
Promotion Effect of the Keggin Structure on the Sulfur and Water Resistance of Pt/CeTi Catalysts for CO Oxidation


Abstract
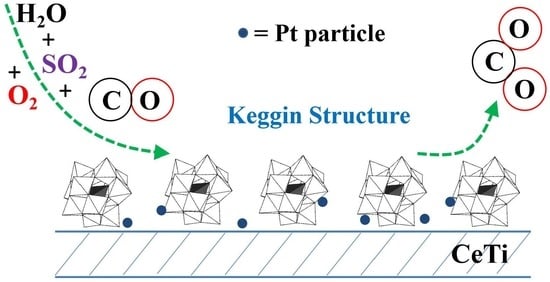
:
1. Introduction
2. Results and Discussion
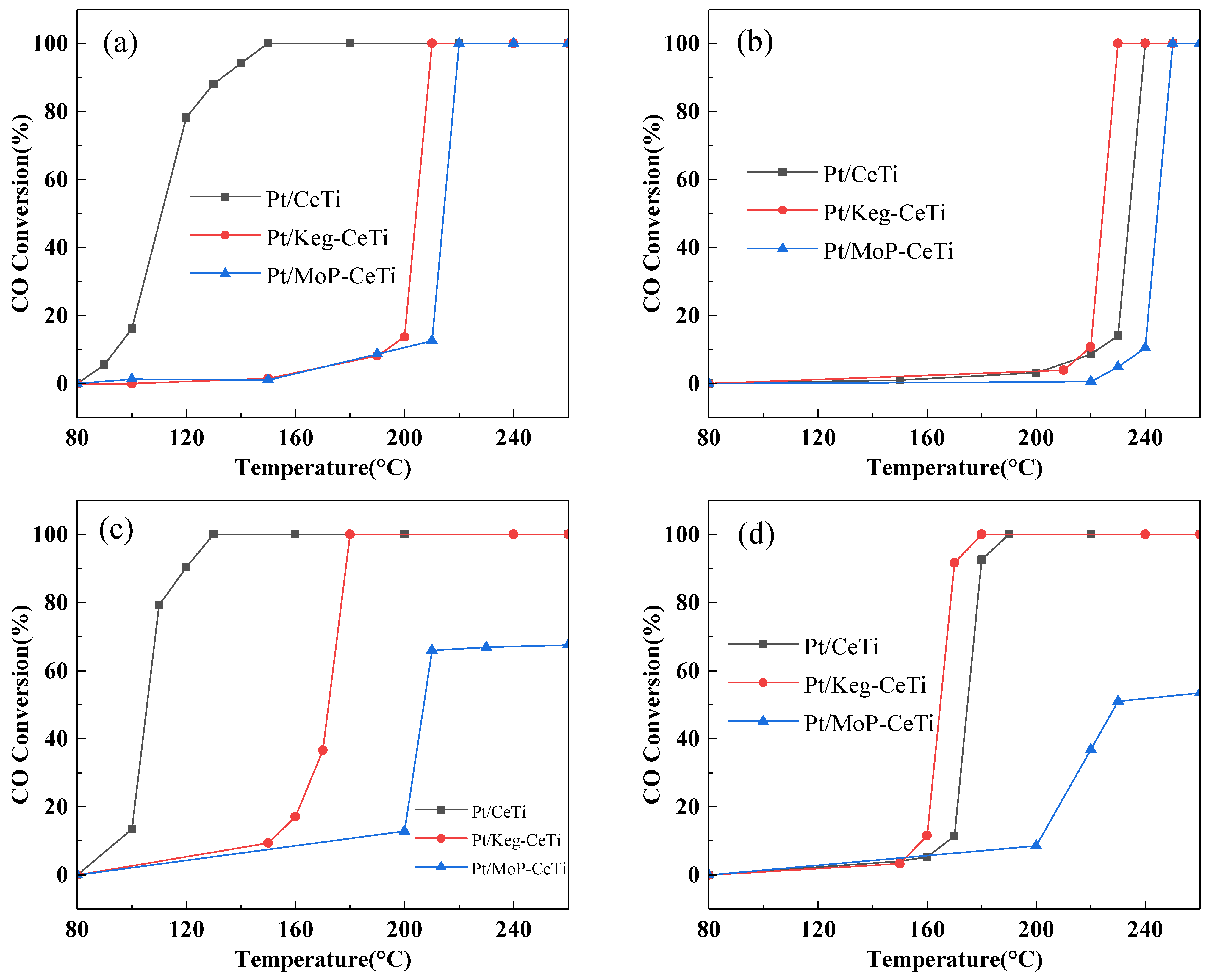
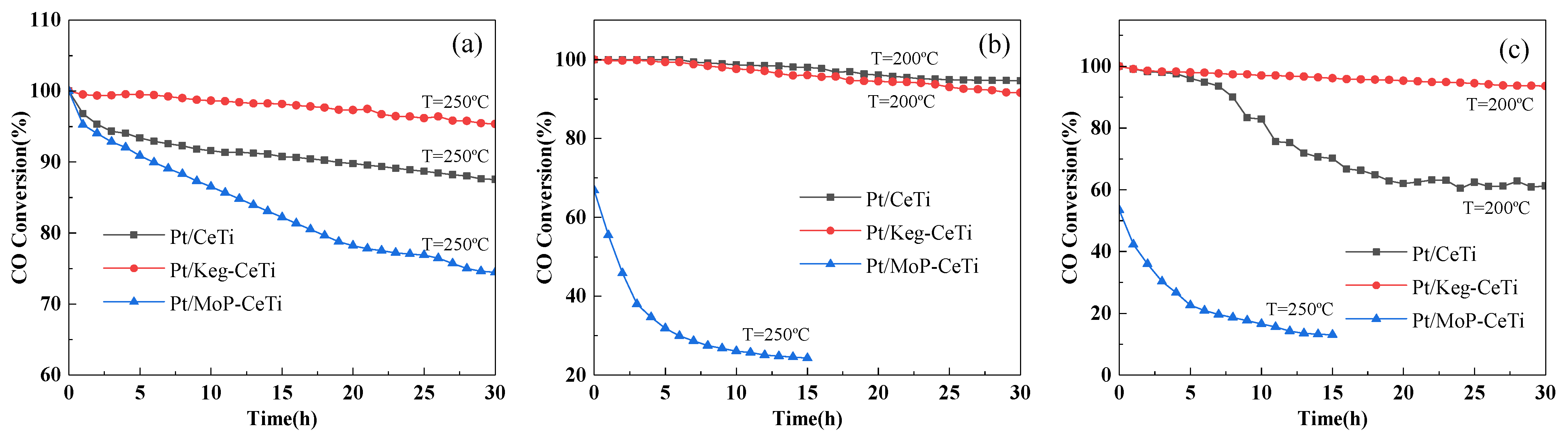
2.1. Catalytic Activity and SO2/H2O Durability
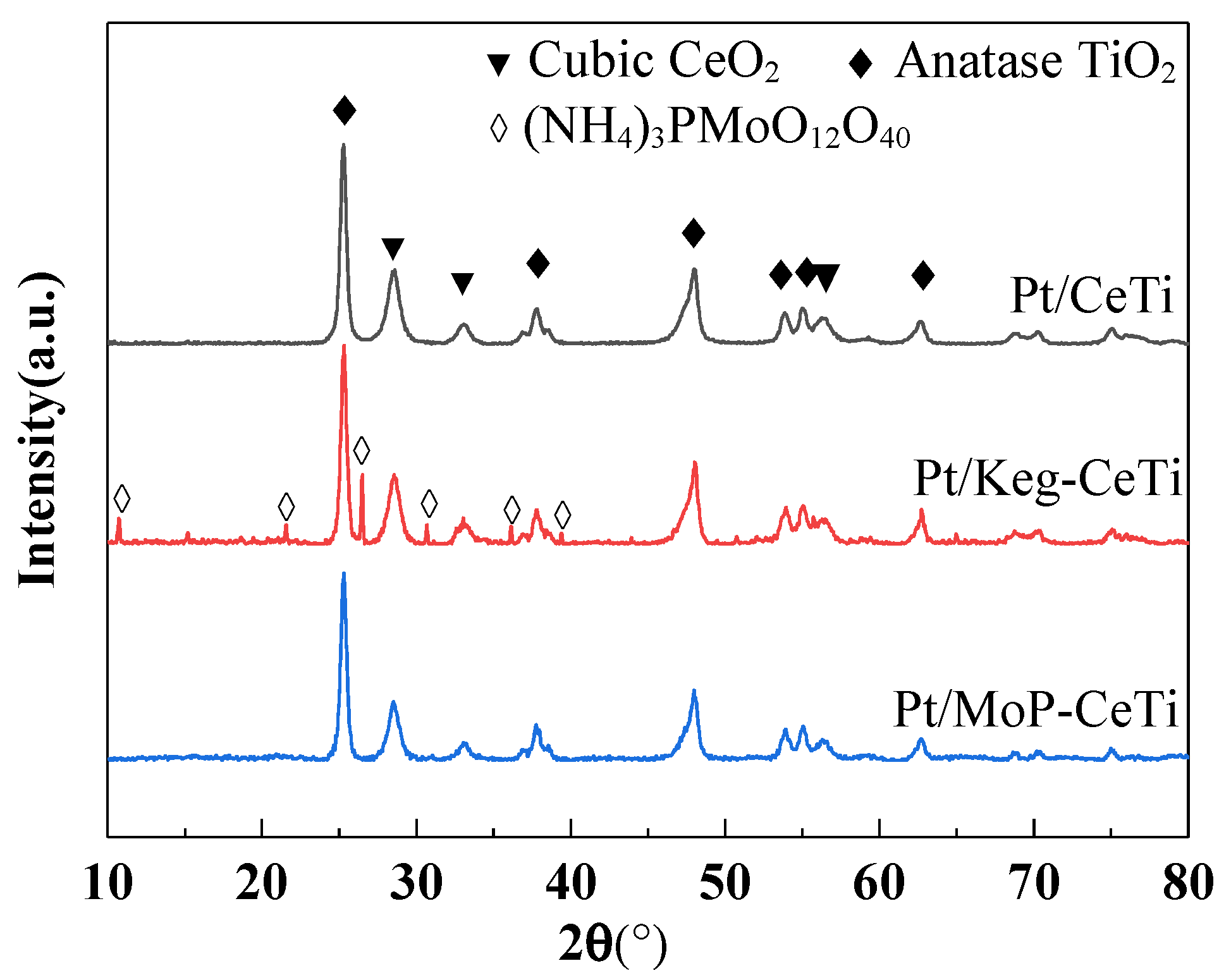
2.2. Structure and Morphology
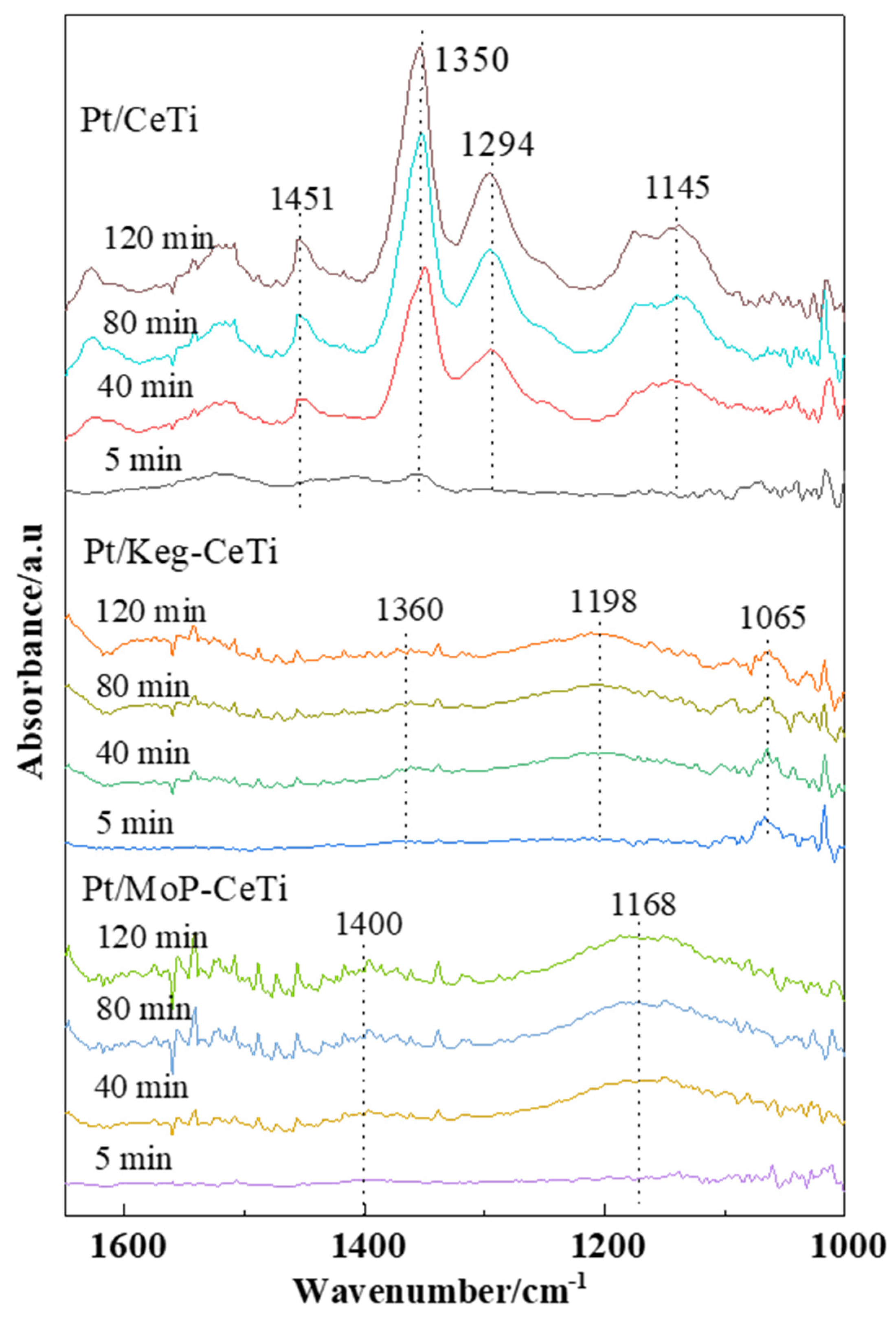
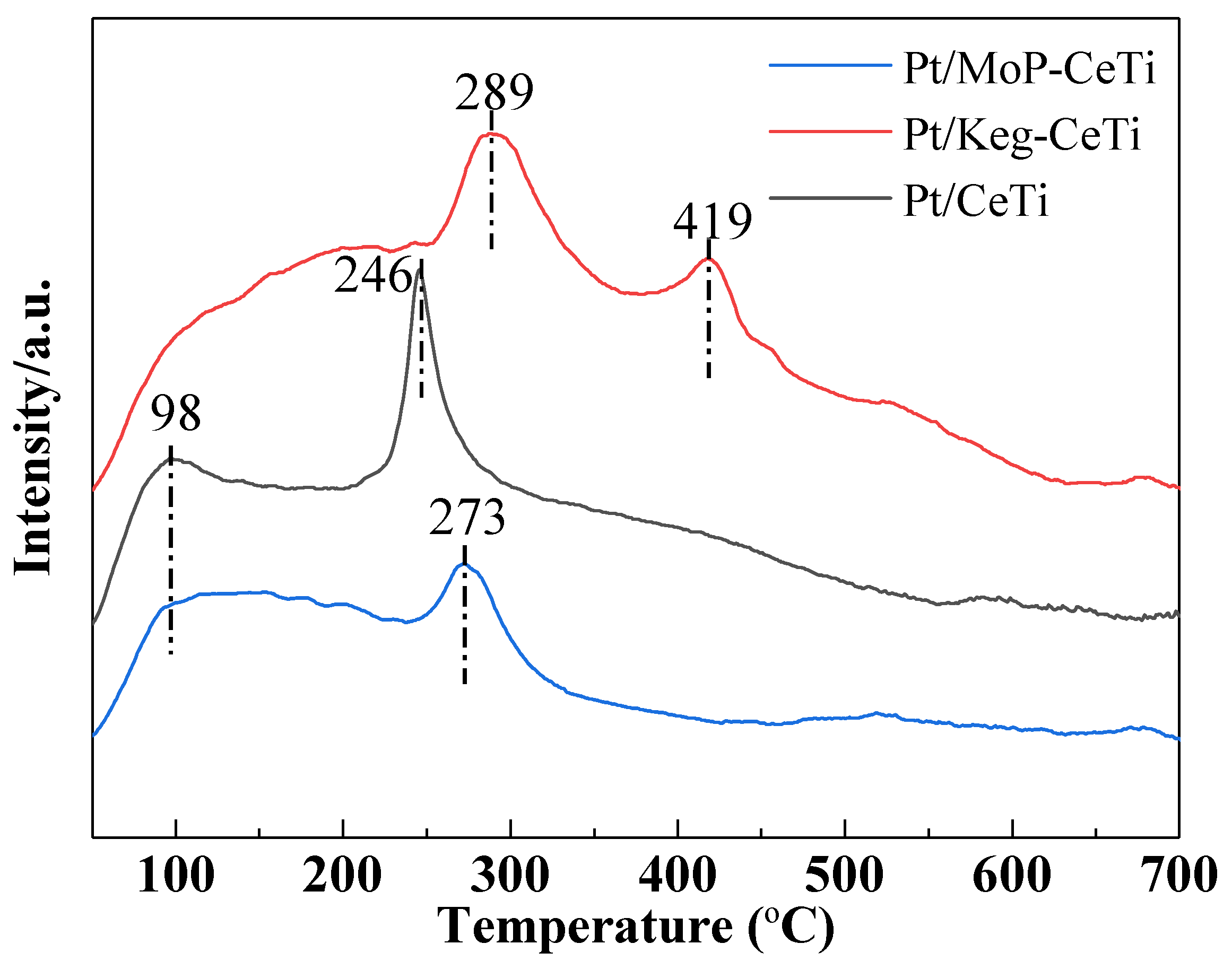
2.3. Adsorption and Desorption of SO2 on the Catalysts
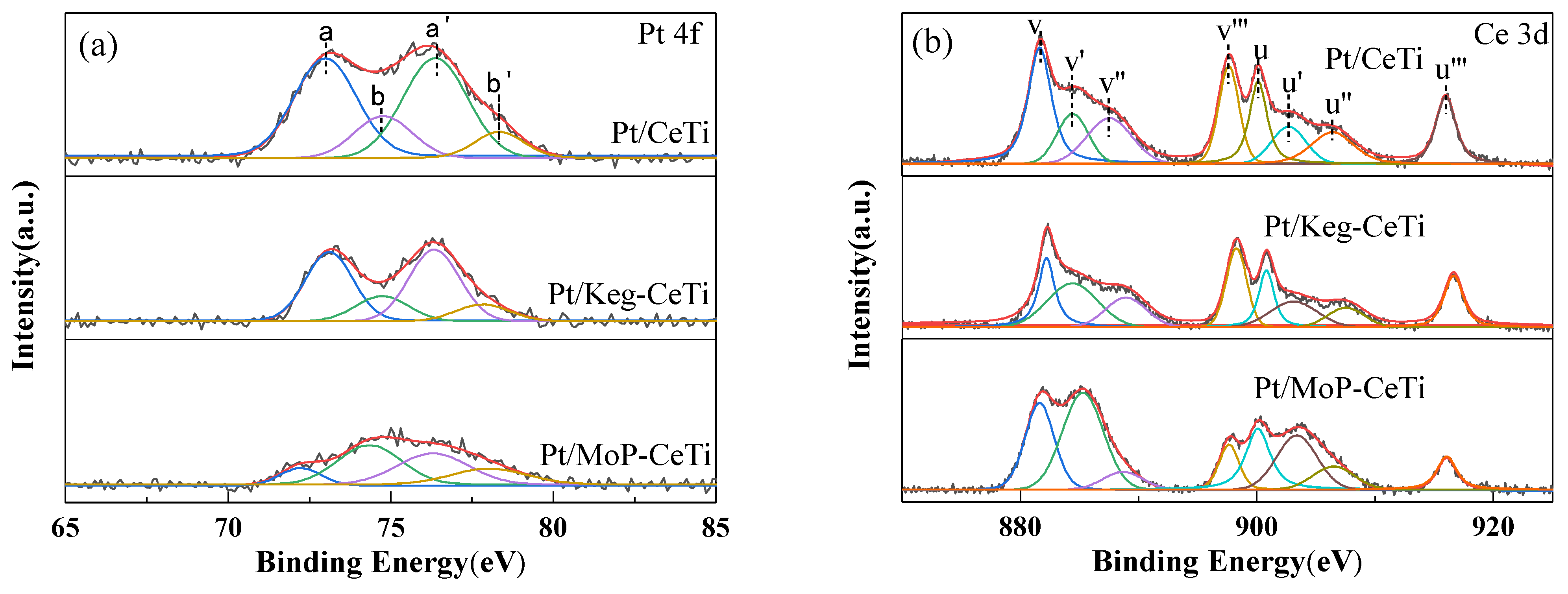
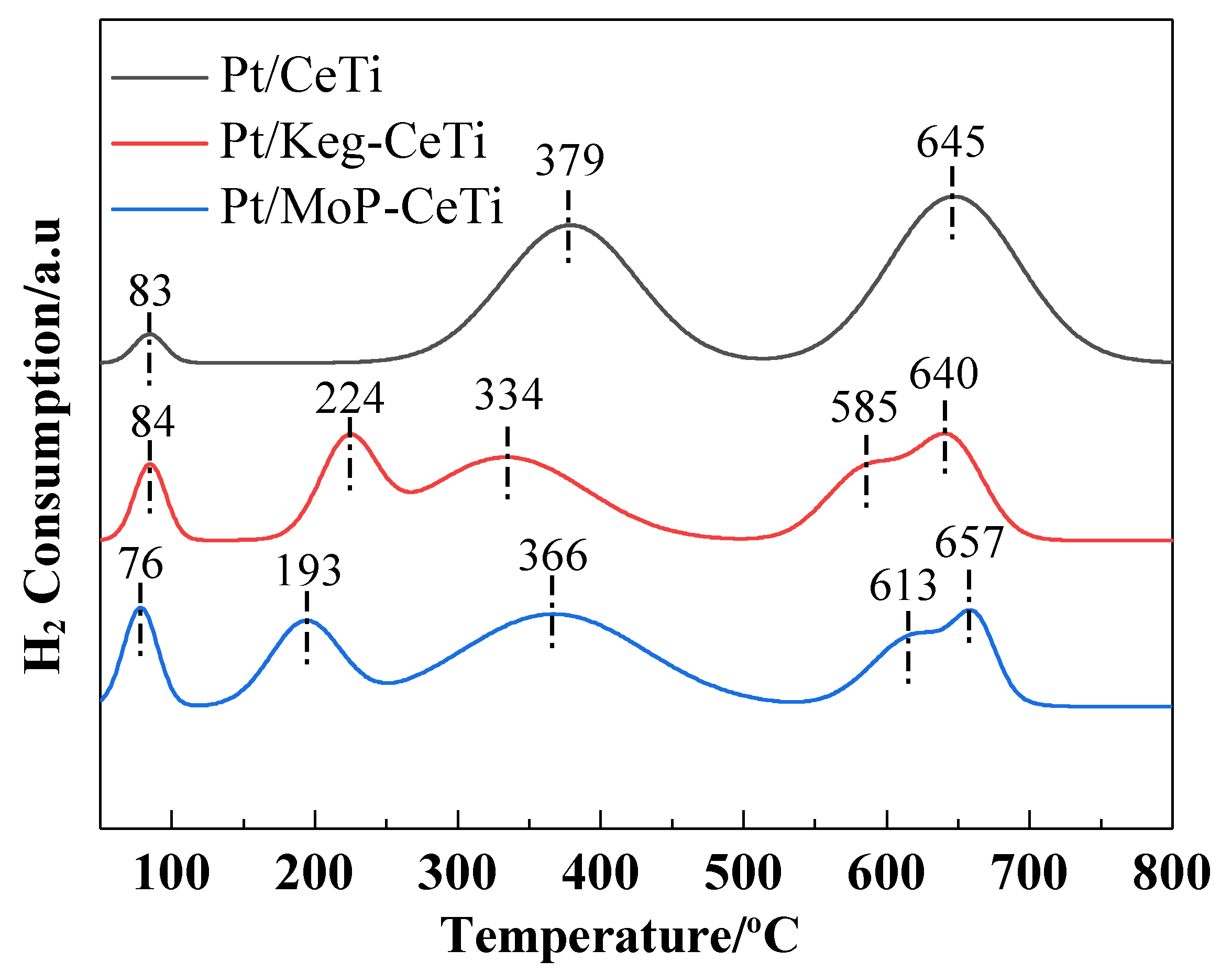
2.4. Surface Properties and Redox Property
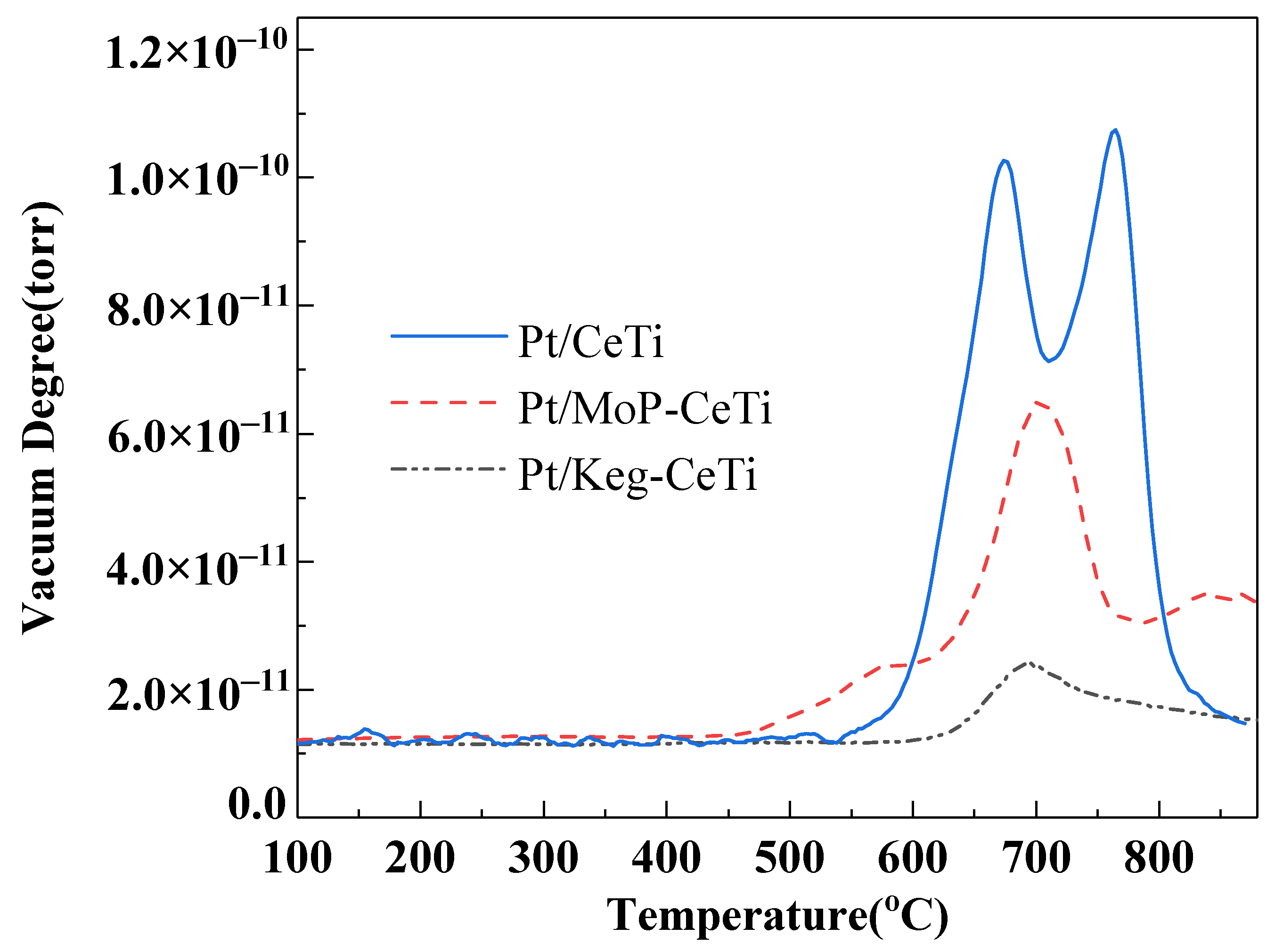
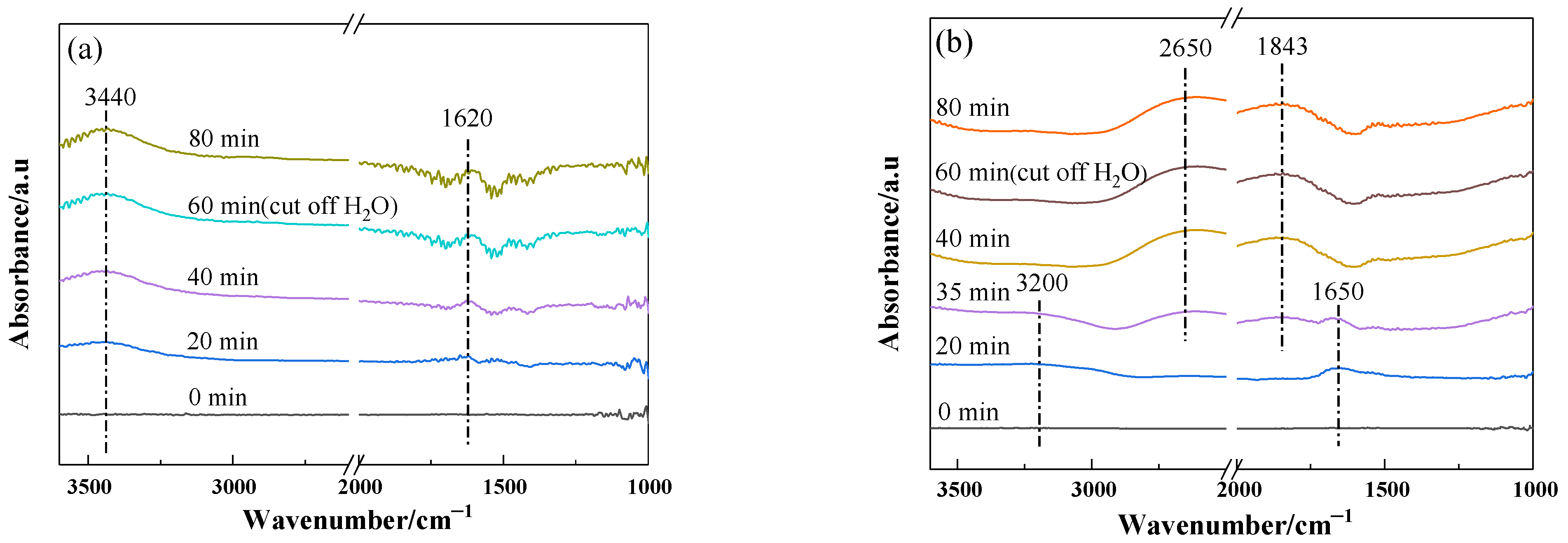
2.5. Water Adsorption on the Catalysts
3. Experimental
3.1. Chemicals
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Catalytic Activity Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kumar, G.M.; Sampath, S.; Jeena, V.S.; Anjali, R. Carbon monoxide pollution levels at environmentally different sites. J. Ind. Geophys. Union. 2008, 12, 31–40. [Google Scholar]
- DeCoste, J.B.; Peterson, G.W. Metal-organic frameworks for air purification of toxic chemicals. Chem. Rev. 2014, 114, 5695–5727. [Google Scholar] [CrossRef]
- Wang, W.; Gong, J.L. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar]
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Patel, D.M.; Kodgire, P.; Dwivedi, A.H. Low temperature oxidation of carbon monoxide for heat recuperation: A green approach for energy production and a catalytic review. J. Clean Prod. 2020, 245, 118838. [Google Scholar] [CrossRef]
- Soubaihi, R.M.A.; Saoud, K.M.; Dutta, J. Critical review of low-temperature CO oxidation and hysteresis phenomenon on heterogeneous catalysts. Catalysts 2018, 8, 660. [Google Scholar] [CrossRef] [Green Version]
- Dobrosz-Gomez, I.; Gomez-Garcia, M.-A.; Rynkowski, J.M. The origin of Au/Ce1-xZrxO2 catalyst’s active sites in low-temperature CO oxidation. Catalysts 2020, 10, 1312. [Google Scholar] [CrossRef]
- Prasad, R.; Singh, P. A review on CO oxidation over copper chromite catalyst. Catal. Rev. Sci. Eng. 2012, 54, 224–279. [Google Scholar] [CrossRef]
- Lin, J.; Wang, X.D.; Zhang, T. Recent progress in CO oxidation over Pt-group-metal catalysts at low temperatures. Chin. J. Catal. 2016, 37, 1805–1813. [Google Scholar] [CrossRef]
- Dey, S.; Dhal, G.C. Property and structure of various platinum catalysts for low temperature carbon monoxide oxidations. Mater. Today Chem. 2020, 16, 100228. [Google Scholar] [CrossRef]
- Beniya, A.; Higashi, S.; Ohba, N.; Jinnouchi, R.; Hirata, H.; Watanabe, Y. CO oxidation activity of non-reducible oxide-supported mass-selected few-atom Pt single-clusters. Nat. Commun. 2020, 11, 1888. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayahsi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of Carbon Monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Haruta, M. Gold rush. Nature 2005, 437, 1098–1099. [Google Scholar] [CrossRef]
- Liu, G.; Walsh, A.G.; Zhang, P. Synergism of iron and platinum species for low-temperature CO oxidation: From two-dimensional surface to nanoparticle and single-atom catalysts. J. Phys. Chem. Lett. 2020, 11, 2219–2229. [Google Scholar] [CrossRef]
- Lamb, A.B.; Bray, W.C.; Frazer, J. The removal of carbon monoxide from air. Ind. Eng. Chem. 1920, 12, 13–221. [Google Scholar] [CrossRef]
- Dey, S.; Dhal, G.C. Deactivation and regeneration of hopcalite catalyst for carbon monoxide oxidation: A review. Mater. Today Chem. 2019, 14, 100180. [Google Scholar] [CrossRef]
- Pillai, U.R.; Deevi, S. Room temperature oxidation of carbon monoxide over copper oxide catalyst. Appl. Catal. B Environ. 2006, 64, 146–151. [Google Scholar] [CrossRef]
- Abdul-Kareem, H.K.; Hudgins, R.R.; Silveston, P.L. Forced cycling of the catalytic oxidation of CO over a V2O5 catalyst II Temperature cycling. Chem. Eng. Sci. 1980, 35, 2085–2088. [Google Scholar] [CrossRef]
- Dey, S.; Dhal, G.C. Cerium catalysts applications in carbon monoxide oxidations. Mat. Sci. Energy Technol. 2020, 3, 6–24. [Google Scholar] [CrossRef]
- Xie, X.W.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W.J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Langmuir, I. The mechanism of the catalytic action of platinum in the reactions 2CO + O2= 2CO2 and 2H2+ O2= 2H2O. Trans. Faraday Soc. 1922, 17, 621–654. [Google Scholar] [CrossRef]
- Kummer, J. Use of noble metals in automobile exhaust catalysts. J. Phys. Chem. 1986, 90, 4747–4752. [Google Scholar] [CrossRef]
- Gandh, H.S.; Graham, G.W.; McCabe, R.W. Automotive exhaust catalysis. J. Catal. 2003, 216, 433–442. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Ramalingam, S.; Rajendran, S.; Ganesan, P. Performance improvement and exhaust emissions reduction in biodiesel operated diesel engine through the use of operating parameters and catalytic converter: A review. Renew. Sustain. Energy Rev. 2018, 81, 3215–3222. [Google Scholar] [CrossRef]
- Shin, H.; Baek, M.; Kim, D.H. Sulfur resistance of Ca-substituted LaCoO3 catalysts in CO oxidation. Mol. Catal. 2019, 468, 148–153. [Google Scholar] [CrossRef]
- Yan, D.; Li, Q.; Zhang, H.; Zhou, X.; Chen, H. A highly dispersed mesoporous zeolite@TiO2-supported Pt for enhanced sulfur-resistance catalytic CO oxidation. Catal. Commun. 2020, 142, 106042. [Google Scholar] [CrossRef]
- Shin, H.; Baek, M.; Ro, Y.; Song, C.; Lee, K.-Y.; Song, I.K. Improvement of sulfur resistance of Pd/Ce-Zr-Al-O catalysts for CO oxidation. Appl. Surf. Sci. 2018, 429, 102–107. [Google Scholar] [CrossRef]
- Jiang, Q.; Huang, M.; Qian, Y.; Miao, Y.; Ao, Z. Excellent sulfur and water resistance for CO oxidation on Pt single-atom-catalyst supported by defective graphene: The effect of vacancy type. Appl. Surf. Sci. 2021, 566, 150624. [Google Scholar] [CrossRef]
- Hu, J.; Burns, R.C. Homogeneous-phase catalytic H2O2 oxidation of isobutyraldehyde using Keggin, Dawson and transition metal-substituted lacunary heteropolyanions. J. Mol. Catal. A Chem. 2002, 184, 451–464. [Google Scholar] [CrossRef]
- Golodov, V.A.; Jumakaeva, B.S. Catalytic oxidation of CO by heteropolyacids (HPA) and dioxygen in the presence of Pd(II) salt-HPA-H2O system. J. Mol. Catal. 1986, 35, 309–315. [Google Scholar] [CrossRef]
- Zhizhina, E.G.; Kuznetsova, L.I.; Maksimovskaya, R.I.; Pavlova, S.N.; Maweev, K.I. Oxidation of CO to CO2 by heteropolyacids in the presence of Palladium. J. Mol. Catal. 1986, 38, 345–353. [Google Scholar] [CrossRef]
- Zhizhina, E.G.; Matveev, K.I. Low-temperature oxidation of CO to CO2 in solutions of halide complexes of Pt and heteropolyacids (HPA). React. Kinet. Catal. Lett. 1992, 47, 255–262. [Google Scholar] [CrossRef]
- Stanis, R.J.; Kuo, M.-C.; Turner, J.A.; Herring, A.M. Use of W, Mo, and V substituted heteropolyacids for CO mitigation in PEMFCs. J. Electrochem. Soc. 2008, 155, B155–B162. [Google Scholar] [CrossRef]
- Yoshida, T.; Murayama, T.; Sakaguchi, N.; Okumura, M.; Ishida, T.; Haruta, M. Carbon monoxide oxidation by polyoxometalate-supported gold nanoparticulate catalysts: Activity, stability, and temperature dependent activation properties. Angew. Chem. Int. Ed. 2018, 130, 1539–1543. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, N.Q.; Liu, X.J.; Li, L.C.; Song, L.Y.; Qiu, W.G.; He, H. The Keggin structure: An important factor in governing NH3-SCR activity over the V2O5-MoO3/TiO2 catalyst. Catal. Lett. 2018, 148, 1228–1235. [Google Scholar] [CrossRef]
- Simsek, E.; Ozkara, S.; Aksoylu, A.E.; Onsan, Z.I. Preferential CO oxidation over activated carbon supported catalysts in H2-rich gas streams containing CO2 and H2O. Appl. Catal. A 2007, 316, 169–174. [Google Scholar] [CrossRef]
- Trimm, D.L. Minimisation of carbon monoxide in a hydrogen stream for fuel cell application. Appl. Catal. A 2005, 296, 1–11. [Google Scholar] [CrossRef]
- Gao, S.; Wang, P.L.; Yu, F.X.; Wang, H.Q.; Wu, Z.B. Dual resistance to alkali metals and SO2: Vanadium and cerium supported on sulfated zirconia as an efficient catalyst for NH3-SCR. Catal. Sci. Technol. 2016, 6, 8148–8157. [Google Scholar] [CrossRef]
- Smirnov, M.Y.; Kalinkin, A.V.; Pashis, A.V.; Sorokin, A.M.; Noskov, A.S.; Bukhtiyarov, V.I.; Kharas, K.C.; Rodkin, M.A. Comparative XPS study of Al2O3 and CeO2 sulfation in reactions with SO2, SO2 + O2, SO2 + H2O, and SO2 + O2 + H2O. Kinet. Catal. 2003, 44, 575–583. [Google Scholar] [CrossRef]
- Luo, T.; Gorte, R. Characterization of SO2-poisoned ceria-zirconia mixed oxides. Appl. Catal. B 2004, 53, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Waqif, M.; Bazin, P.; Saur, O.; Lavalley, J.C.; Blanchard, G.; Touret, O. Study of ceria sulfation. Appl. Catal. B 1997, 11, 193–205. [Google Scholar] [CrossRef]
- Kwon, D.W.; Park, K.H.; Hong, S.C. Enhancement of SCR activity and SO2 resistance on VOx/TiO2 catalyst by addition of molybdenum. Chem. Eng. J. 2016, 284, 315–324. [Google Scholar] [CrossRef]
- Tumuluri, U.; Li, M.J.; Cook, B.G.; Sumpter, B.; Dai, S.; Wu, Z.L. Surface structure dependence of SO2 interaction with ceria nanocrystals with well-defined surface facets. J. Phys. Chem. C 2015, 119, 28895–28905. [Google Scholar] [CrossRef]
- Goodman, A.L.; Bernard, E.T.; Grassian, V.H. Spectroscopic study of nitric acid and water adsorption on oxide particles: Enhanced nitric acid uptake kinetics in the presence of adsorbed water. J. Phys. Chem. A 2001, 105, 6443–6457. [Google Scholar] [CrossRef]
- Tan, W.; Wang, J.M.; Yu, S.H.; Liu, A.N.; Li, L.L.; Guo, K.; Luo, Y.D.; Xie, S.H.; Gao, F.; Liu, F.D.; et al. Morphology-Sensitive sulfation effect on ceria catalysts for NH3-SCR. Top. Catal. 2020, 63, 932–943. [Google Scholar] [CrossRef]
- Bazin, P.; Sauf, O.; Lavalley, J.C.; Blanchard, G.; Visciglio, V.; Touret, O. Influence of platinum on ceria sulfation. Appl. Catal. B 1997, 13, 265–274. [Google Scholar] [CrossRef]
- Xu, W.Q.; He, H.; Yu, Y.B. Deactivation of a Ce/TiO2 Catalyst by SO2 in the Selective Catalytic Reduction of NO by NH3. J. Phys. Chem. C 2009, 113, 4426–4432. [Google Scholar] [CrossRef]
- Pei, W.B.; Liu, Y.X.; Deng, J.G.; Zhang, K.F.; Hou, Z.Q.; Zhao, X.T.; Dai, H.X. Partially embedding Pt nanoparticles in the skeleton of 3DOM Mn2O3: An effective strategy for enhancing catalytic stability in toluene combustion. Appl. Catal. B 2019, 256, 117814. [Google Scholar] [CrossRef]
- Tsunekawa, S.; Fukuda, T.; Kasuya, A. X-ray photoelectron spectroscopy of monodisperse CeO2−x nanoparticles. Surf. Sci. 2000, 457, L437–L440. [Google Scholar] [CrossRef]
- Liu, C.; Chen, L.; Li, J.; Ma, L.; Arandiyan, H.; Du, Y.; Xu, J.; Hao, J. Enhancement of activity and sulfur resistance of CeO2 supported on TiO2-SiO2 for the selective catalytic reduction of NO by NH3. Environ. Sci. Technol. 2012, 46, 6182–6189. [Google Scholar] [CrossRef] [PubMed]
- Sellick, D.R.; Aranda, A.; Garcia, T.; Lopez, J.M.; Solsona, B.; Mastral, A.M.; Morgan, D.J.; Carley, A.F.; Taylor, S.H. Influence of the preparation method on the activity of ceria zirconia mixed oxides for naphthalene total oxidation. Appl. Catal. B 2013, 132–133, 98–106. [Google Scholar] [CrossRef]
- Chmielarz, L.; Dziembaj, R.; Grzybek, T.; Klinik, J.; Lojewski, T.; Olszewska, D.; Wegrzyn, A. Pillared smectite modified with carbon and manganese as catalyst for SCR of NOx with NH3. Catal. Lett. 2000, 70, 51–56. [Google Scholar] [CrossRef]
- Wu, J.; Jin, S.; Wei, X.; Gu, F.; Han, Q.; Lan, Y.; Qian, C.; Li, J.; Wang, X.; Zhang, R.; et al. Enhanced sulfur resistance of H3PW12O40-modifed Fe2O3 catalyst for NH3-SCR: Synergistic effect of surface acidity and oxidation ability. Chem. Eng. J. 2021, 412, 128712. [Google Scholar] [CrossRef]
- Lee, J.; Ryou, Y.S.; Chan, X.J.; Kim, T.J.; Kim, D.H. How Pt interacts with CeO2 under the reducing and oxidizing environments at elevated temperature: The origin of improved thermal stability of Pt/CeO2 compared to CeO2. J. Phys. Chem. C 2016, 120, 25870–25879. [Google Scholar] [CrossRef]
- Damyanova, S.; Perez, C.A.; Schmal, M.; Bueno, J.M.C. Characterization of ceria-coated alumina carrier. Appl. Catal. A 2002, 234, 271–282. [Google Scholar] [CrossRef]
- Cheng, K.; Song, W.Y.; Cheng, Y.; Liu, J.; Zhao, Z.; Wei, Y.C. Selective catalytic reduction over size-tunable rutile TiO2 nanorod microsphere-supported CeO2 catalysts. Catal. Sci. Technol. 2016, 6, 4478–4490. [Google Scholar] [CrossRef]
- North, J.; Poole, O.; Alotaibi, A.; Bayahia, H.; Kozhevnikova, E.F.; Alsalme, A.; Siddiqui, M.R.H.; Kozhevnikov, I.V. Efficient hydrodesulfurization catalysts based on Keggin polyoxometalates. Appl. Catal. A 2015, 508, 16–24. [Google Scholar] [CrossRef]
- Feng, C.L.; Liu, X.L.; Zhu, T.Y.; Hu, Y.T.; Tian, M.K. Catalytic oxidation of CO over Pt/TiO2 with low Pt loading: The effect of H2O and SO2. Appl. Catal. A 2021, 622, 118218. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2/g) | Smic (m2/g) | Vtot (cm3/g) | Pore Size (nm) |
|---|---|---|---|---|
| Pt/CeTi | 63 | 0.0 | 0.210 | 9.6 |
| Pt/Keg-CeTi | 53 | 7.4 | 0.152 | 12.3 |
| Pt/MoP-CeTi | 41 | 3.5 | 0.152 | 9.6 |
| Samples | Surface Atomic Concentration (Atom%) | Atomic Ratio (%) | ||||
|---|---|---|---|---|---|---|
| Pt | Ce | O | Pt4+/(Pt2+ + Pt4+) | Ce3+/(Ce3+ + Ce4+) | Oads/(Oads + Olatt) | |
| Pt/CeTi | 0.23 | 1.78 | 54.39 | 23.3 | 18.0 | 10.1 |
| Pt/Keg-CeTi | 0.16 | 1.33 | 45.86 | 24.7 | 31.0 | 22.2 |
| Pt/MoP-CeTi | 0.11 | 1.61 | 44.54 | 55.7 | 42.3 | 20.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Qiu, W.; Zhu, H.; Ding, X.; Wu, R.; He, H. Promotion Effect of the Keggin Structure on the Sulfur and Water Resistance of Pt/CeTi Catalysts for CO Oxidation. Catalysts 2022, 12, 4. https://doi.org/10.3390/catal12010004
Zhang T, Qiu W, Zhu H, Ding X, Wu R, He H. Promotion Effect of the Keggin Structure on the Sulfur and Water Resistance of Pt/CeTi Catalysts for CO Oxidation. Catalysts. 2022; 12(1):4. https://doi.org/10.3390/catal12010004
Chicago/Turabian StyleZhang, Tong, Wenge Qiu, Hongtai Zhu, Xinlei Ding, Rui Wu, and Hong He. 2022. "Promotion Effect of the Keggin Structure on the Sulfur and Water Resistance of Pt/CeTi Catalysts for CO Oxidation" Catalysts 12, no. 1: 4. https://doi.org/10.3390/catal12010004
APA StyleZhang, T., Qiu, W., Zhu, H., Ding, X., Wu, R., & He, H. (2022). Promotion Effect of the Keggin Structure on the Sulfur and Water Resistance of Pt/CeTi Catalysts for CO Oxidation. Catalysts, 12(1), 4. https://doi.org/10.3390/catal12010004