
Transition Metal-Catalyzed and MAO-Assisted Olefin Polymerization; Cyclic Isomers of Sinn’s Dimer Are Excellent Ligands in Iron Complexes and Great Methylating Reagents


Abstract
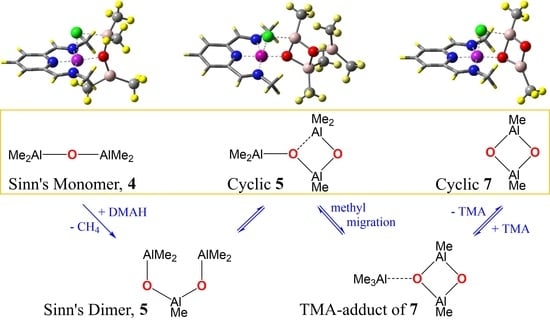
:
1. Introduction
2. Results and Discussion
2.1. Formation of Sinn Dimer 5 and Its Structural Isomerization
2.2. Iron-Complexes: Dichlorides and Monochlorides
2.3. Iron-Complexation by Sinn Monomer 4, Cyclic Aluminoxane 7, and Sinn Dimer 5
2.4. Kinetic vs. Thermodynamic Control of Complex Formation
2.5. Absolute and Relative Oxophilicities and MAO Topology
2.6. Mechanistic Hypothesis and Suggested Experimental Approach
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziegler, K. Folgen und Werdegang einer Erfindung Nobel-Vortrag am 12. Dezember 1963. Angew. Chem. 1964, 76, 545–553. [Google Scholar] [CrossRef]
- Natta, G. Von der Stereospezifischen Polymerisation zur Asymmetrischen Autokatalytischen Synthese von Makromolekülen Nobel-Vortrag am 12. Dezember 1963. Angew. Chem. 1964, 76, 553–566. [Google Scholar] [CrossRef]
- Nowlin, T.E.; Mink, R.I.; Kissin, Y.V. Supported Magnesium/Titanium-Based Ziegler Catalysts for Production of Polyethylene. In Handbook of Transition Metal Polymerization Catalysts; John Wiley & Sons, Inc.: St. Hoboken, NJ, USA, 2010; pp. 131–155. [Google Scholar]
- Severn, J.; Jones, R.L. Stereospecific α-Olefin Polymerization with Heterogeneous Catalysts. In Handbook of Transition Metal Polymerization Catalysts; John Wiley & Sons, Inc.: St. Hoboken, NJ, USA, 2010; pp. 157–230. [Google Scholar]
- Sinn, H.; Kaminsky, W. Ziegler-Natta Catalysis. Adv. Organomet. Chem. 1980, 18, 99–149. [Google Scholar]
- Andresen, A.; Cordes, H.G.; Herwig, J.; Kaminsky, W.; Merck, A.; Mottweiler, R.; Pein, J.; Sinn, H.; Vollmer, H.J. Halogen-Free Soluble Ziegler Catalysts for the Polymerization of Ethylene. Control of Molecular Weight by Choice of Temperature. Angew. Chem. 1976, 88, 689–690. [Google Scholar] [CrossRef]
- Severn, J. Methylaluminoxane (MAO), Silica and a Complex: The “Holy Trinity” of Supported Single-Site Catalyst. In Tailor-Made Polymers: Via Immobilization of Alpha-Olefin Polymerization Catalysts; John Wiley & Sons, Inc.: St. Hoboken, NJ, USA, 2008; pp. 95–138. [Google Scholar]
- Zaccaria, F.; Budzelaar, P.; Zuccaccia, C.; Cipullo, R.; Macchioni, A.; Busico, V.; Ehm, C. Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts 2021, 11, 215. [Google Scholar] [CrossRef]
- Kumar, S.; Dholakiya, B.; Jangir, R. Role of Organometallic Complexes in Olefin Polymerization: A Review Report. J. Organomet. Chem. 2021, 953, 122066. [Google Scholar] [CrossRef]
- Köppl, A.; Alt, H.H.; Phillips, M.D. Partially Hydrolyzed Trimethylaluminum (PHT) as Heterogeneous Cocatalyst for the Polymerization of Olefins with Metallocene Complexes. J. Appl. Polym. Sci. 2001, 80, 454–466. [Google Scholar] [CrossRef]
- Small, B.L.; Brookhart, M.; Bennett, A.M.A. Highly Active Iron and Cobalt Catalysts for the Polymerization of Ethylene. J. Am. Chem. Soc. 1998, 120, 4049–4050. [Google Scholar] [CrossRef]
- Small, B.L.; Brookhart, M. Iron-Based Catalysts with Exceptionally High Activities and Selectivities for Oligomerization of Ethylene to Linear α-Olefins. J. Am. Chem. Soc. 1998, 120, 7143–7144. [Google Scholar] [CrossRef]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100, 1169–1203. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, W.H.; Li, T.; Yang, X. Influence of Electronic Effect on Catalytic Activity of Bis(imino)pyridyl Fe(II) and Bis(imino)pyrimidyl Fe(II) Complexes. J. Mol. Catal. A 2004, 218, 119–124. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, W.-H.; Xiao, T.; Hao, X. Ferrous and Cobaltous Chlorides Bearing 2,8-Bis(imino)quinolines: Highly Active Catalysts for Ethylene Polymerization at High Temperature. Organometallics 2010, 29, 1168–1173. [Google Scholar] [CrossRef]
- Guo, D.; Han, L.; Zhang, T.; Sun, W.H.; Li, T.; Yang, X. Temperature Dependence of the Activity of a Late Transition Metal Catalyst by Molecular Modeling. Macromol. Theory Simul. 2002, 11, 1006–1012. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, D.; Jie, S.; Sun, W.H.; Li, T.; Yang, X. Influence of Electronic Effect on Catalytic Activity of Salicylaldiminato Nickel(II) Complexes. J. Polym. Sci. Part A 2004, 42, 4765–4774. [Google Scholar] [CrossRef]
- Kong, S.; Guo, C.Y.; Yang, W.; Wang, L.; Sun, W.H.; Glaser, R. 2,6-Dibenzhydryl-N-(2-phenyliminoacenaphthylenylidene)-4-Chloroaniline nickel Dihalides: Synthesis, Characterization and Ethylene Polymerization for Polyethylenes with High Molecular Weights. J. Organomet. Chem. 2013, 725, 37–45. [Google Scholar] [CrossRef]
- Lai, J.; Zhao, W.; Yang, W.; Redshaw, C.; Liang, T.; Liu, Y.; Sun, W.H. 2-[1-(2,4-Dibenzhydryl-6-methylphenylimino)ethyl]-6-[1-(arylimino)ethyl]pyridylcobalt(II) Dichlorides: Synthesis, Characterization and Ethylene Polymerization Behavior. Polym. Chem. 2012, 3, 787–793. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Bruce, M.; Gibson, V.C.; Kimberley, B.S.; Maddox, P.J.; Mastroianni, S.; McTavish, S.J.; Redshaw, C.; Solan, G.A.; Strömberg, S.; et al. Iron and Cobalt Ethylene Polymerization Catalysts Bearing 2,6-Bis(Imino)Pyridyl Ligands: Synthesis, Structures, and Polymerization Studies. J. Am. Chem. Soc. 1999, 121, 8728–8740. [Google Scholar] [CrossRef]
- Zuo, W.; Sun, W.-H.; Zhang, S.; Hao, P.; Shiga, A. Highly Active Ethylene Polymerization and Copolymerization with Norbornene Using Bis(imino-indolide) Titanium Dichloride–MAO System. J. Polym. Sci. Part A 2007, 45, 3415–3430. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kinoshita, S.; Shibahara, A.; Ishii, S.; Kawamura, K.; Inoue, Y.; Fujita, T. Trimerization of Ethylene to 1-Hexene with Titanium Complexes Bearing Phenoxy−Imine Ligands with Pendant Donors Combined with MAO. Organometallics 2010, 29, 2394–2396. [Google Scholar] [CrossRef]
- Velthoen, M.; Muñoz-Murillo, A.; Bouhmadi, A.; Cecius, M.; Diefenbach, S.; Weckhuysen, B. The Multifaceted Role of Methylaluminoxane in Metallocene-Based Olefin Polymerization Catalysis. Macromolecules 2018, 51, 343–355. [Google Scholar] [CrossRef]
- Barron, A.R. A New Understanding of the Co-Catalytic Activity of Alumoxanes: The Opening of a Black Box! Macromol. Symp. 1995, 97, 15–25. [Google Scholar] [CrossRef]
- Watanabi, M.; McMahon, C.N.; Harlan, C.J.; Barron, A.R. Reaction of Trimethylaluminum with [(tBu)Al(μ3-O)]6: Hybrid tert-Butylmethylalumoxanes as Cocatalysts for Olefin Polymerization. Organometallics 2001, 20, 460–467. [Google Scholar] [CrossRef]
- Pasynkiewicz, S. Alumoxanes: Synthesis, Structures, Complexes and Reactions. Polyhedron 1990, 9, 429–453. [Google Scholar] [CrossRef]
- Sinn, H.J. Proposals for Structure and Effect of Methylalumoxane Based on Mass Balances and Phase Separation Experiments. Macromol. Symp. 1995, 97, 27–52. [Google Scholar] [CrossRef]
- Zurek, E.; Woo, T.K.; Firman, T.K.; Ziegler, T. Modeling the Dynamic Equilibrium between Oligomers of (AlOCH3)n in Methylaluminoxane (MAO). A Theoretical Study Based on a Combined Quantum Mechanical and Statistical Mechanical Approach. Inorg. Chem. 2001, 40, 361–370. [Google Scholar] [CrossRef]
- Negureanu, L.; Hall, R.W.; Butler, L.G.; Simeral, L.A. Methyaluminoxane (MAO) Polymerization Mechanism and Kinetic Model from Ab Initio Molecular Dynamics and Electronic Structure Calculations. J. Am. Chem. Soc. 2006, 128, 16816–16826. [Google Scholar] [CrossRef]
- Linnolahti, M.; Severn, J.R.; Pakkanen, T.A. Formation of Nanotubular Methylaluminoxanes and the Nature of the Active Species in Single-Site α-Olefin Polymerization Catalysis. Angew. Chem. Int. Ed. 2008, 47, 9279–9283. [Google Scholar] [CrossRef]
- Linnolahti, M.; Severn, J.R.; Pakkanen, T.A. Are Aluminoxanes Nanotubular? Structural Evidence from a Quantum Chemical Study. Angew. Chem. Int. Ed. 2006, 45, 3331–3334. [Google Scholar] [CrossRef]
- Falls, Z.; Tyminska, N.; Zurek, E. The Dynamic Equilibrium Between (AlOMe)n Cages and (AlOMe)n·(AlMe3)m Nanotubes in Methylaluminoxane (MAO): A First-Principles Investigation. Macromolecules 2014, 47, 8556–8569. [Google Scholar] [CrossRef]
- Tyumkina, T.V.; Islamov, D.N.; Kovyazin, R.V.; Parfenovam, L.V. Chain and Cluster Models of Methylaluminoxane as Activators of Zirconocene Hydride, Alkyl, and Metallacyclopropane Intermediates in Alkene Transformation. Mol. Catal. 2021, 512, 111768. [Google Scholar] [CrossRef]
- Glaser, R.; Sun, X. Thermochemistry of the Initial Steps of Methylaluminoxane Formation. Aluminoxanes and Cycloaluminoxanes by Methane Elimination from Dimethylaluminum Hydroxide and Its Dimeric Aggregates. J. Am. Chem. Soc. 2011, 133, 13323–13336. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, H.S.; Harder, S. Methylalumoxane-History, Production, Properties, and Applications. Eur. J. Inorg. Chem. 2015, 19–43. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09. 2016. Available online: https://gaussian.com/g09citation (accessed on 1 March 2022).
- RCSS Documentation. Available online: http://docs.rnet.missouri.edu/lewis-and-clark-clusters (accessed on 1 March 2022).
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 0618–0622. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. I. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Atoms. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A Complete Basis Set Model Chemistry. II. Open-shell Systems and the Total Energies of the First-Row Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction/Process | E | ΔH0 | ΔH298 | ΔG298 |
|---|---|---|---|---|
| 5a → 5b (Cyclization) | −9.86 | −8.73 | −10.25 | −0.64 |
| 5b → 5c | −0.95 | −0.67 | −1.04 | 0.78 |
| 5a → 5c (Cyclization) | −10.82 | −9.40 | −11.29 | 0.14 |
| Eact, TSRF(5a,5b) vs. 5a | 1.57 | 1.67 | 0.80 | 5.63 |
| Eact, TSMT(5b,5c) vs. 5b | 0.12 | 0.06 | −0.56 | 1.57 |
| Eact, ATS(5c) vs. 5c | 21.58 | 20.39 | 20.93 | 16.74 |
| DMAH (2) + TMA (1) → 4 + CH4 | −49.14 | −48.20 | −47.63 | −46.16 |
| 4 + 2 → 5a + CH4 | −48.05 | −47.26 | −46.64 | −45.38 |
| 4 + 2 → 5c + CH4 | −58.87 | −56.66 | −57.93 | −45.24 |
| 4 + MeAlO (3) → 5c | −98.87 | −96.27 | −97.03 | −79.69 |
| 4 + 0.5 (MeAlO)2 (7) → 5c | −32.22 | −30.78 | −31.58 | −18.71 |
| Rxn. | Reaction/Process | ΔE | ΔH0 | ΔH298 | ΔG298 |
|---|---|---|---|---|---|
| N-phenyl ligands L(Ph)2 | |||||
| 1 | L(Ph)2FeCl2, C2 → Cs | 0.02 | 0.03 | 0.03 | −0.18 |
| 2 | [L(Ph)2FeCl]+, I → II | 3.58 | 0.43 | 0.47 | 0.62 |
| 3 | [L(Ph)2FeCl]+, I → III | 0.59 | 0.43 | 0.47 | 0.62 |
| 4 | [L(Ph)2FeClax(4)eq]+ → [L(Ph)2FeCleq(4)ax]+ | 4.15 | 4.06 | 4.14 | 3.84 |
| 5 | [L(Ph)2FeClax(7)eq]+ → [L(Ph)2FeCleq(7)ax]+ | 3.83 | 3.65 | 3.82 | 2.82 |
| 6 | [L(Ph)2FeClax(5)eq]+ → [L(Ph)2FeCleq(5)ax]+ | −4.66 | −4.72 | −4.74 | −4.57 |
| 7 | [L(Ph)2FeCl]+ + 4 → [L(Ph)2FeCleq(4)ax]+ | −34.60 | −33.00 | −32.68 | −14.96 |
| 8 | [L(Ph)2FeCl]+ + 4 → [L(Ph)2FeClax(4)eq]+ | −38.75 | −37.06 | −36.82 | −18.80 |
| 9 | [L(Ph)2FeCl]+ + 7 → [L(Ph)2FeCleq(7)ax]+ | −50.29 | −49.02 | −48.49 | −32.56 |
| 10 | [L(Ph)2FeCl]+ + 7 → [L(Ph)2FeClax(7)eq]+ | −54.12 | −52.67 | −52.31 | −35.38 |
| 11 | [L(Ph)2FeCl]+ + 5 → [L(Ph)2FeCleq(5)ax]+ | −54.22 | −53.40 | −52.29 | −39.55 |
| 12 | [L(Ph)2FeCl]+ + 5 → [L(Ph)2FeClax(5)eq]+ | −49.56 | −48.69 | −47.55 | −34.97 |
| N-methyl ligands L(Me)2 | |||||
| 13 | [L(Me)2FeClax(4)eq]+ → [L(Me)2FeCleq(4)ax]+ | 5.06 | 4.62 | 4.90 | 4.62 |
| 14 | [L(Me)2FeClax(7)eq]+ → [L(Me)2FeCleq(7)ax]+ | −0.33 | −0.62 | −0.43 | −1.73 |
| 15 | [L(Me)2FeClax(5)eq]+ → [L(Me)2FeCleq(5)ax]+ | −3.10 | −3.39 | −3.29 | −3.41 |
| 16 | [L(Me)2FeCl]+ + 4 → [L(Me)2FeCleq(4)ax]+ | −40.51 | −38.87 | −38.57 | −21.44 |
| 17 | [L(Me)2FeCl]+ + 4 → [L(Me)2FeClax(4)eq]+ | −45.57 | −43.49 | −43.47 | −26.06 |
| 18 | [L(Me)2FeCl]+ + 7 → [L(Me)2FeCleq(7)ax]+ | −54.33 | −53.17 | −52.53 | −38.27 |
| 19 | [L(Me)2FeCl]+ + 7 → [L(Me)2FeClax(7)eq]+ | −53.99 | −52.54 | −52.09 | −36.53 |
| 20 | [L(Me)2FeCl]+ + 5 → [L(Me)2FeCleq(5)ax]+ | −58.65 | −57.83 | −56.69 | −44.70 |
| 21 | [L(Me)2FeCl]+ + 5 → [L(Me)2FeClax(5)eq]+ | −55.55 | −54.44 | −53.41 | −41.28 |
| Oxophilicity Advantage | ΔE | ΔH0 | ΔH298 | ΔG298 |
|---|---|---|---|---|
| N-phenyl ligands L(Ph)2 | ||||
| Difference Ωr(8,10) | 15.36 | 15.60 | 15.48 | 16.58 |
| Difference Ωr(8,12) | 10.81 | 11.62 | 10.72 | 16.17 |
| Difference ΔΩr(10,12) | −4.55 | −3.98 | −4.76 | −0.41 |
| N-methyl ligands L(Me)2 | ||||
| Difference Ωr(17,19) | 8.42 | 9.05 | 8.62 | 10.47 |
| Difference Ωr(17,21) | 9.98 | 10.96 | 9.94 | 15.22 |
| Difference ΔΩr(19,21) | 1.56 | 1.90 | 1.31 | 4.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Glaser, R. Transition Metal-Catalyzed and MAO-Assisted Olefin Polymerization; Cyclic Isomers of Sinn’s Dimer Are Excellent Ligands in Iron Complexes and Great Methylating Reagents. Catalysts 2022, 12, 312. https://doi.org/10.3390/catal12030312
Yang K, Glaser R. Transition Metal-Catalyzed and MAO-Assisted Olefin Polymerization; Cyclic Isomers of Sinn’s Dimer Are Excellent Ligands in Iron Complexes and Great Methylating Reagents. Catalysts. 2022; 12(3):312. https://doi.org/10.3390/catal12030312
Chicago/Turabian StyleYang, Kaidi, and Rainer Glaser. 2022. "Transition Metal-Catalyzed and MAO-Assisted Olefin Polymerization; Cyclic Isomers of Sinn’s Dimer Are Excellent Ligands in Iron Complexes and Great Methylating Reagents" Catalysts 12, no. 3: 312. https://doi.org/10.3390/catal12030312
APA StyleYang, K., & Glaser, R. (2022). Transition Metal-Catalyzed and MAO-Assisted Olefin Polymerization; Cyclic Isomers of Sinn’s Dimer Are Excellent Ligands in Iron Complexes and Great Methylating Reagents. Catalysts, 12(3), 312. https://doi.org/10.3390/catal12030312