1. Introduction
The increased amount of anthropogenic CO
2 emissions since the beginning of the industrial era (starting around 1750) has significantly affected the natural biogeochemical carbon cycle. An analysis of the components of the anthropogenic “CO
2 budget”, as defined in Reports released by the Intergovernmental Panel on Climate Change (IPCC) [
1] and the Global Carbon Budget Project [
2] (GCB Project,
Table S1), show that, since 1750, global CO
2-emissions amounted to about 700 GtC compared to 400 GtC fixed by CO
2-natural sinks (
Table S1). Therefore, approximately 285 GtC has accumulated in the atmosphere causing an increase to the actual value of 407 ppm.
However, estimates of carbon fluxes between different compartments of terrestrial ecosystems (
Figure S1, Table S2, Supplementary Materials) evidence that, during the last six decades, oceans and land have adapted to such “CO
2 pressure”, increasing CO
2 natural sink [
2].There is a consensus that terrestrial carbon sink (S
LAND, see
Section S1) has constantly increased in the Northern Hemisphere largely due to the “CO
2-fertilization effect”. This effect accounts for higher rates of photosynthesis and, consequently, for increase of plant Gross Primary Production (GPP) due to rising atmospheric CO
2 concentration [
3,
4] strengthened by concomitant increased N-deposition [
5].
At the biochemical level Rubisco is the key enzyme in the Calvin cycle (see
Section S2), responsible for photosynthetic CO
2-fixation, and is considered the most abundant protein of the biosphere [
6]. Considering the extensive literature dealing with the rate of photosynthetic carbon assimilation [
7,
8], plant growth and biomass production [
9], the main focus of this review is to provide as complete a framework as possible of the mechanism of the Rubisco carboxylation reaction. Throughout the paper, particular attention is placed on the structure and functioning of the active site as well on the functioning of the protein “molecular machine” including its physical molecular properties underlying CO
2 vs. O
2 selection. An overview of stepwise progresses achieved in elucidation of the carboxylase reaction mechanism since 2008 is presented, including a very recent model proposed by the Gready’s Group in 2020. Finally, recent advances revealing the existence of strong biochemical links between the C- and N-fixation processes by plants will be reported.
The review includes a short summary of the literature dealing with the Rubisco protein structure, with the aim of providing information needed for the subsequent description of the reaction mechanism.
3. The Rubisco’s Secondary, Tertiary and Quaternary Structure
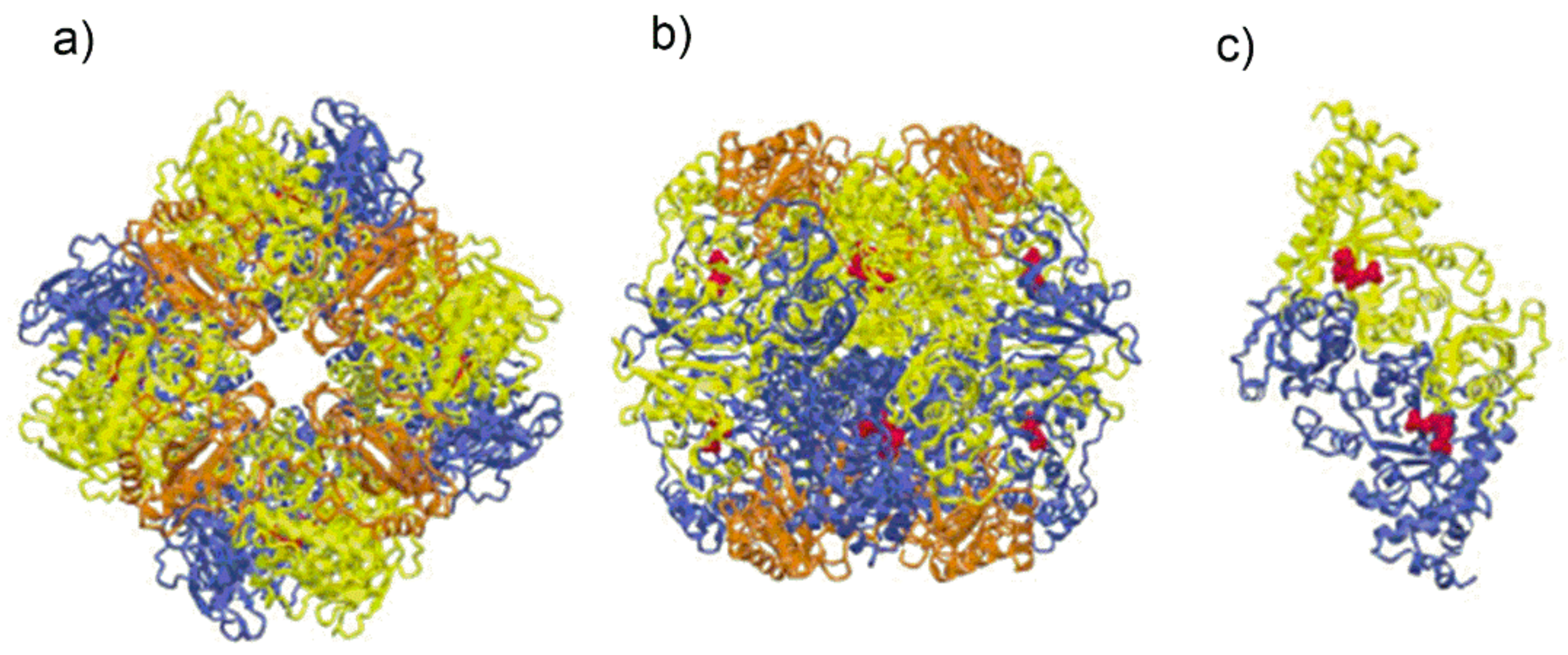
Due to the wide range of protein structures solved by X-ray crystallography and the availability of genomic and metabolomic data, four different Rubisco molecular isoforms have been identified in nature that essentially differ in the number of subunits and their arrangement within the macromolecular complex (
Figure 1) [
13].
Isoforms I and II, whose structural features are discussed in this section, play a central role in carbon fixation via the CBB cycle. The phylogenetic distribution of form I Rubisco encompasses all higher plants, red and brown algae, cyanobacteria and most chemoautotrophic bacteria while form II is found in photosynthetic bacteria and several methanogenic Archaea.
Mentioning briefly the remaining isoform of Rubisco proteins, form III Rubisco is found exclusively in Archaea and isoform IV (also named Rubisco-like protein, RLP) is found in both Bacteria and Archaea where it may have a role in sulfur metabolism.
This section analyzes the structural features of Rubisco proteins to provide a molecular basis for subsequent description of the carboxylase reaction mechanism. A detailed overview of the Rubisco’s structure and function can be found in the review published by Andersson and Backlund in 2008 covering structures deposited to the PDB (Protein Data Bank) archive over approximately a 20-year period (including Rubisco-like proteins) [
13]. Since 2008, several additional Rubisco structures have been solved by X-ray as, for example, the crystal structure of Rubisco from
Pisum sativum, [
14]
Galderia sulphuraria, [
15]
Rhodopseudomonas palustris [
16] and
Arabidopsis thaliana [
17].
The pioneering work published by Smith et al., in 1987 [
18] describes Rubisco from
Nicotidiana tabacum (form I Rubisco) as a globular protein, barrel-shaped (“a keg 105 Å along the 4-fold axis and 132 Å in diameter at the widest point of the keg”), showing a L
8S
8 quaternary structure where L denotes the “large” catalytic subunit and S denotes the “small” subunit (L: 52.9 kDa; S: 14.8 kDa). The eight L subunits are arranged as a toroid around a
z-axis of four-fold symmetry. Along this axis a solvent channel transverses the molecular complex. The molecular symmetry of the L
8S
8 hexa-decamer of Rubisco from spinach is shown in
Figure 1a [
19].
Figure 1a is a view along the four-fold axis, while
Figure 1b shows the protein viewed along one of the two-fold axes. In
Figure 1b the two large subunits of the front-facing L
2 dimer are shown in yellow and blue color. The polypeptide chains of each L
2 dimer assume an antiparallel arrangement so that, the N-terminal domain of the yellow L subunit is close to the C-terminal domain of the blue L subunit.
Figure 1.
(
a) Schematic drawing of Form I Rubisco from spinach showing the spatial arrangement of the L
8S
8 hexa-decamer view along the four-fold axis. The L subunits appearing in the figure are represented in yellow and blue. The figure also evidences the S subunits (represented in orange) delimiting the solvent channel; (
b) schematic drawing of the Rubisco from spinach, view along one of the two-fold axes; (
c) Form II Rubisco from
Rhodospirillum rubrum showing the two-fold symmetry of the L2 dimer. The substrate mimic 2-carboxy-D-arabinitol 1,5-bisphosphate (2CABP), displayed as red spheres (Figure from ref. [
19], adapted and used by permission of Elsevier (copyright © 2003).
Figure 1.
(
a) Schematic drawing of Form I Rubisco from spinach showing the spatial arrangement of the L
8S
8 hexa-decamer view along the four-fold axis. The L subunits appearing in the figure are represented in yellow and blue. The figure also evidences the S subunits (represented in orange) delimiting the solvent channel; (
b) schematic drawing of the Rubisco from spinach, view along one of the two-fold axes; (
c) Form II Rubisco from
Rhodospirillum rubrum showing the two-fold symmetry of the L2 dimer. The substrate mimic 2-carboxy-D-arabinitol 1,5-bisphosphate (2CABP), displayed as red spheres (Figure from ref. [
19], adapted and used by permission of Elsevier (copyright © 2003).
Figure 1a,b also show the arrangement of the S subunits (represented in orange) clustered around the four-fold axis.
Figure 1c represents a schematic drawing of the quaternary structure of type II Rubisco from
Rhodospirillum rubrum consisting of a L
2 dimer [
19].
The secondary structure elements (α-helices, β-strands and loops) of the polypeptide chain of Rubisco from spinach are shown in the connectivity diagram reported in
Scheme S2 [
20]. Interestingly, despite differences in the primary structure of different Rubisco proteins, the secondary structure of L subunits is highly conserved in the phylogenetic tree of the enzyme including isoforms I–III and Rubisco-like proteins (form IV).
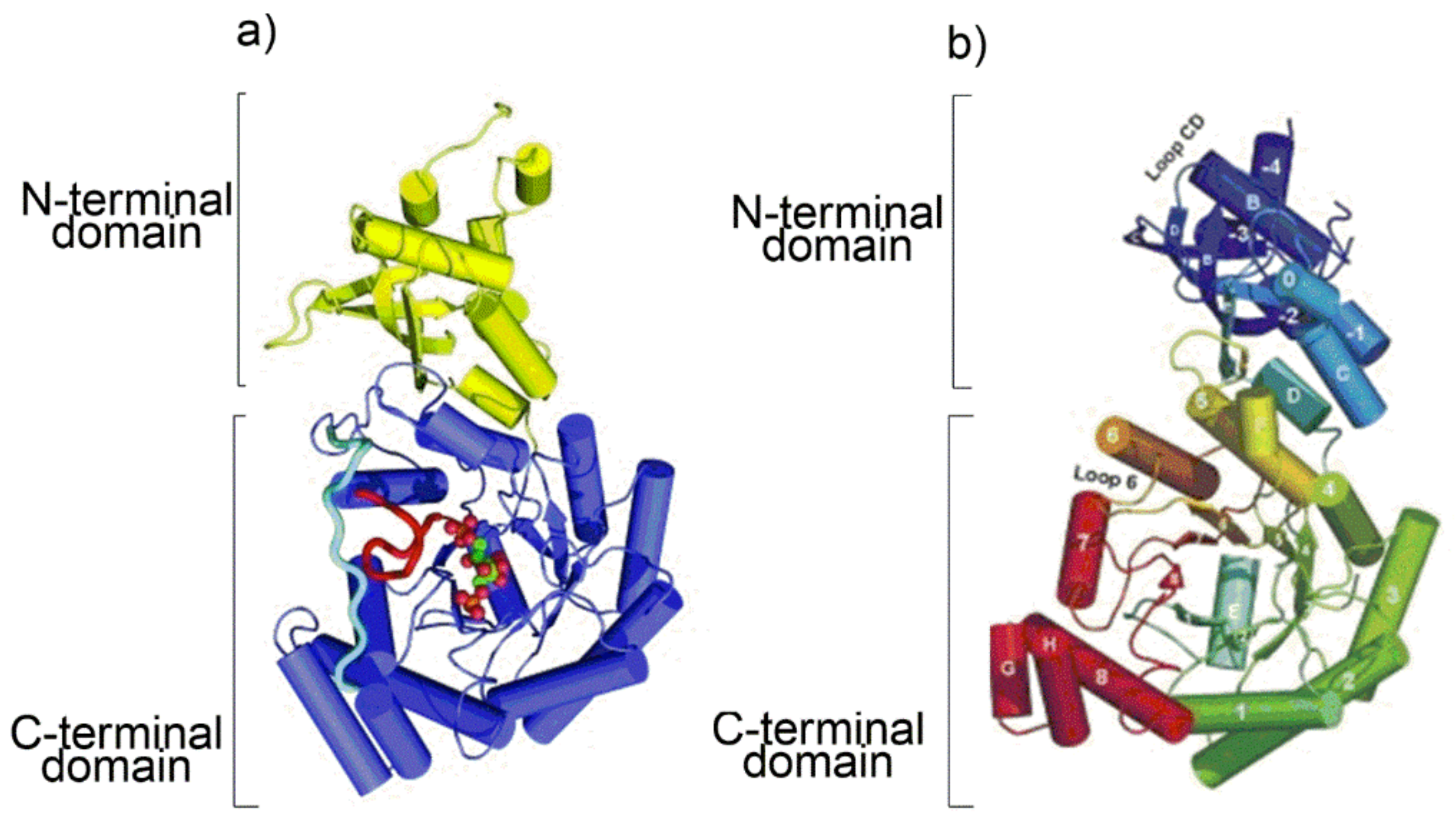
As the protein secondary structure dictates the subsequent tertiary fold, a cartoon representation of the tertiary folding of Rubisco from
Arabidopsis thaliana (form I Rubisco) and
Chlorobium tepidum (Rubisco-like protein) obtained from the X-ray crystal structure is shown in
Figure 2 [
17,
21].
At the C-terminal domain of each L subunit eight β-strands (from β1 to β8 in the topologic diagram shown in
Figure S2) separated by eight α-helices (from α1 to α8) build up a very common tertiary fold currently named “TIM barrel” (TIM: triose phosphate isomerase). Interestingly, although RLP do not catalyze the RuBP carboxylation/oxygenation reactions, they share with the Rubisco enzymes substantial similarity in secondary and tertiary structure, probably because of a common archaeal ancestor.
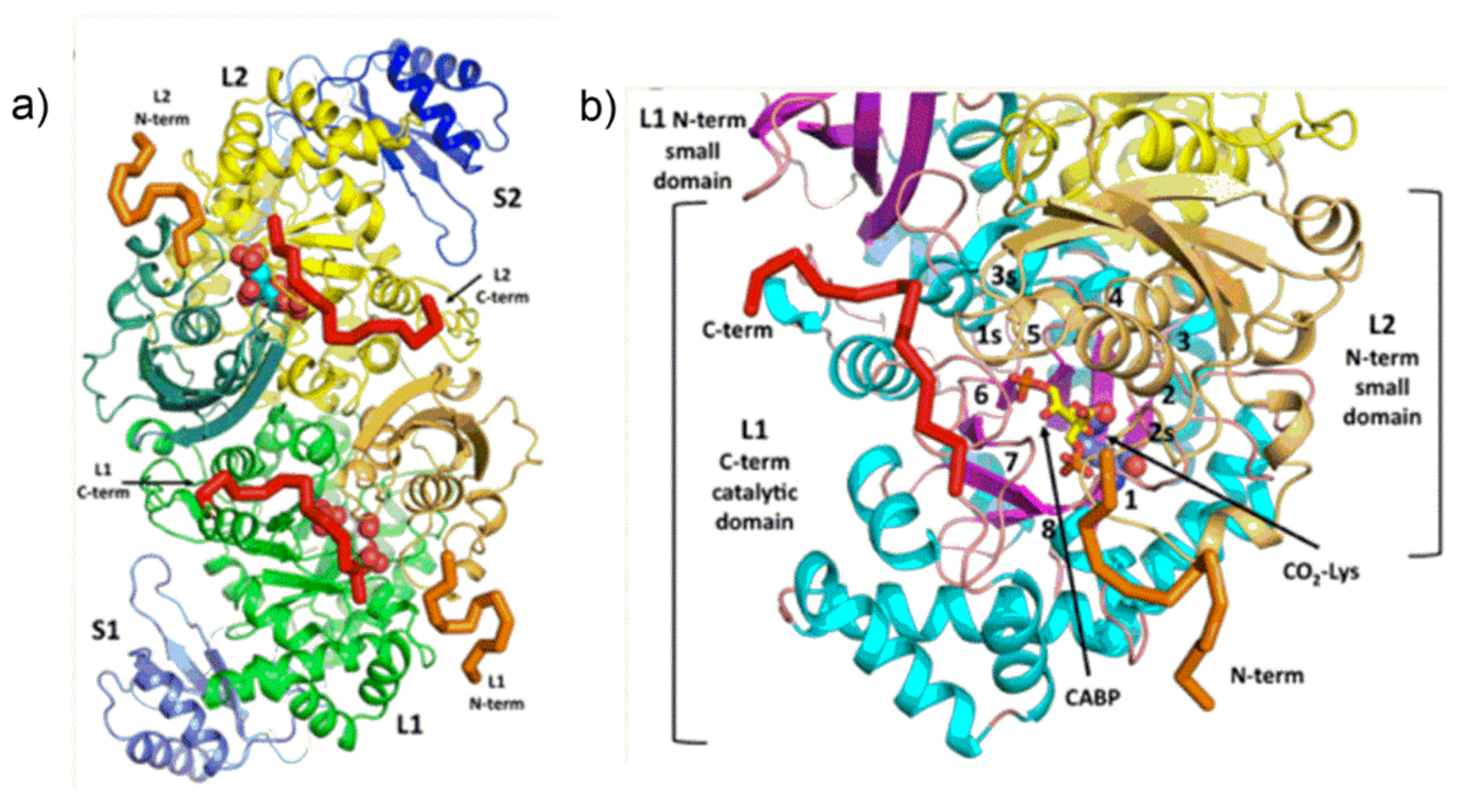
More details about the quaternary structure of the L
2S
2 unit of
Galdieria sulphuraria [
15] Rubisco are shown in
Figure 3. The L
2S
2 dimer is composed of two identical “head to tail” oriented L subunits viewed down the two-fold axis. The two S subunits are represented in blue.
As shown in
Figure 3a,b, each L subunit hosts an active site which is located at the mouth of the TIM barrel. The N-terminal domain of the anti-paralleled L subunit in the L
2 dimer “covers” the TIM barrel contributing to the active site of the enzyme.
The Rubisco Small Subunit
As the L subunit guests the enzyme active pocket, the S-subunit (ranging about 12–18 kDa among different Rubisco enzyme) is found only in form I Rubisco and is proposed to play both a structural and a functional role.
The primary structure for a series of S subunits from different organisms is reported in
Figure S2 including the sequence of the secondary structure elements shown in ribbon representation in
Figure S4 (orange ribbon) [
22]. The figure evidences location of the βA-βB loop while the secondary structure elements of the L subunit functionally related to the S subunit are evidenced in green and yellow color. The βA-βB loop of each S subunit constitute an extension forming a β-hairpin. The βA-βB loops of four S subunits surround the opening of the solvent channel that transverses the L
8 octamer along the axis of four-fold symmetry (
Figure 1a).
Structurally the S-subunit is involved in stabilizing the holoenzyme quaternary structure [
23] and possibly modifying the conformation of the enzyme. In this respect, Hartl et al., have shown that the S subunit play a decisive role in assembly of the red-type Rubisco from
Rhodobacter sphaeroides [
24]. Concerning its functional role, the S-subunit is shown to affect several kinetic properties as the enzymatic catalytic efficiency and S
C/O specificity (see
Table S3). Moreover, Fukayama and co-workers have recently shown that overexpression of S subunit from C4 plants (
Pennisetum purpureum and
Megathyrsus maximus) in rice (a C3 plant) significantly enhances the
kcat value of the C3 Rubisco [
25]. Analogously, expression of the small subunit of
Sorghum (a C4 plant) in rice produces a Rubisco with higher catalytic rate when compared to rice [
26]. Very recently Hanson et al., achieved expression of tobacco Rubisco in
E.coli assembling enzyme complexes with different small subunits including a small subunit from trichome. The new technique allowed precise evaluation of the effects of different small subunits on the Rubisco kinetic parameters [
27]. The Whitney group has produced new tobacco genotypes encoding both large and small subunits in the chloroplast genome, producing a bioengineering chassis useful in evaluation of novel Rubisco complexes [
28].
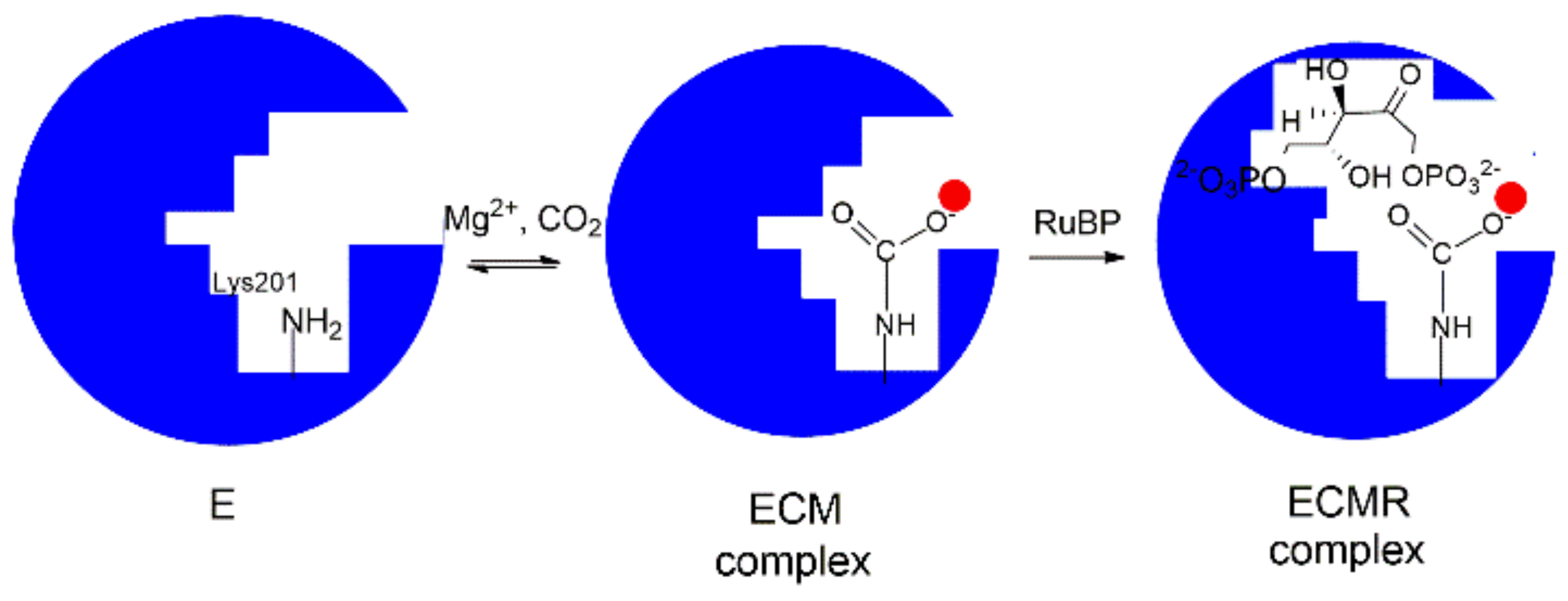
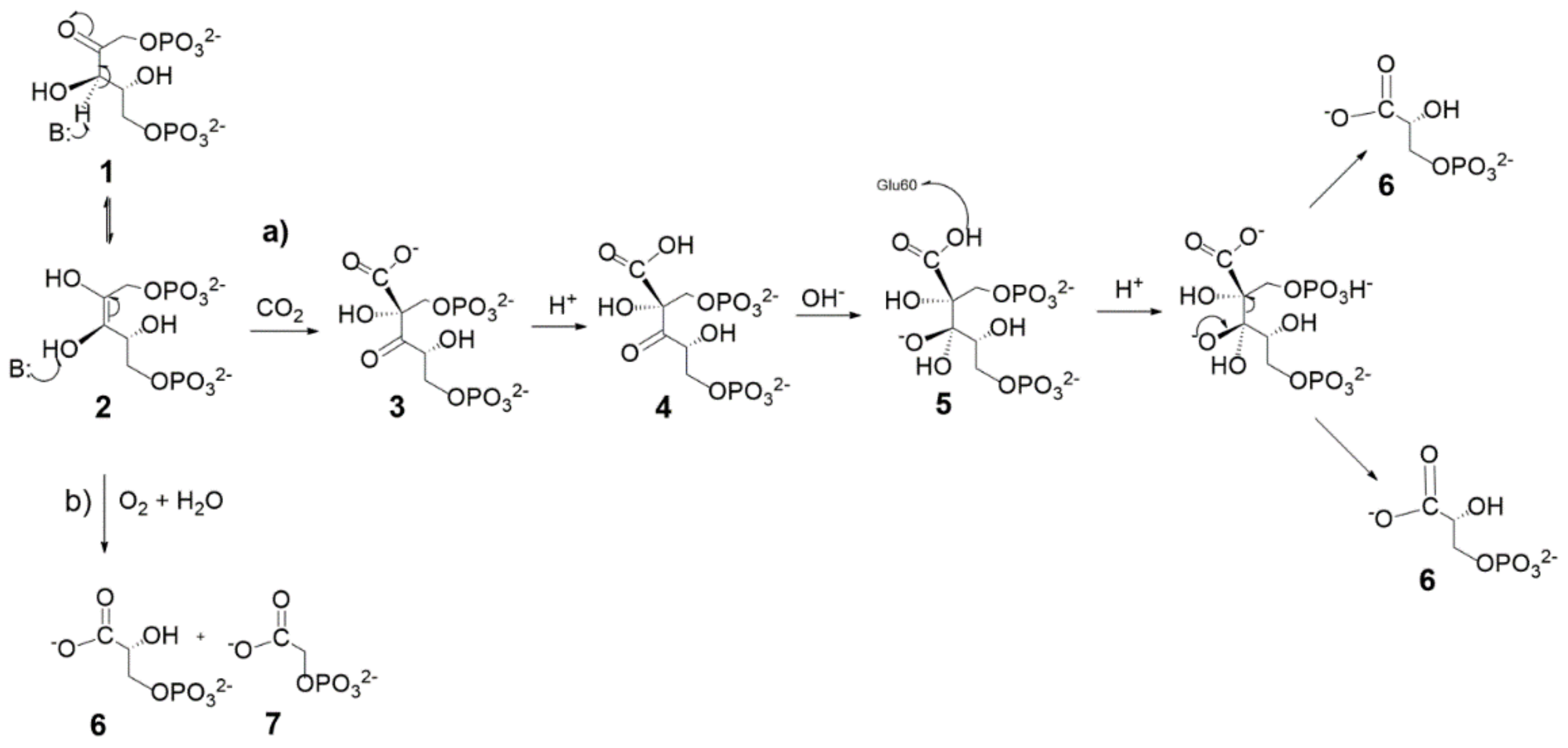
4. Mechanism of Enzyme Activation by Lys201-Carbamoylation
The information drawn from X-ray crystal structures and kinetic studies has allowed the proposal of a model describing the Rubisco physiological function as a sequence of “enzyme activation” and “enzyme catalysis” [
29]. According to the model, the “enzyme activation” step consists in carbamylation of Lys201 affording an adduct stabilized by Mg
2+ cation (ECM complex).
In particular, as depicted in
Figure 4, the binding pocket of the unbound enzyme (represented as E) binds CO
2 (C) on Lys201 and the carbamylated-Lys201 is stabilized by the binding of Mg
2+ (M) to give the ternary ECM complex Lys-201-NH-COO
− Mg
2+. This reaction allows the transition from a “catalytic inactive” to a “catalytic active” enzyme prone to tightly binds RuBP to give the ECMR complex. The ECMR complex may begin the “enzyme catalysis”, reacting with CO
2 or O
2 according to the RuBP “carboxylation” or “oxygenation” reactions. Interestingly, it has been demonstrated that the “activating CO
2 molecule” (i.e., the CO
2 molecule binding Lys201) is not the CO
2 molecule involved in the RuBP carboxylation reaction (
Scheme 1a).
The apparently “simple” Lys201-carbamylation reaction underlines a quite complex reaction mechanism recently disclosed by Stec [
15]. The author succeeded in obtaining a high resolution structure (1.9 Å) of Rubisco from
Galdieria sulphuraria bearing O
2 at the active site (in the center of the TIM barrel fold). Subsequently O2 was replaced by CO
2 by incubation of crystals at 40 °C in the presence of CO
2 and Mg
2+. After incubation, harvested crystals showed evidence of ECM complex formation. Comparison of the two crystal structures bearing O
2 or the Lys201-CO
2−Mg
2+ complex allowed describing the structure of the “activation binding site” (
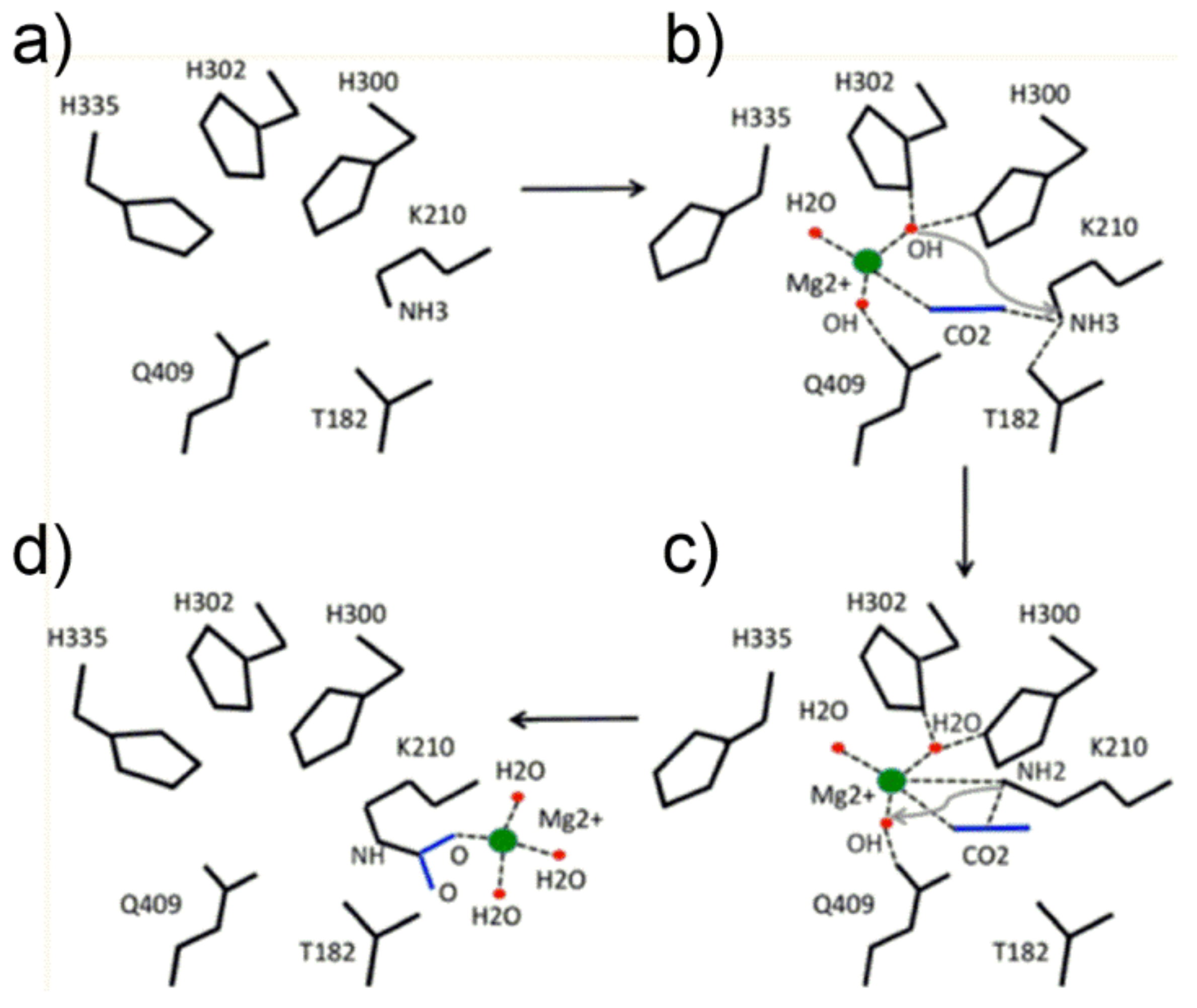
Figure 5a) evidencing its different location with respect to the “catalytic binding site” described in the next section.
The proposed mechanism for the Lys201 carbamoylation reaction starts with a conformation change of His335 (comparing
Figure 5a,b, note the conformational flexibility of the His335 side chain) allowing Mg
2+ to locate in the transient binding site shown in
Figure 5b. Interestingly, the metal ion was modelled to coordinate two OH
− groups, one water molecule and CO
2 (the latter in the end-on mode) displaying a pseudo-tetrahedral geometry with the linear CO
2 molecule pointing towards the ε-NH
3+ group of Lys201. Subsequently, one metal-bound hydroxide promotes the proton abstraction of the ε-NH
3+ group of Lys201 (see
Figure 5c), producing the ε-NH
2 functionality prone to nucleophilic attack of the CO
2 molecule to give the ε-NH
2(+)-CO
2(−) moiety. Finally, further proton abstraction from the ε-NH
2(+)-CO
2(−) moiety produces the Mg
2+-stabilized aci-carbamate complex that relocates in a new binding site (shown in
Figure 5d) where an octahedral coordination sphere around Mg
2+ is completed with Glu213 and Asp212 (not shown in
Figure 5d). Relocation of the Mg
2+-complex allows His335-side chain return to its original conformation.
5. CO2/O2 Molecular Recognition Strategy
In the same study [
15], Stec was able to propose a mechanistic explanation for the Rubisco ability to discriminate between CO
2 and O
2 at “RuBP-binding site”. As intracellular (CO
2) is estimated approximately 7–8 μM in C3 plants and 80 μM in C4 plants, while [O
2] is about 260 μM, [
30] a key point in Rubisco catalysis is the CO
2/O
2 competition for the RuBP-binding active site. To compensate for the low CO
2 intracellular level, Rubisco shows K
C values ranging from 8 to 19 μM and K
O values ranging from 230 to 620 μM while S
C/O values range between 70 and 100 in plants (considering both C3 e C4 plants) and can reach values as high as 222 in
Galderia suphuraria (a red algae).
Considering that Rubisco does not have specific binding sites for CO
2 or O
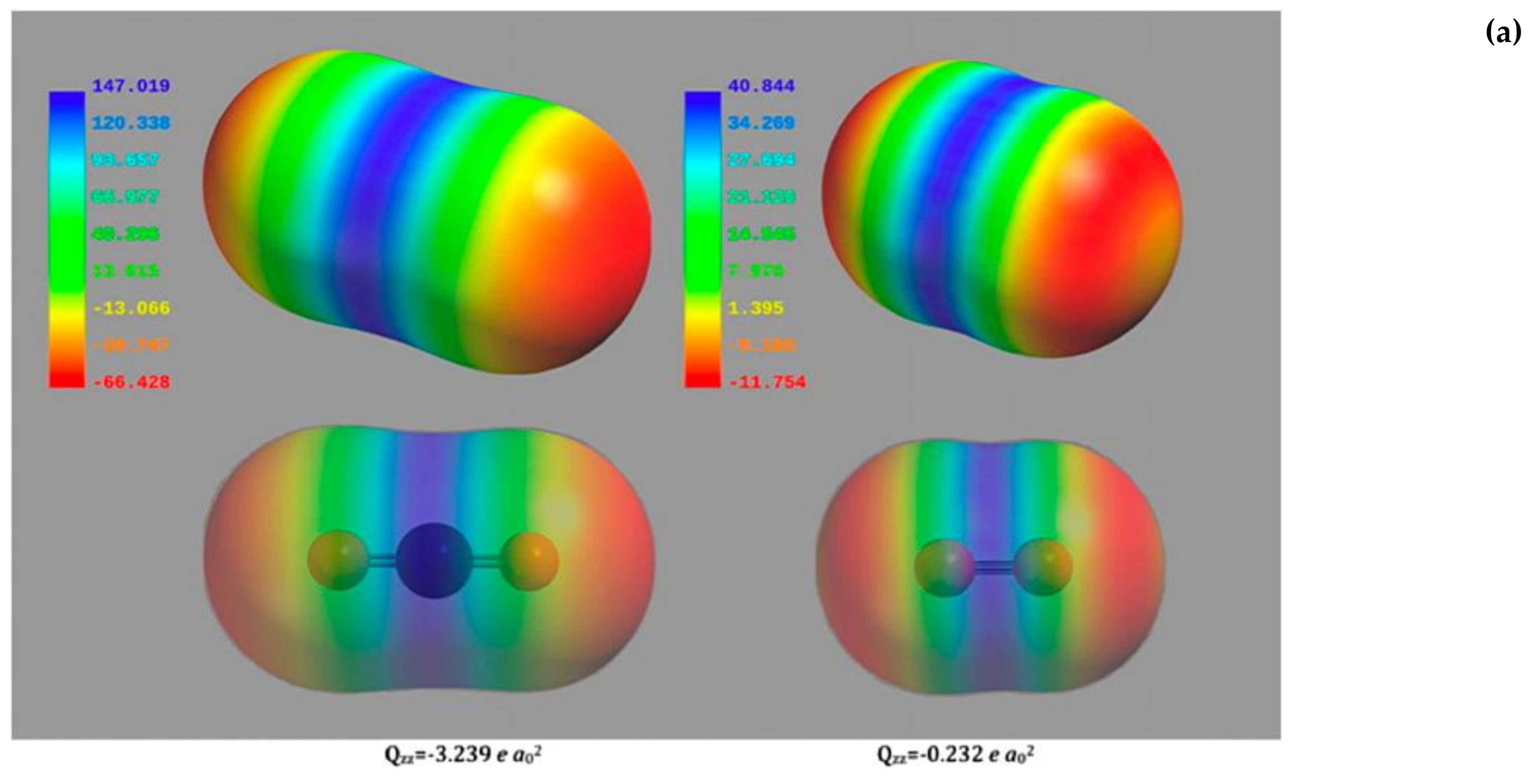
2 and the two gaseous molecules are not so different as far as dimensions and electrostatic potential surfaces are concerned (
Figure 6a), the Rubisco higher specificity for CO
2 is not easily explained.
Stec [
15] proposes that the two gaseous molecules “bind” to the same region of the active site, at the mouth of the TIM barrel, covered as a lid by the N-terminal domain of the adjacent L subunit (see
Figure 3), where a high electrostatic field gradient is set up by the closeness of negative and positive surfaces potential.
Figure 6b shows a negative surface potential (represented in red) at the N-terminal domain and a positively charged cavity (represented in blue) at the C-terminal domain. As the quadrupole moment of CO
2 is approximately 15-fold higher than that of O
2 (for CO
2 Qzz = −3.239 e a
02, for O
2 Qzz = −0.232 e a
02 as calculated by Stec), the quadrupole moment of the two gaseous ligands interacting with the very strong electric field built across the active site allows discrimination between the two species.
The hypothesis advanced by Stec appears correct and provided a rationale for the minimal requirement of a L2 dimer as a basic functional unit found across the Rubisco protein family. In fact, contact between the surfaces of the two subunits (facing each other) is needed to build up the gradient of the electric potential.
Andersson et al. [
31] have also faced the problem concerning the favourable competition of CO
2 vs. O
2 by investigating, via molecular dynamics simulations, the interaction of the two gaseous molecules with the protein. A modelled L
8S
8 hexa-decamer was solvated in a dodecahedron cell with an edge length of 12 nm and the desired amount of gas molecules was created to model the CO
2 and O
2 migration around the protein. The L
8S
8 models reproduced the protein structure of Rubisco from spinach (PDB access code 1aus, 1ruc and 1rcx) and from
Chlamidomonas reinhardtii (PDB access code 1gk8). On the basis of the partition of the two gases between the protein and the surrounding solution, the authors were able to calculate the intrinsic affinity of Rubisco for CO
2 as +0.3 kJ/mol compared to +10 kJ/mol calculated for O
2. These data indicate that the whole protein binds CO
2 stronger that O
2. In agreement with Stec, the authors support the stronger CO
2 protein-binding with the difference in molecular quadrupoles of the two ligands. Moreover, by modelling the CO
2 and O
2 densities around the active site of Rubisco from
Clamidomonas reinhardtii, the authors found that CO
2 has a preference for binding a region around the active site that extends to the closest S subunit (
Figure S4). The simulation showed that the overall S subunit has a higher affinity for CO
2 than the overall L subunit suggesting that the S subunit plays a role as “CO
2 reservoir” for catalysis.
The hypothesis advanced by Stec is supported to some degree by a recent study by Falkowsky et al. [
32] which analyzed the high resolution crystal structures of 11 Rubisco proteins stored in the PDB. The structural study was integrated with a phylogenetic analysis constructed using 55 Rubisco proteins sequences that confirmed that Rubisco homologs are assigned into four different phyletic groups named G-I, G-II, GIII and G-IV (the four forms are usually designated as form I, II, III and IV as mentioned in
Section 3). Moreover the study demonstrated that: (i) G-II enzymes evolved from proteobacteria; (ii) G-III Rubisco evolved from the archaeal domain; (iii) G-I Rubisco evolved later being represented among bacteria, eukaryotic algae and higher plants. Distinct subclass of Rubisco proteins were found among the G-I group (i.e., IA, IB, IC and ID) showing a wide range of S
C/O values ranging from 40 to 110.
The 11 high resolution crystal structures belonging to the monophyletic groups G-I and G-II allowed the observation that amino acid residues proximal to the active sites are not strictly conserved among different homologs and are likely sites for evolution. Crystal structures assigned the PBD ID5MAC, 1BXN and 4FOK are related to G-II, G-IC and G-ID Rubisco respectively, and belong to proteins progressively adapted to increasing O2 concentration. Interestingly, comparison of these crystal structures evidenced the presence of higher surface charge potential proximal to the active site and solvent accessible channels connecting the protein surface to the active site. Enzymes bearing these larger positive channels easily “drive” CO2 towards the active site, thanks to the electrostatic steering with the CO2 quadrupole coming from a partial negative charge on the two oxygen atoms.
Most important is that the extension of the positive charged areas progressively increases from 5MAC to 1BXN structures reaching the maximum extension in the 4FOK structure. Interestingly, increasing extension of the positive charged areas within the three proteins parallels increased SC/O selectivity.
6. Structure of the Rubisco “Catalytic Active Site”
The inspection of the list of Rubisco’s structure published by Andersson and Backlund in 2008 [
13] evidences that a majority of the structural studies have dealt with Rubisco in complex with RuBP and Ca
2+ [
33] or in complex with 2CABP or XuRB (
Scheme 2) and Mg
2+. Only one crystal structure of Rubisco in complex with RuBP and the Mg
2+ ion has been solved, probably because of an inherent instability of the RuBP-Mg
2+-protein complex [
34]. By contrast, 2-carboxy-D-arabinitol-1,5-bisphosphate (2CABP) forms with the Mg
2+ ion an exchange-resistant complex which turned out to be useful in crystallographic studies [
35].
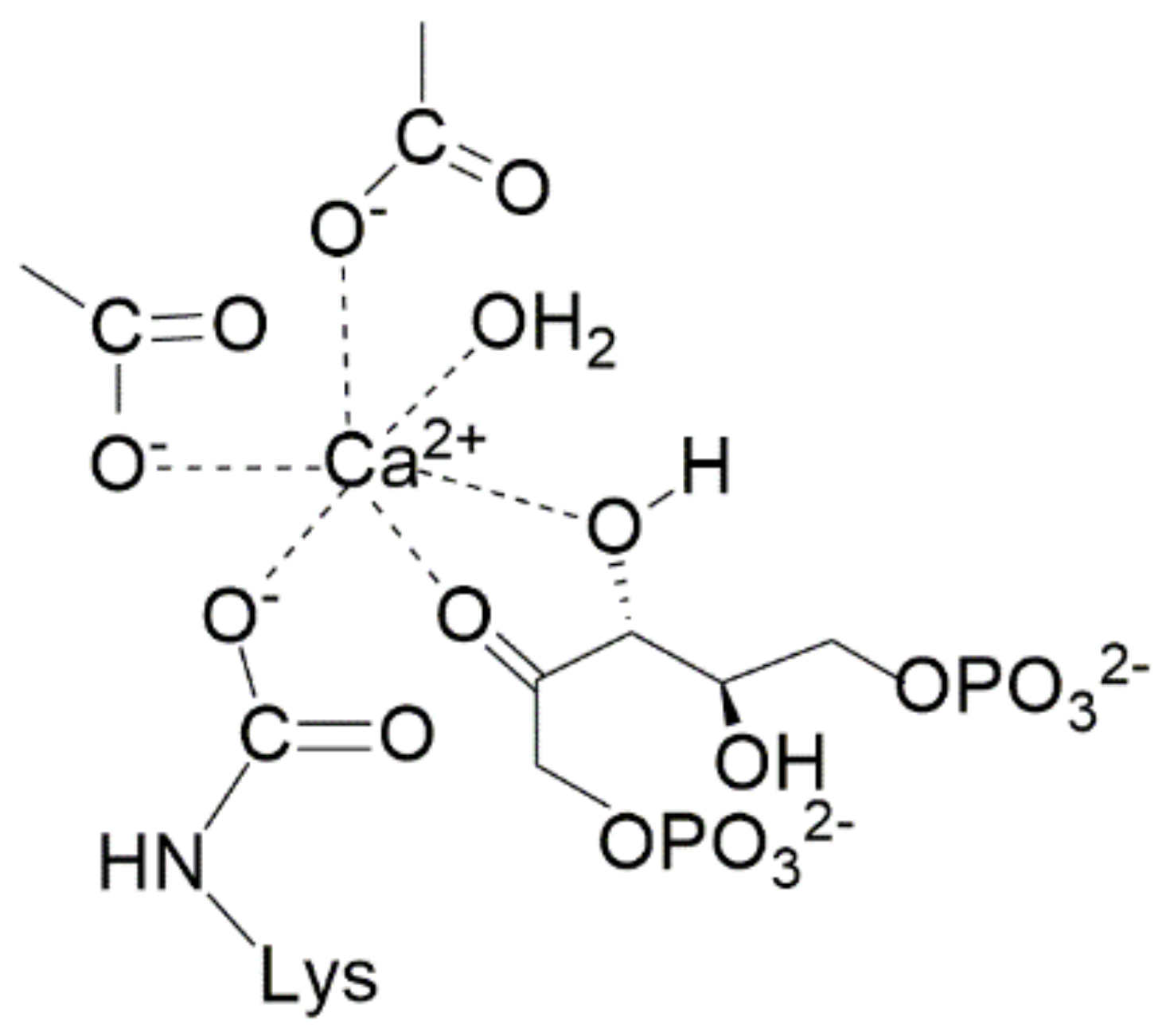
The structure of 2-carboxy-D-arabinitol-1,5-bisphosphate (2CABP) shown in
Scheme 2 closely resembles the six-carbon intermediate 2-carboxy-3-keto-D-arabinitol-1,5-bisphosphate (3-keto-2-CABP) assumed to be the transient intermediate in the carboxylation reaction. Therefore, for a long time it was considered that the interaction of 2CABP with Mg
2+ mimics the transition state of the metal-bound β-keto acid intermediate.
Two capstone publications reporting the structure of Rubisco from spinach in complex with 2-carboxyarabinitol-bisphosphate (2CABP) are those of Knight et al., in 1990 [
36] and Anderson et al., in 1996 (PDB code number 8ruc) [
35]. The two structures were resolved at 2.4 and 1.6 Å, respectively. As amino acids taking part in catalysis are often directly involved in substrate interactions, a distance <4.0 Å between amino acid residues and 2CABP was used to identify amino acids as catalytically competent.
As shown in
Table 1 [
36], amino acids contributing to the catalytic pocket of Rubisco are located at the loops following the C-terminal ends of the β-strands (from β1 to β8 of the C-terminal domain) and at the N-terminal domain of the antiparallel L subunit. Amino acid residues represented in bold are strongly conserved among Rubiscos from diverse species.
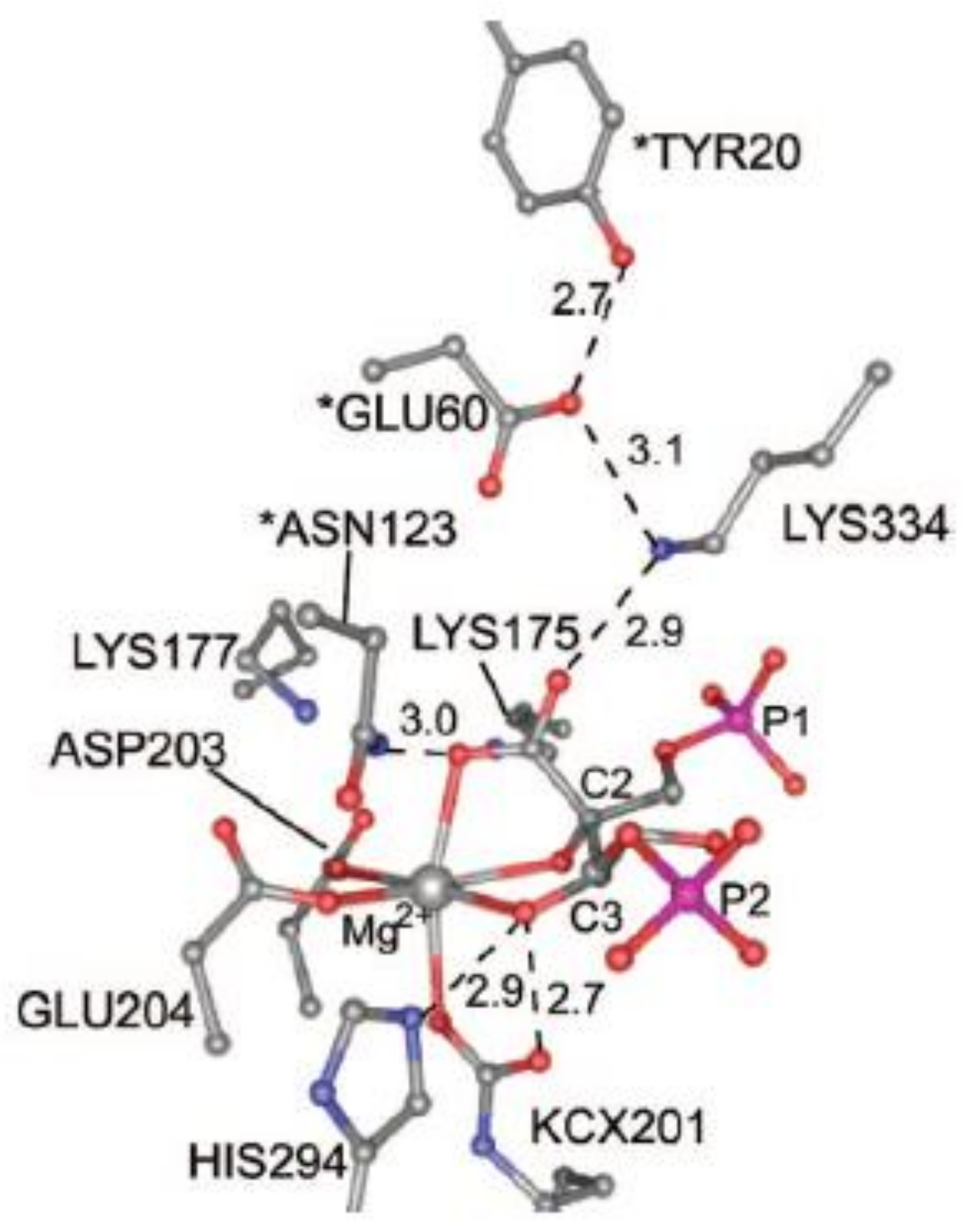
Figure 7 shows the structure of the active site of activated 2CABP-bound Rubisco as proposed by Gready et al., in 2008 [
37] (the structure has been plotted from the file 8RUC deposited at the Protein Data Bank, Anderson, 1996 [
35]). According to this model, carbamylated Lys201 (KCX201), Asp203, Glu204, 2-carboxy-D-arabinitol-1,5-bisphosphate (2CABP) and H
2O complete the octahedral distorted geometry around Mg
2+.
Moreover,
Figure 7 evidences Asn123, Glu60 and Tyr20 residues (marked with an asterisk), located at loopA and loopB of the N-terminal domain of the L-partner subunit, interacting with the ligands of the first coordination sphere. Lys334 is an amino acid residue of loop 6 (i.e., the loop connecting β-strand 6 with α-helix 6 of the TIM barrel) undergoing conformational changes during catalysis (see
Section 7).
Concerning the binding of the actual RuBP ligand to Rubisco, the crystal structure resolved by Andersson in 1997 [
33] (refined to 2.1 Å) contained the catalytically inactive Ca
2+ interacting with the sugar phosphate. RuBP is proposed to bind the metal ion through the O
2 and O
3 to give a five-member metallacycle. This structure has been considered by several authors as the starting structure to model the mechanism of the carboxylation reaction (see
Section 8).
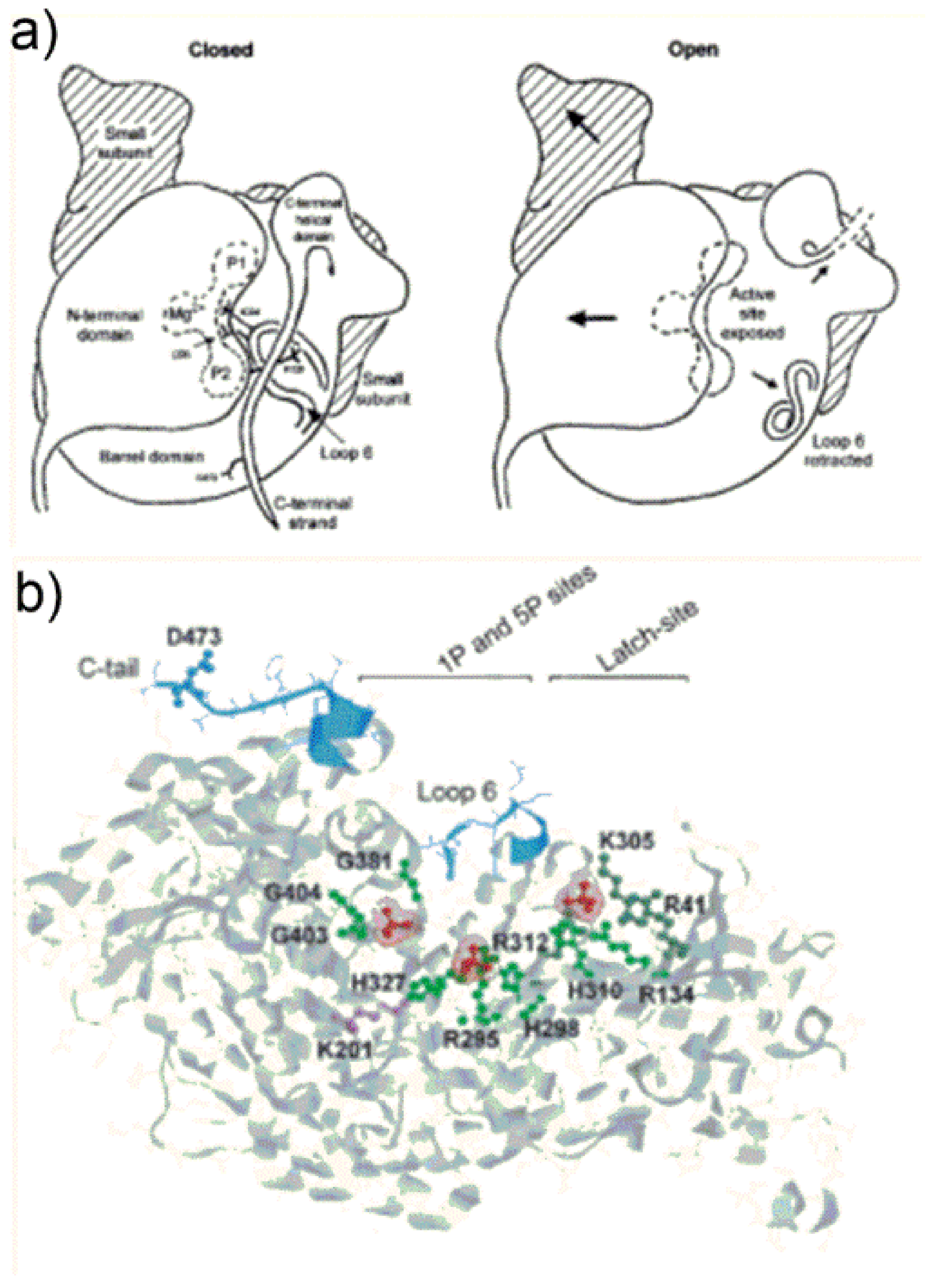
7. Conformational Changes during Catalysis: “Open” and “Closed” Form of the Enzyme
In an interesting study Curmi et al. [
38] have compared X-ray crystal structures of Rubisco from spinach, tobacco and
Synecococcus considering either “empty active site” proteins (i.e., polypeptides bearing the ECM complex but lacking substrate binding) and sugar phosphate-bound forms of Rubisco (including RuBP, 2CABP or XuBP ligands). Careful comparison of structures allowed identification of a complex sequence of conformational changes occurring during transition from the so called “open” (with the active site accessible to solvent) to “closed” states of the protein. Occurrence of catalysis requires the enzyme to lie in the “closed” state with the active site not accessible to the solvent.
After enzyme activation (i.e., ECM-complex formation as described in
Section 4) Rubisco remains in an “open” conformation schematically depicted in
Figure 8a.
Figure 8a (open state) shows a view from the central axis of the TIM barrel of the L1-subunit (referred as “active site exposed”) which is partially “covered” by the N-terminal domain of the juxtaposed L2 subunit. In the “open state”,
Figure 8a shows loop 6 is in a retracted position, and shows the C-terminal helical domain (composed of amino acid residues from Trp462 to the C-terminus) pointing far from the active site.
Once RuBP binds tightly to the active site, the enzyme transits from the “open” to the “closed” conformation shown in
Figure 8a. The catalytic site is schematized evidencing location of the Mg
2+ as well as the P1- and P2-binding sites of the sugar phosphates.
The “open” to “closed” transition occurs through a sequence of three main conformational changes: (i) first the N-terminal domain of the L subunit rotates as a semi-rigid body occluding the entrance of the TIM barrel (i.e., the sugar phosphates binding pocket); (ii) loop 6 extends in a position that occludes the active site; (iii) the C-terminal strand overlaps with loop 6.
It is worth noting that “open” state conformation is also observed in X-ray crystal structures of proteins bearing 2CABP bound to the inactivated (metal-free) active site or RuBP bound to the catalytically inert Ca2+. In the latter cases, sugar phosphates bind the active site quite weakly.
Taking a closer look at the tertiary structure conformational changes occurring during open-to-closed transition, Curmi et al., propose that a central role is played by binding of the two phosphate anions of ribulose-1,5-bisphosphate to specific active site positions. A very important point noted by Curmi et al., is that all Rubisco crystallized in the “closed” state show 1P and 5P at a distance less than 9.1 Å, while all proteins crystallized in the “open” state show the two phosphates at a distance higher than 9.4 Å, pointing out that decrease of the 1P-5P distance triggers important conformational changes.
Figure 8b evidences in red the 1P- and 5P-group binding sites of RuBP in Rubisco from the cyanobacterium
Synechocystis sp. [
39]. In the “open” state, 1P binds the main-chain NH-groups of Gly381, Gly404 and Gly403 (located at β-strand-7 and loop-8 of the TIM barrel) and engages in hydrogen bonding with Trp66 located at the N-terminal domain of the antiparallel L-subunit (not shown in
Figure 8b). The 5P-anion engages in hydrogen bonding with Arg295 and His298-H
+ occupying the so called “upper subsite” located at β-strand-5 of the TIM barrel. The third phosphate group shown in
Figure 8b interacts with the latch-site which is assigned a particular role in regulation of catalysis.
During the open-to-closed transition the 5P-group displaces, occupying the so called “lower subsite” upon interaction with Arg295 and His327 (
Figure 8b). Moreover, the 1P-anion reaches a position closer to the 5P-group engaging in hydrogen bonding with Thr65 of the N-terminal domain of the adjacent L2 subunit (not shown in
Figure 8b). A change of the backbone ψ torsion angle of Thr65 moving towards the 1P-group seems to be the event triggering the rotation of the N-domain to cover the C-terminal domain TIM barrel. It is important to note that amino acid residues from Trp66 to Thr75 are located in the middle of the loop connecting α-helix-B to β-strand-C of the N-terminal domain (see
Figure S2). This loop constitutes the major interface between the N-terminal domain of one L subunit and the TIM-barrel of the antiparallel L subunit and does not alter in position relative to the TIM barrel during open-to-closed transition. In contrast, the remainder of the N-terminal subunit is triggered to rotate.
Conformational changes occurring at loop 6 enable loop mobility between two well defined states i.e., “retracted” (while the enzyme is “open state”) and “extended” (while the enzyme is “closed state”): extension of loop 6 over the active site (to reach the enzyme closed state) is due to dramatic changes in the φ and ψ angles of Gly333 and Gly337. These residues provide two flexible elements favoring the rearrangement. Moreover, the 5P-anion binding to the “upper sub-site” exerts steric hindrance toward Leu335 of loop 6 favoring the “retracted” vs. “extended” conformation of the loop.
As the enzyme needs to reach the “closed state” before initiating the carboxylation/oxygenation reactions, conformational changes significantly affect the “entropic cost” of enzyme functioning. Stec points out that the required conformational changes may explain, in part, the Rubisco’s low catalytic turnover rates. Moreover, Gready et al. [
37] point out the low
kcat,C of Rubisco enzymes may be due to the complexity of the reaction mechanism with all reaction steps taking place at a single active site. The following section will focus on the description of the reaction mechanism as recently proposed by Gready’s research group.
8. Mechanism of the Rubisco Carboxylation Reaction: The Role of Broensted Acid-Base Catalysis
By integrating information from the body of existing literature concerning crystallographic and mutational studies, Cleland et al., proposed in 1998 the first consensus mechanism for the Rubisco carboxylase reaction [
40]. The model encompasses RuBP-enediolate coordination to the Mg
2+ ion undergoing subsequent transformation implying, in specific steps, proton-transfer reactions and water addition.
From this study it became clear that elucidation of the Rubisco carboxylation mechanism requires large reliance on computational methods, as elementary steps consisting in proton exchanges and water addition cannot be disclosed by analysis of X-ray crystal structures. A decade later, Gready et al. [
37] were able to propose a detailed reaction mechanism for the Rubisco carboxylase reaction by integrating computational studies with a density functional theory calculation on active-site fragment models. To allow DFT analysis, active site fragment models of a size of up to 77 atoms were built by using the coordinates of Rubisco crystal structures. In particular the authors have used the crystal structure of the activated spinach Rubisco complexed with Ca
2+ and RuBP (PDB code number 1RXO [
33] showing Ca
2+ coordination as depicted in
Figure 9) and that of spinach Rubisco complexed with Mg
2+ and 2CABP (PDB code number 8RUC [
35]). The building of approximately twenty fragment models enabled mapping of structures of reaction intermediates, and the computational analysis (based on the use of the B3LYP functional) allowed optimizing of their geometries. Validation of the optimized model structures was carried out comparing calculated interatomic distances with the active-site structures revealed by X-ray diffraction. For 75% of the compared distances, deviation of less than 0.6 Å was obtained.
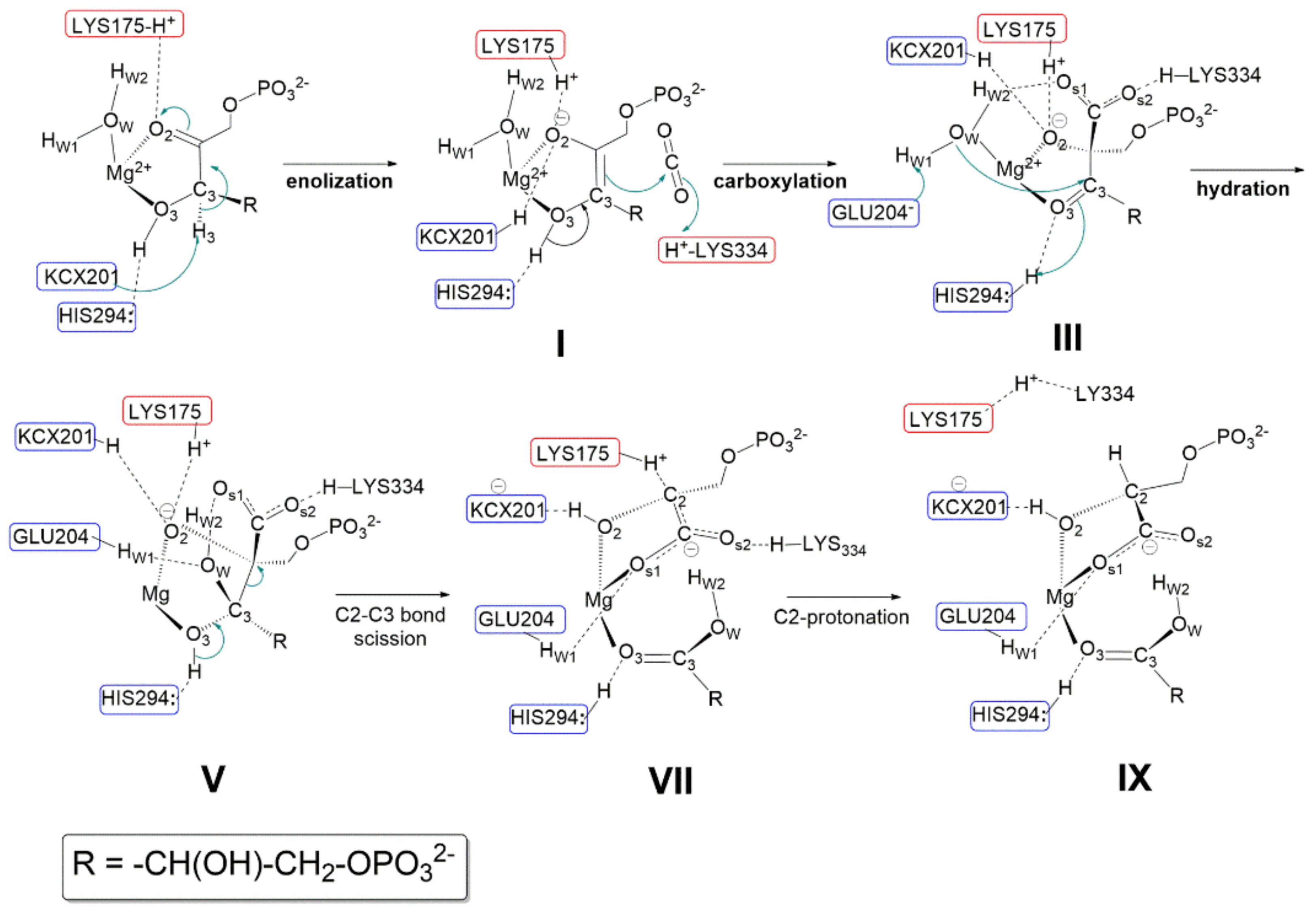
Relevant features of the reaction mechanism as proposed by Gready et al., in 2008 [
37] are shown in
Figure 10 (amino acid sequence numbering refers to Rubisco from spinach). The initial model fragment along the reaction coordinate shown in
Figure 10 was modelled using the active site structure shown in
Figure 9, but replacing Ca
2+ with Mg
2+. However,
Figure 10 does not show the pseudo-octahedral coordination around Mg
2+ as, for a simplified view, only the RuBP ligand and the water molecule (H
w1-O
w-H
w2) are highlighted. According to Gready’s description, every single step of the reaction involves proton-transfer process. Therefore, amino acid residues acting as Lewis base are highlighted in blue boxes, while amino acid residues stabilizing anions (or the negative charge of dipolar groups) through hydrogen bond are shown in red boxes.
Relevant steps involving acid-base catalysis are summarized below:
- (1)
Enolization step: Following RuBP binding to Mg2+ through O2 and O3, Lys175-H+ is proposed to engage in hydrogen bonding with the carbonyl C2=O2. The carbamoyl moiety Lys201-COO− (also noted as KCX201) acts as a base abstracting the C3-H3 proton allowing RuBP enolization to the 2,3-cis-enediol(ate) form (shown in structure I). Both KCX201-H and LYS175-H+ are proposed to engage in hydrogen bonding with O2 partially neutralizing its negative charge. His294 is proposed to play an important role in RuBP enolization, engaging in hydrogen bonding with H-O3 and preventing, thus, its interaction with KCX201−. As a matter of fact, it is assumed that in the absence of the His294-H-O3 interaction the carbamoyl group would abstract H-O3 more easily than H3-C3.
- (2)
Carboxylation step: following RuBP enolization, His294 is proposed to abstract the H3-O3 proton allowing ketonisation at C3 followed by negative charge localisation at C2 (structure I). The Si face of the incipient C2-carbanion attacks CO2 forming the 2-carboxy-3-keto-D-arabinitol-1,5-bisphophate intermediate. Structure III evidences the hydrogen-bonding network, stabilizing the geometry of the carboxylate intermediate within the fragment model. Relevant interactions are: (i) the protonated Lys334-H+ and the water molecule stabilise the negative charge of the carboxylate moiety; (ii) His294-H+ maintains hydrogen bonding with O3; (iii) Lys175-H+ and KCX201-H keep in hydrogen bonding with O2 atom.
- (3)
Hydration step: to start the hydration step (structure III) Hw1 is proposed to be abstracted by Glu204− anion allowing nucleophilic attack by OH− onto the C3 carbon. His294-H+ assists the hydration reaction by proton transfer to O3 allowing C3 to reach a sp3-hybridization to form a six-carbon gem-diolate intermediate (see structure V).
- (4)
C2-C3 bond scission and C2 protonation: Structure V shows the six-carbon gem-diolate intermediate which is proposed to undergo proton abstraction from H-O3 by His294 with ketonization at the C3 carbon followed by C2-C3 bond scission. As a consequence two molecular fragments are produced:(i) one 3-phospho-D-glyceric acid molecule (lower part of the structure); (ii) a C2-carbanion that delocalizes the negative charge over the carboxylate moiety producing an aci-acid intermediate (it is important to note that the C2=COs1Os2− fragment is planar), and this charge is stabilized by Lys334-H+ and Lys175-H+ (see structure VII). Finally Lys175-H+ promotes stereospecific protonation of the C2 carbanion to yield the 3-phospho-D-glycerate. Only at this stage does Lys334-H+ engage in hydrogen bonding with Lys175.
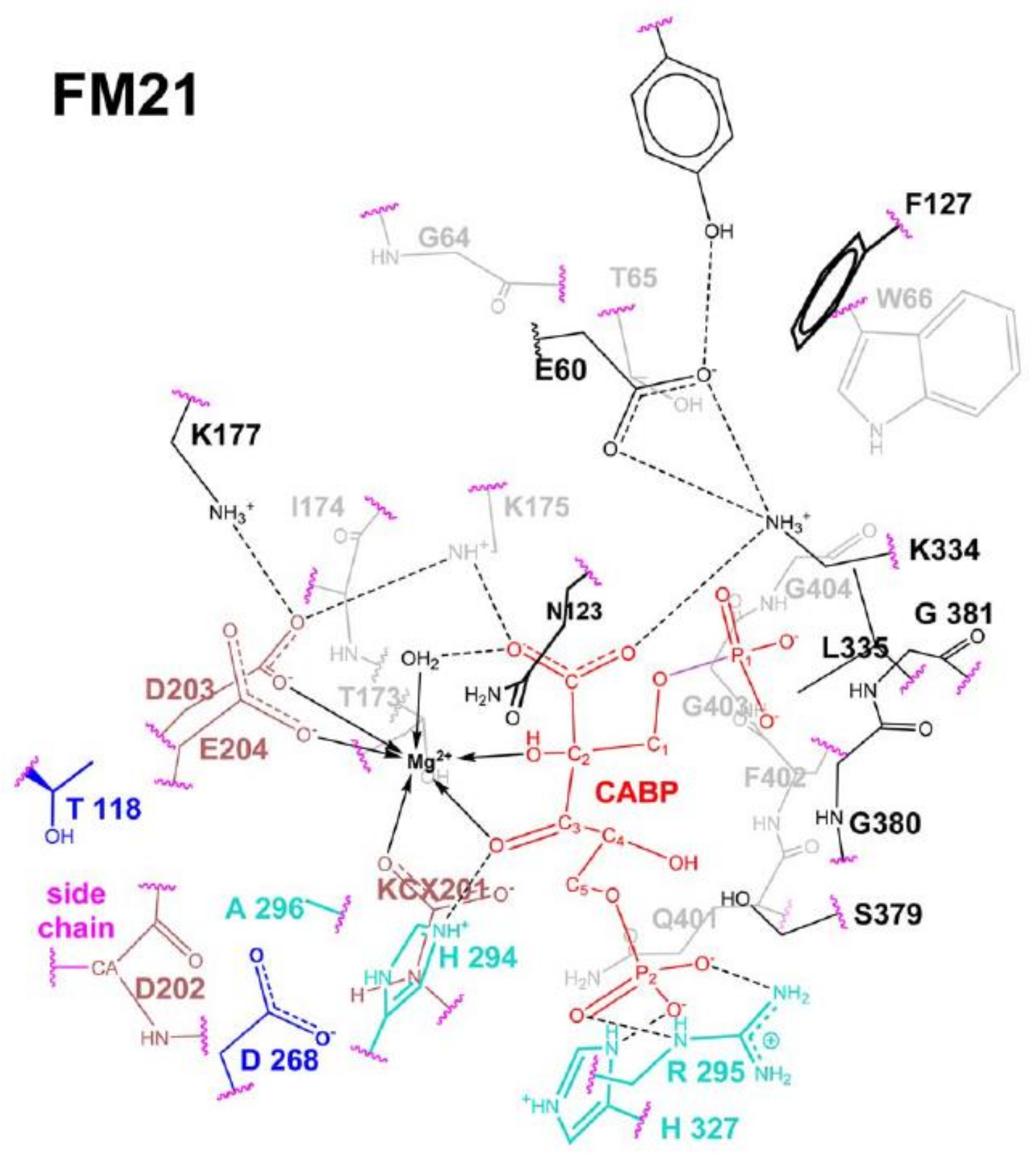
The Rubisco reaction model summarized in
Figure 10 has been “reshaped” by using recently available computational methods (as hybrid semi-empirical (SE) quantum mechanics, molecular mechanics (QM/MM) plus molecular dynamics (MD) calculations (QN/MM + MD)] in conjunction with energy-minimization calculations. The new computational methods allowed the building of active-site fragment models (FM21 shown in
Figure 11) consisting of many hundreds of atoms [
35]. FM21 was built based on coordinates from the crystal structure of spinach (PDB code 8RUC, [
29]) and includes 343 atoms within a range of 24 Å from Mg
2+.
The new study [
41] does not consider the RuBP enolization step but focuses on the subsequent elementary steps of the reaction coordinate (i.e., carboxylation, hydration, C2-C3 bond scission and C-2 protonation) shown in
Figure 12.
Results of the study [
41] provide new insight into the following reaction steps which confirm the relevance of the acid-base chemistry: (i) the 2-carboxy-3-keto-arabinitol-1,5-diphosphate (3 in
Figure 12) was shown to undergo protonation at the carboxylic group (affording the C2-carboxylic acid) to energetically facilitate the subsequent nucleophilic attack of OH
− onto the C3 carbon leading the C3-gemdiolate 5; (ii) the C3-gemdiolate was found to the most stable intermediate bound to the Mg
2+-cation along the reaction coordinate, as shown from Relative Potential Energy calculations; (iii) the C2-C3 scission/C2-protonation events were described occurring in a concerted mechanism; (iv) Lys175-H
+ was established acting as proton source in step (iii).
It is worth noting that all the previously proposed reaction mechanisms have considered a two-step reaction consisting in the cleavage of the C2-C3 carbon bond, affording a C2-carbanion intermediate followed by stereospecific C2-carbon protonation to give 3-phospho-D-glycerate.
Due to the novelty of the conclusion drawn in the previous work concerning the C2-C3 scission/C2-protonation steps occurring in a concerted mechanism, a more careful investigation was developed by Gready et al., in 2019 [
42] by applying ab initio molecular dynamic simulations on the molecular fragment FM21 (
Figure 11 by using the Kohn-Sham density functional theory (
Figure 13b).
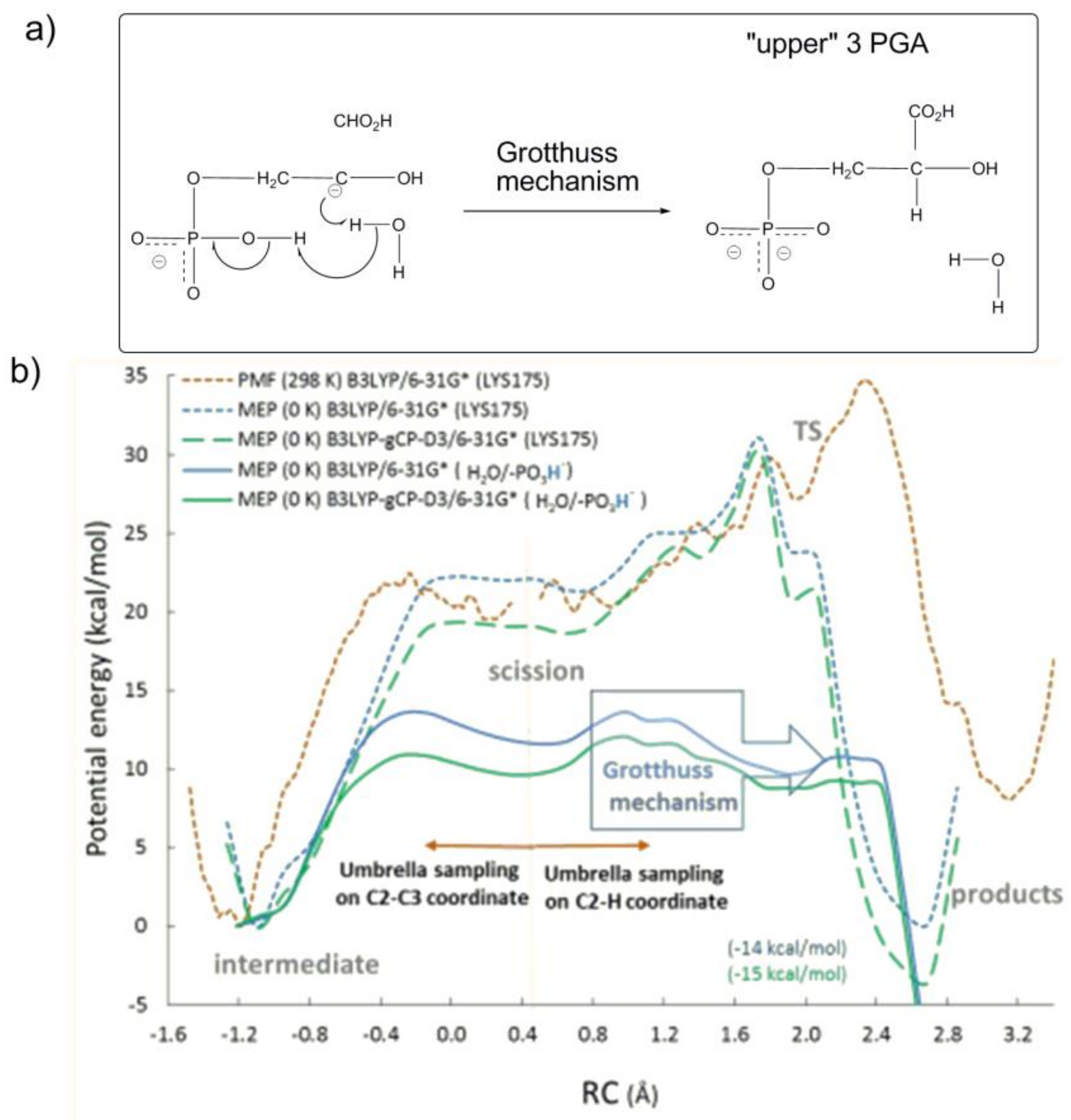
By computing the variation of potential mean force (PMF at 298 K) and minimum potential energy (MPE at 0 K) along the reaction coordinate, the study intended to assess whether protonated Lys175-H
+ or a proton translocation across a network of H-bonded water molecules with P1-group as proton donor (the so called phosphate mediated Grotthus mechanism,
Figure 13a) is involved in the stereospecific protonation of the C3-gemdiolate (“intermediate” in
Figure 13b) to afford 3PGA (“products” in
Figure 13b).
Interestingly, the computational study revealed that the protonation of the C3-gemdiolate from Lys175-H
+ (represented as the orange dotted line in
Figure 13b) is not favoured, as products are located 5 kcal/mol higher than the C3-gemdiolate intermediate. In contrast, the proton transfer process occurring through the Grotthuss mechanism (represented as the blue and green continue lines in
Figure 13b) shows an activation energy of 15.6 kcal/mol (in close agreement with experimental data) and obtains products 14 kcal/mol more stable with respect to C3-gemdiolate intermediate.
All of the studies carried by the Gready group summarized above indicate that the last reaction steps concerning C2−C3 scission (of the C3-gemdiolate) and stereospecific C2 protonation described in
Figure 13a proceed through preliminary protonation of the P1-group of the C3-gemdiolate. Subsequently, in a concerted reaction mechanism, a proton at the P1-group induces stereospecific protonation of the C2-carbon via the Grotthuss mechanism.
Due to the rapidly evolving view of the Rubisco reaction mechanism, in a very recent publication, Cummins and Gready [
43] have considered the reactional events from a completely new perspective.
The rationale behind the reaction mechanism proposed in
Figure 10 is that RuBP strongly coordinates Mg
2+ and most parts of the Brœnsted acid-base reactions imply “movement” of amino acid side chains approaching the substrate. As a consequence, the requirement for conformational chain reorganization implies a considerable amount of strain energy. In a very recent publication, Cummins and Gready [
43] have refined the model of the Rubisco catalytic site using a complex of approximately 340 atoms, as in previous studies, but introducing new structural details. Considering that the structure of Rubisco from spinach (PDB 8RUC) gives a snapshot of the enzyme in its “closed” conformation, a careful analysis of the water molecules identified by X-ray crystallography around RuBP and Mg
2+ suggests the formation of short water wires. The introduction of the “water wire” concept into the model allows proton transfer processes to occur through the Grotthuss mechanism (at several steps along the reaction coordinate) as an alternative to the previous amino acids side-chain conformation change mechanism. Notably, the new reaction model does not imply a direct coordination of RuBP to Mg
2+. Interestingly, the new model allows the calculation of accurate potential energy values for intermediates and transition states along the reaction coordinate by using the B3LYP/6-31G* and corresponding corrected B3LYP-gCP-D3 functional.
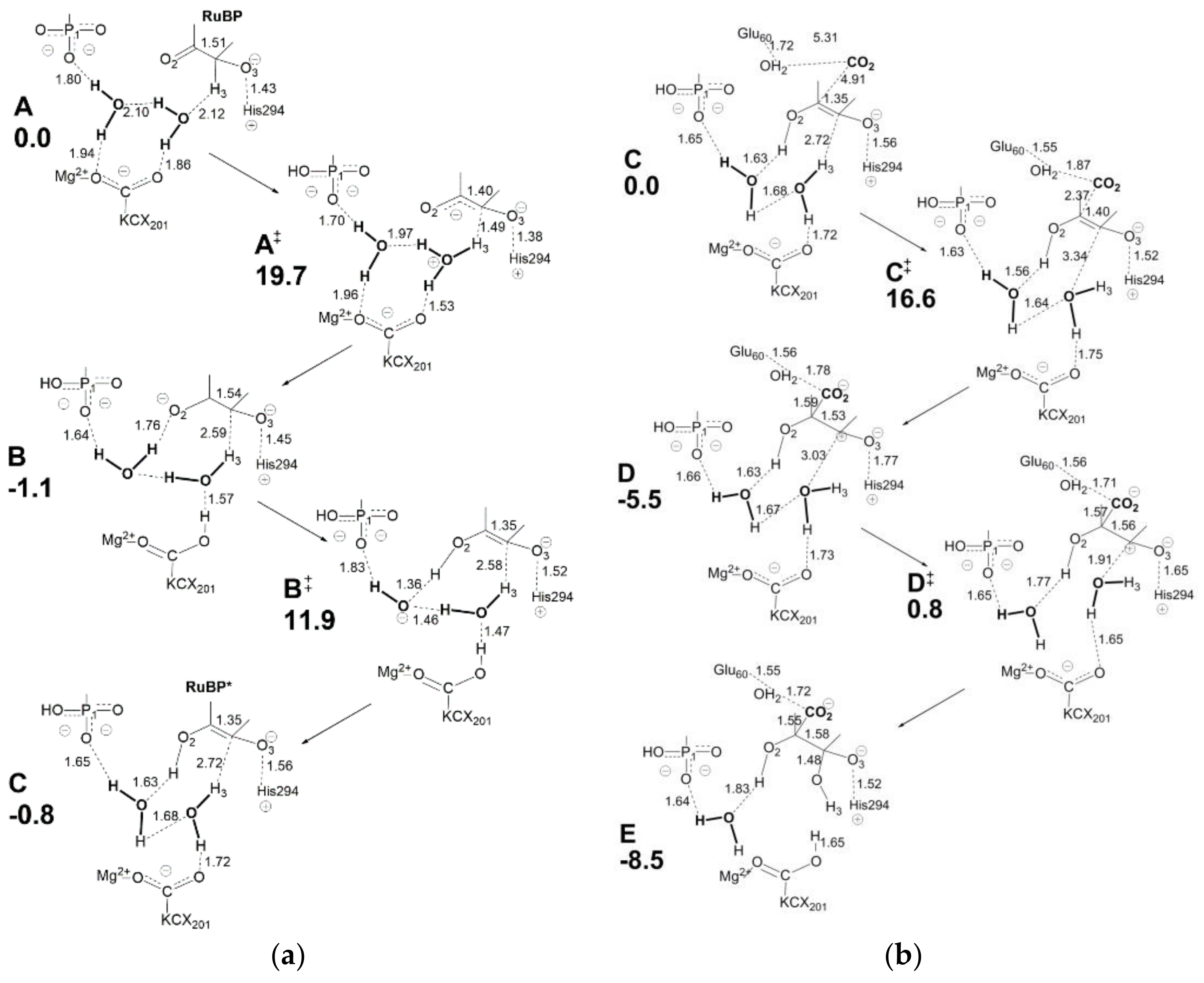
Figure 14a displays relevant structural features of the initial molecular complex (
A) considered in the new model. Although the choice of molecular arrangement within the active site is somehow arbitrary, the authors consider this a logical choice based on the information available. Due to the complexity of the overall RuBP carboxylation reaction, only the enolization and carboxylation/hydration steps are analyzed here.
Relevant features of
Figure 14a are: (i) Mg
2+ is coordinated to the LYS201-carbamylate moiety (usually referred as KCX201) in the initial molecular complex A; (ii) two water molecules are shown engaging in a network of hydrogen bonding with KCX201 and RuBP in A; (iii) His294 is assumed to be unprotonated in the activated RuBP unbound active site, so that it can interact with O3-H once RuBP enters the active site to give the protonated form His294
+. Proton transfer processes required for RuBP enolization are mediated by two proton wires acting in sequence: H3-H
2O-KCX201 (see transition state A
≠) and KCX201-H
2O-H
2O-O2 (see intermediate B). Enolization is started by assistance of the H3-H
2O-KCX201 proton wire acting in H3 abstraction to form H
3O
+ (in A
≠) followed by H
+ transfer to KCX201 to give KCX201-H in intermediate B. It is important to note that H3 abstraction causes negative charge localization at O2 in intermediate B. The assistance of proton wire KCX201-H
2O-H
2O-O2 (in transition state B
≠) allows proton transfer to O2 to give the RuBP enolate shown in intermediate C.
Considering
Figure 14b, intermediate C shows a C2=C3 distance of 1.35 Å in agreement with formation of a double bond. The electron density associated with the C2=C3 bond is largely employed in a nucleophilic attack onto CO
2 affording intermediate
D where C2-C3 distance (1.53 Å) and C2-CO2 distance (1.59 Å) agree with single bond formation. At the same time, a positive charge located at C3 triggers hydration by the vicinal water molecule (transition state D
≠) while KCX201 accepts the excess proton producing the C3-gemdiolate in intermediate E. The model considers Glu60 (located at the loop 6) as being in protonated form and playing a role in stabilizing the 2-carboxy moiety through interaction with an additional water molecule.
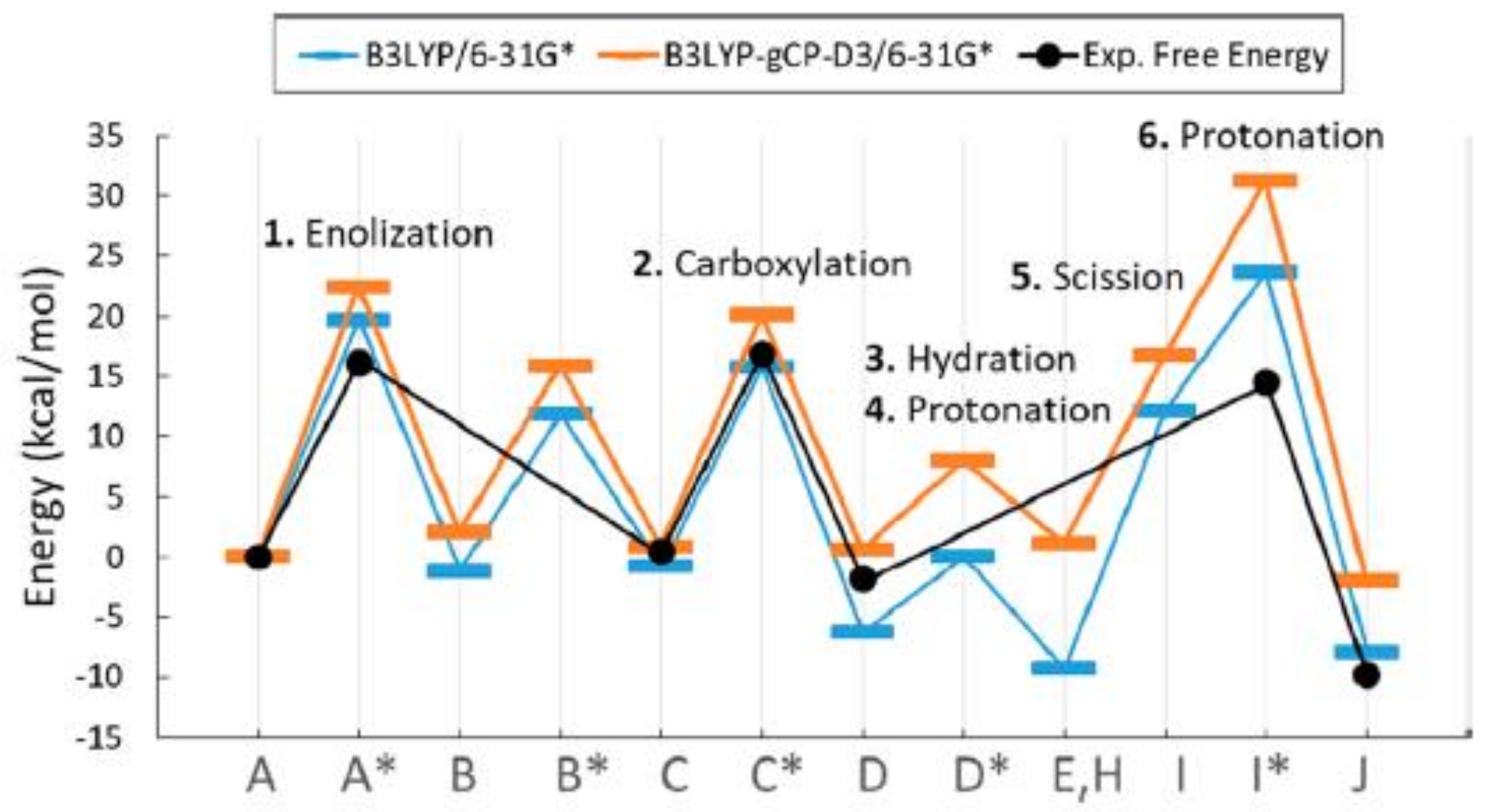
Figure 15 displays the potential energy profile along the reaction coordinate calculated by using the B3LYP/6-31G* and B3LYP-gCP-D3 functional (represented in blue and orange color, respectively), compared with the experimental estimations of free-energy barriers (represented in black).
As shown in
Figure 15, the experimental activation free energy (related to
kcat,C) is estimated at 16 kcal/mol [
44,
45] while calculated potential energy barriers are consistently higher, probably due to the error range for the approximate DFT methods.
Although not all the aspects of the proposed reaction mechanism find good interpretation, the model seems to provide a unique explanation for the a set of experimental evidence.
Mulholland et al. [
46] tried to obtain more accurate estimation for the reaction electronic energy involved in the Rubisco carboxylation reaction by using the projector-based embedding technique (SCS-MP2/aug-cc-pVDZ method) applied to the model fragments constructed by the Gready group in 2008 (each model fragment composed of 77 atoms and 316 electrons). Although the selected computational method usually provides estimation of the activation and reaction energy close to that of the CCSD(T) method [
47] when applied to enzyme active sites, the energy profile produced for the complete reaction coordinate (including carboxylation, hydration, dissociation and protonation reaction steps) seems not to agree with experimental results.
9. Rubisco Kinetics
From a bioenergetic point of view, the RuBP conversion into two 3PGA molecules catalyzed by Rubisco is spontaneous as the Gibbs free energy change (ΔG° as calculated by Faber et al. [
48] at about −30 kcal mol
−1) allows the reaction to proceed without energy supply in the form of ATP, NAD(P)H or other reducing agents. Concerning the enzyme kinetics, experimental estimation of the activation free-energy barrier for the rate-limiting transition state is, as cited above, around 16 kcal/mol (at 298 K, for Rubisco from spinach). The corresponding maximal catalytic rate observed in vitro (
Kcat,C) is 3–4 s
−1. However, the potential energy profile of the overall reaction appears quite complex (see
Figure 15), therefore, over the last two decades, considerable effort has been made towards a complete description of the elementary steps and associated microscopic rate constants to assist in understanding enzyme kinetics.
Very recently Cummins and Gready [
49] have described a detailed multistep mechanism for the Rubisco carboxylation and oxygenation reactions including microscopic rate constants associated with each elementary step (see
Section S6, Figure S5). On the basis of the proposed mechanism the authors were able to derive equations that relate all measurable kinetic parameters such as S
C/O,
kcat,C, K
C,
kcat,O, and K
O (defined in
Table S3) to the microscopic rate constants. Moreover, the authors have calculated the expected values of dissociation rate constants for CO
2 and O
2 bound to the RuBP-enzyme complex. The kinetic model described by Cummins and Gready has been used by Milo et al., to draw important information about the variability of the S
C/O selectivity factor among a large series of Rubisco enzymes (see later).
Before describing recent advances achieved by the Gready and Milo Research Groups, it is worthwhile citing previous studies developed by Tcherkez [
50] and Tlusty [
30], which started evidencing specific correlations between measurable kinetic parameters with the aim of finding specific adaptations during Rubisco evolution.
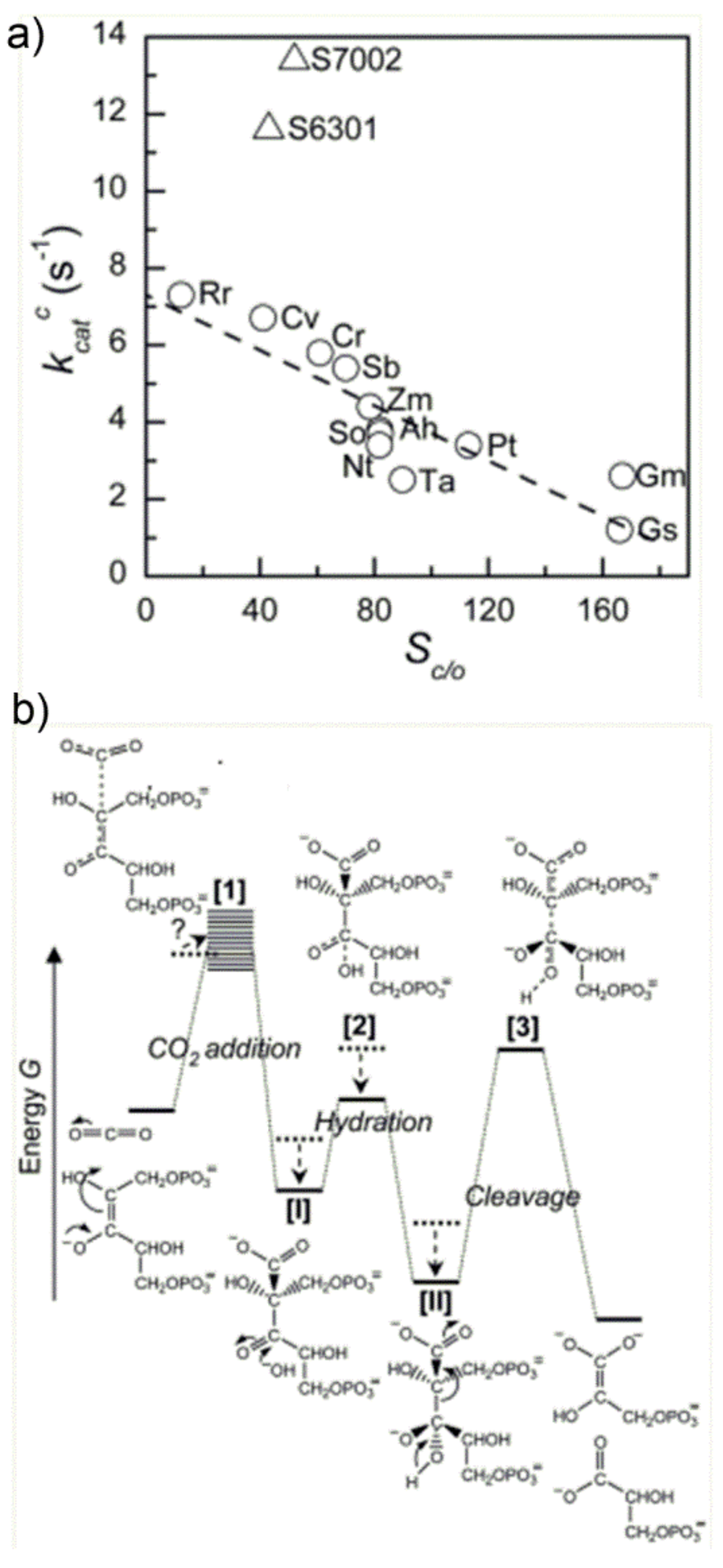
In an interesting study, Tcherkezet al. [
50] have analyzed the
kcat,C value of diverse Rubisco enzymes (see
Figure 16a), evidencing an inverse relationship between S
C/O and
kcat,C. This observation could be rationalized by proposing the qualitative energy profile along the reaction coordinate shown in
Figure 16b.
The major feature of the diagram shown in
Figure 16b is the appearance of three energetic barriers (for the CO
2-addition reaction, carboxylic acid hydration and C2-C3-gemdiolate cleavage, respectively), with the “C2-C3-gemdiolate cleavage” barrier being the higher, which identifies the rate determining step. Most important, the authors have proposed that different Rubisco enzymes have evolved by optimizing the energetic profile in response to the CO
2:O
2 ratio available at the subcellular environment, which is characteristic of each species. Moreover, in order to increase the ability of Rubisco to discriminate between CO
2 and O
2 (enhancing thus the CO
2/O
2 specificity) the authors propose that the TS 1 for the CO
2-addition reaction (first energetic barrier in
Figure 16b) must be product-like. In other words, the enzyme active site must possess a high affinity for the enediolate carboxylate TS 1 and, therefore, the ability to stabilize it in the energetic diagram. Under this hypothesis, the carboxy-ketone intermediate [
I] and its gem-diol hydrate [
II], possessing a carboxylate-moiety, are proposed to bind tightly the active site undergoing energy stabilization. As a consequence, the energy stabilization of intermediate II causes an increase in the activation barrier for the subsequent C2-C3-gemdiolate cleavage-step, which is related to k
cat,
C. On the whole, according to the author’s hypothesis, an increase in CO
2/O
2 specificity is paid for with a slower rate of C2-C3 bond cleavage and, ultimately, with a slower overall reaction. The occurrence of these two coupled effects is cited in the literature as the “trade-off” between CO
2-specificity and reaction rate: the more the enzyme is specific for CO
2 (vs. O
2), the slower the overall carboxylation/hydration reaction and vice versa.
In 2010 Tlusty et al. [
30] simplified the energetic profile proposed by Tcherkez et al., identifying two energetic “effective barriers” along the reaction coordinate corresponding to two irreversible steps which were identified as: (i) “effective gas binding” (encompassing RuBP enolization and subsequent CO
2/O
2 addition) described by the
kon,C kinetic constants; (ii) “effective catalysis” (encompassing the C2-carboxylic acid hydrolysis and C2-C3 gemdiolate bond cleavage) described by
kcat,C kinetic constant. Overall, Tlusty’s study confirmed the negative power low correlation between specificity S
C/O and
kcat,C.
The postulated “trade-off” between CO
2-specificity and carboxylation reaction rate of Rubisco was criticized by the capstone study published by Cummins and Gready in 2018 [
49]. Giving a complete description of the reaction elementary steps and related microscopic kinetic constants, the study aimed at evaluating whether the Rubisco enzyme is tightly constrained by its active-site chemistry or whether the implementation of powerful tools as directed evolution techniques may have the opportunity to break the above cited “trade-off” and improve simultaneously both
kcat,C and S
C/O parameters. Results of the analysis point out that simultaneous improvement of CO
2-specificity and turnover rate is possible considered that, more likely, the stronger molecular constraint relay on the “activity-molecular stability” trade off. Moreover, the authors gave a complete derivation of the Michaelis-Menthen type rate, law for the Rubisco catalytic activity.
Very recently, the increased availability of measured kinetic parameters for the RuBP carboxylation and oxygenation reactions from a variety of C3- and C4-plants, algae (both green and red algae), bacteria and cyanobacteria allowed the analysis of a greater collection of data among different species.
Milo et al., have investigated the possible relationship between kinetic parameters measured for approximately 325 species at 25 °C and pH = 8 [
51]. The analysis was developed by using the Rubisco reaction mechanism and nomenclature described by Cummins and Gready in 2018 [
49] (see
Section S6).
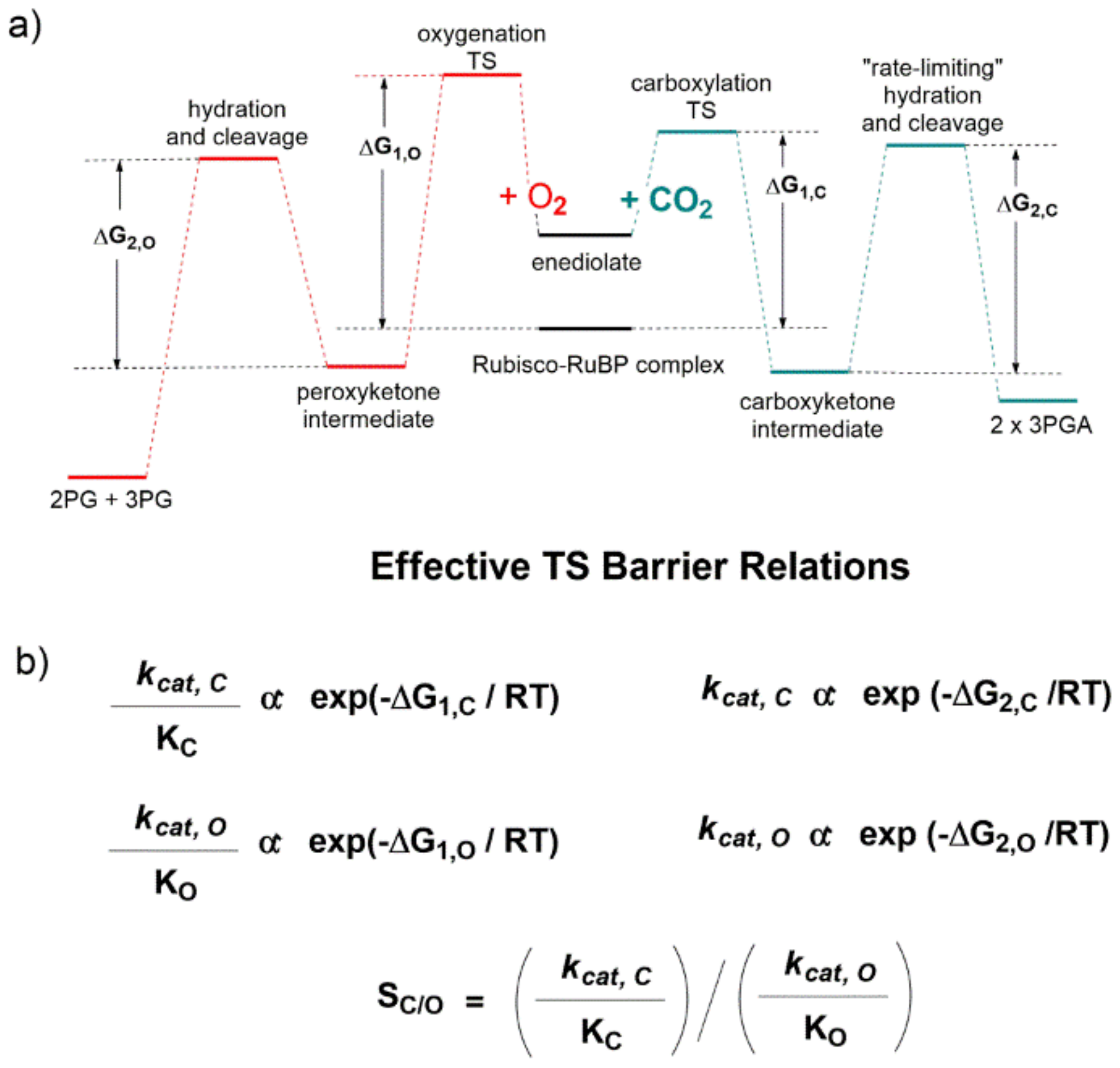
The qualitative energetic diagram shown in
Figure 17a evidences that the carboxylation (line green profile) and oxygenation (line red profile) reactions proceed through two “effective” energetic barriers. Considering the carboxylation reaction after RuBP enolization, the first energetic barrier is related to CO
2-addition to the enediolate, while the second “rate limiting” energetic barrier is related to the C2-carboxylic acid hydration and C2-C3-gemdiolate bond cleavage (
Figure 17a). Given standard assumptions, the authors derived the catalytic efficiency (defined as
kcat/K
M) is related to the height of the first effective barrier, while
kcat is related to the second (
Figure 17b).
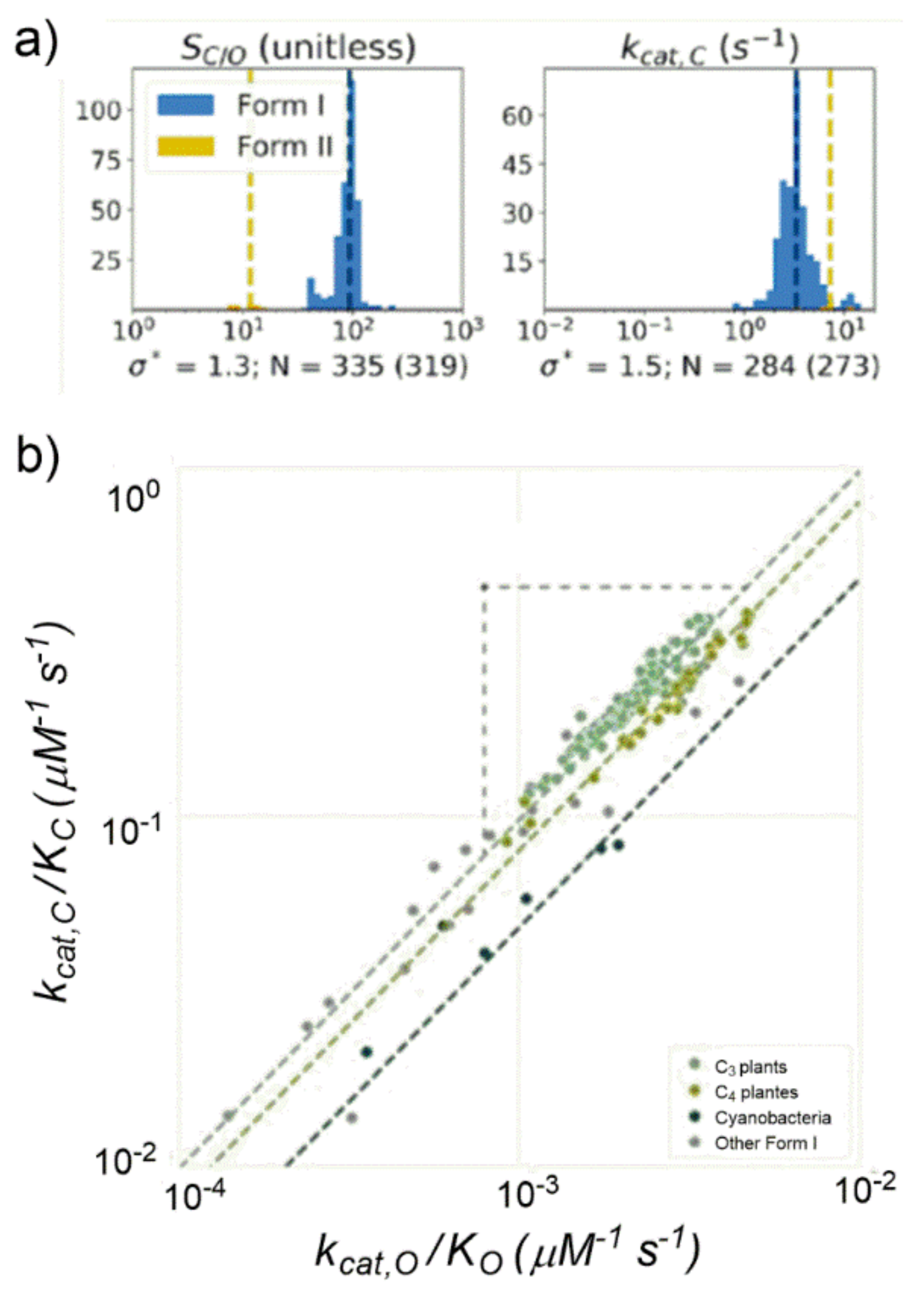
Thanks to the increased amount of kinetic data analyzed, Milo observed that the previously assessed strong
kcat,C-S
C/O negative correlation was significantly weakened. The previously proposed correlation between
kcat,C and K
C or
kcat,C and S
C/O appeared inconsistent if analyzed among a larger amount of data. An important outcome of the study was that a short range of variations clearly emerged for most kinetic parameters if analysed among organisms with similar physiology. For example,
kcat,C showed variation by only 50% among Form I Rubiscos and S
C/O showed variation of less than 30% among Form I Rubiscos (
Figure 18a).
Considering the S
C/O definition (see
Table S3) the following power–law relationship could be derived: log(
kcat,C/K
C) = log(
kcat,O/K
O) + log(S
C/O). When considering a set of data grouped for similar cellular physiology such as C3- and C4-plants and cyanobacteria (
Figure 18b), having S
C/O values roughly constant, experimentally a strong positive correlation was found between
kcat,C/K
C and
kcat,O/K
O (in a log–log diagram). As matter of fact, Form I Rubiscos display a high-confidence power-law relationship between
kcat,C/K
C and
kcat,O/K
O (R = 0.94,
p < 10
−10).
Moreover, considering that the catalytic efficiencies for the carboxylation and oxygenation reactions are proportional to ΔG
1,C and ΔG
1,O, respectively, (ΔG
1,C and ΔG
1,O are shown in
Figure 17a), the following relationship must hold: ΔG
1,C = ΔG
1,O + C. As a consequence of this relationship, at the active site molecular level every factor causing a decrease of the CO
2-addition barrier (ΔG
1,C) causes, at the same time, an approximately equal decrease in the O
2-addition barrier (ΔG
1,O).
10. Photorespiration
As described above, according to the recent study published by Milo et al., the catalytic efficiencies of the carboxylation and oxygenation reactions are strictly related. Photosynthetic organisms (such as plants and bacteria) with similar cellular physiology have adapted to share similar SC/O selectivity. This observation can indirectly indicate that efforts aiming at improving SC/O value by directed mutagenesis would probably be unproductive.
However, a recent study from the Farquhar Group [
12] opened a new research scenario, having evidenced the existence of biochemical links between the photosynthesis and photorespiration pathways. The study shed a new light on the photorespiration process and some of its beneficial features, giving a possible explanation as to why photosynthetic organisms have evolved by “tolerating” the oxygenation reaction instead of “eliminating” this side reaction.
Over the last four decades, according to the Farquhar et al., (FvCB model) photosynthesis model, [
11] the RuBP-oxygenation reaction leading to 2-phosphoglycolate production has been considered a deleterious process, as the toxic intermediate 2-phosphoglycolate must be rapidly metabolized by the photorespiratory metabolism causing the following two drawbacks: (i) reduction of RuBP available for the carboxylation reaction; (ii) recycle of glycolate back to RuBP in a process that co-produces CO
2 and NH
3. CO
2 is usually released from leaf while NH
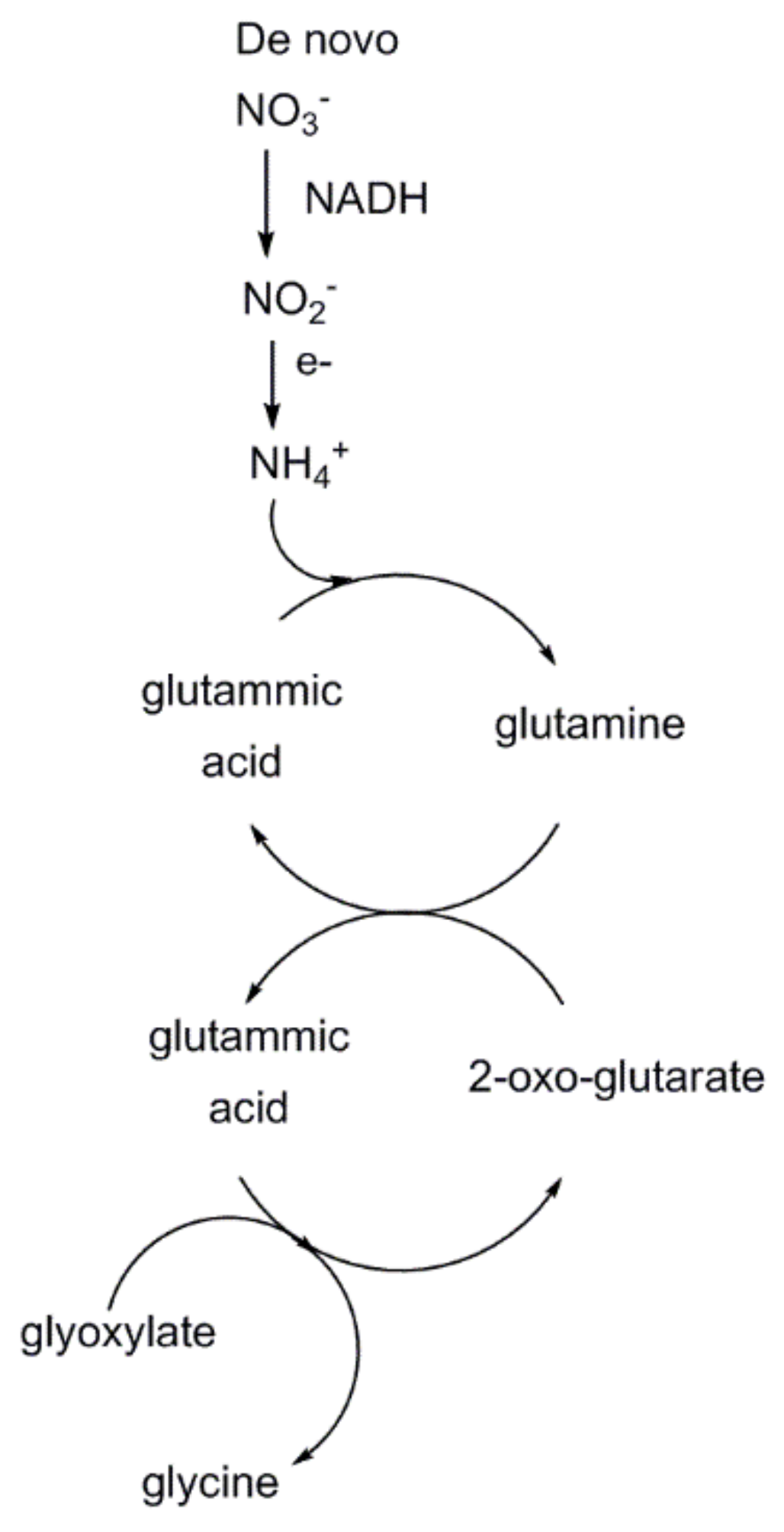
3 recycling is energetically costly. Overall, approximately 2 mol of 2-phosphoglycolate produced undo one carboxylation reaction, causing a decrease of carbon fixation efficiency into hexose sugars by up to 50%. However, the recent study points out that photorespiratory metabolism increases de novo nitrogen uptake (as NO
3−) and reduction (to NH
4+) by terrestrial plants, increasing availability of the precious element that enters the 2-oxo-glutarate/glutamic acid cycle harnessing, thus, glyoxylate conversion into glycine (
Figure 19). Therefore, the negative effect due to reduced CO
2-fixation into hexose sugars is counterbalanced by an increased synthesis of glycine that is partially converted into serine. Overall, photorespiration, despite reducing the Rubisco carboxylation efficiency, seems to improve the overall photosynthetic performance.
11. Conclusions
As mentioned in the introductory section, the steady increase over the last century of the CO2-fertilization effect due to increase of atmospheric CO2 concentration (GATM) is considered the largest contributor (among others) to the modelled GPP increase by terrestrial plants and, ultimately, to the terrestrial CO2-sink (SLAND) ability.
In a recent study Farquhar et al. [
4] on the basis of various Terrestrial Biosphere Models (TBM), have shown that GPP (expressed in units of Gt of carbon atoms fixed annually by plants at global level) has constantly increased from approximately 105 GtCy
−1 estimated in 1900 to approximately 140 GtCy
−1 estimated in 2010. According to these models, approximately 135 GtCy
−1 fixed into plant biomass in 2010 are due to the CO
2-fertilization effect that accounts for a 30% increase with respect to the value estimated in 1900.
Moreover, Farquhar identifies two main components as contributing to the CO2-fertilization effect: (i) the CO2 greening effect; (ii) the CO2 leaf-level effect. The CO2 greening effect is defined as “the increased light harvesting capacity due to the investment of additional plant carbon in a higher leaf area” while the CO2 leaf-level effect is “the CO2-induced increase in GPP that occurs at constant leaf area”. The leaf level effect, in turn, includes the direct biochemical effect of increased atmospheric CO2 on the Rubisco carboxylase activity.
According to the Farquhar’s study and other related research [
52,
53], terrestrial plants have shown a significant natural ability to adapt to increased CO
2-concentration over the last century. The development of more advanced TBMs taking into account the plant coordination of photosynthesis [
54] will probably give a better estimation of plant’s potential for CO
2-removal in the near future.

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}