. Stabilization of a highly active but unstable alcohol dehydrogenase from yeast using immobilization and post-immobilization techniques. Process Biochem. 2012, 47, 679–686. [Google Scholar] [CrossRef]
- Lopez-Gallego, F.; Betancor, L.; Hidalgo, A.; Dellamora-Ortiz, G.; Mateo, C.; Fernández-Lafuente, R.; Guisán, J.M. Stabilization of different alcohol oxidases via immobilization and post immobilization techniques. Enzym. Microb. Technol. 2007, 40, 278–284. [Google Scholar] [CrossRef]
- Trobo-Maseda, L.; Orrego, A.H.; Moreno-Pérez, S.; Fernández-Lorente, G.; Guisan, J.M.; Rocha-Martin, J. Stabilization of multimeric sucrose synthase from Acidithiobacillus caldus via immobilization and post-immobilization techniques for synthesis of UDP-glucose. Appl. Microbiol. Biotechnol. 2018, 102, 773–787. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterlling, R.; Berenguer-Murcia, Á.; Alcántara, A.R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Enzyme co-immobilization: Always the biocatalyst designers’ choice…or not? Biotechnol. Adv. 2020, 107584. [Google Scholar] [CrossRef]
- Zhang, J.; Li, F.; Wang, R.; Tan, X.; Hagedoorn, P.-L. Dialysis membrane enclosed laccase catalysis combines a controlled conversion rate and recyclability without enzyme immobilization. AMB Express 2020, 10, 19. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534. [Google Scholar] [CrossRef]
- Andersson, M.M.; Hatti-Kaul, R. Protein stabilising effect of polyethyleneimine. J. Biotechnol. 1999, 72, 21–31. [Google Scholar] [CrossRef]
- Simpson, R.J. Stabilization of proteins for storage. Cold Spring Harb. Protoc. 2010, 5, pdb.top79. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Support | Retention Time | TA | TA + Rec |
| HFA403-S | 10 min | <5% | 22 ± 2 |
| 20 min | <5% | 50 ± 12 | |
| Ag-Ep | 10 min | 22 ± 5 | 29 ± 5 |
| 20 min | 23 ± 5 | 40 ± 5 | |
| Additive | Concentration (mg/mL) | Accumulated Yield (%) | Relative Improvement |
|---|---|---|---|
| Control | - | 52 | 1 |
| PEI25 | 2.5 | 53 | 1 |
| PEI60 | 48 | 0.9 | |
| PEI270 | 61 | 1.2 | |
| PEI750 | 51 | 1.0 | |
| PEG | 25 | 51 | 1.0 |
| 50 | 65 | 1.3 | |
| 100 | 57 | 1.1 | |
| Glycerol | 100 | 54 | 1.1 |
| 150 | 70 | 1.4 | |
| 200 | 72 | 1.4 | |
| Sucralose | 10 | 55 | 1.1 |
| 50 | 69 | 1.3 | |
| 100 | 57 | 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roura Padrosa, D.; Nisar, Z.; Paradisi, F. Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors. Catalysts 2021, 11, 520. https://doi.org/10.3390/catal11040520
Roura Padrosa D, Nisar Z, Paradisi F. Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors. Catalysts. 2021; 11(4):520. https://doi.org/10.3390/catal11040520
Chicago/Turabian StyleRoura Padrosa, David, Zoya Nisar, and Francesca Paradisi. 2021. "Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors" Catalysts 11, no. 4: 520. https://doi.org/10.3390/catal11040520
APA StyleRoura Padrosa, D., Nisar, Z., & Paradisi, F. (2021). Efficient Amino Donor Recycling in Amination Reactions: Development of a New Alanine Dehydrogenase in Continuous Flow and Dialysis Membrane Reactors. Catalysts, 11(4), 520. https://doi.org/10.3390/catal11040520