

Rational Design of the Catalysts for the Direct Conversion of Methane to Methanol Based on a Descriptor Approach


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
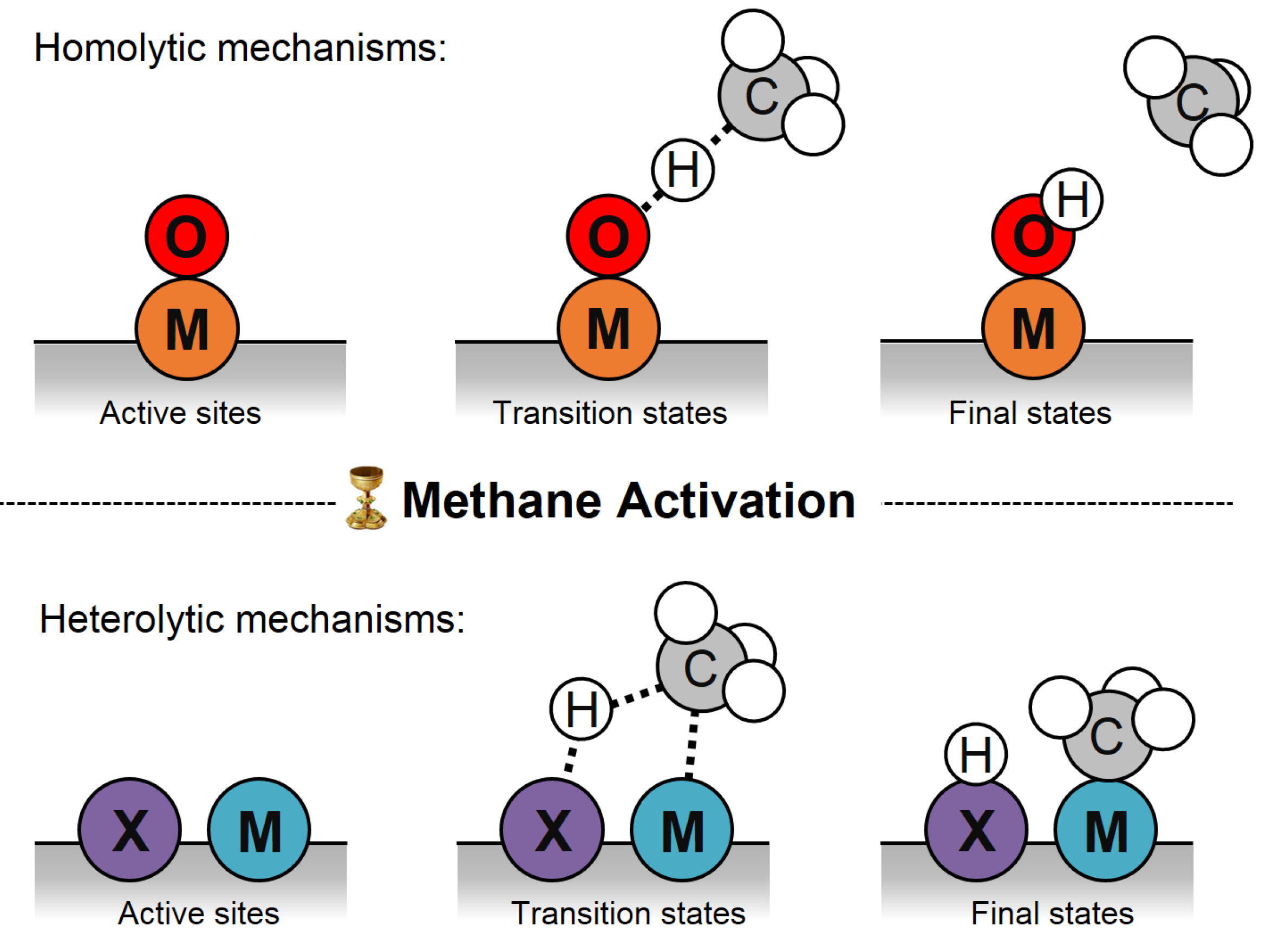
2. Methane Activation Mechanisms
2.1. Homolytic Mechanisms
2.2. Heterolytic Mechanisms
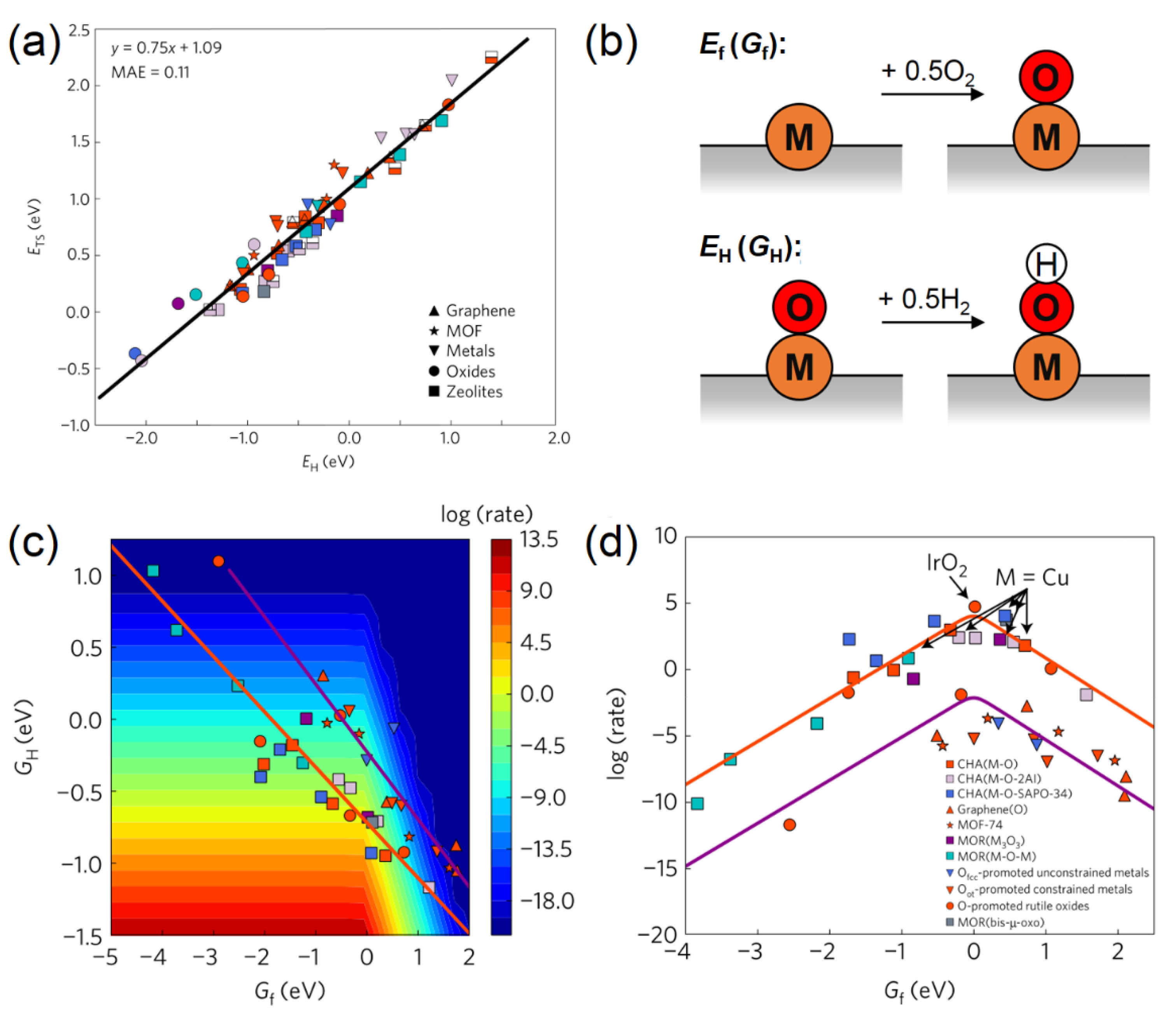
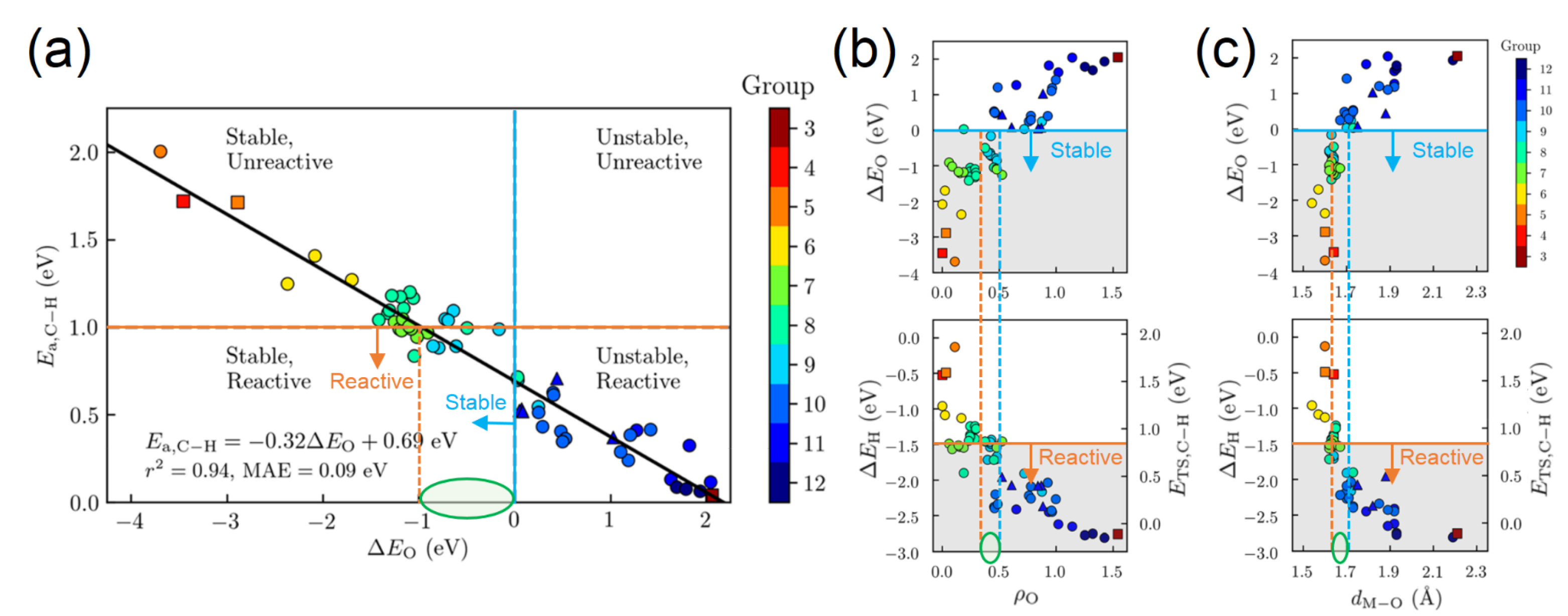
3. Activity Descriptors
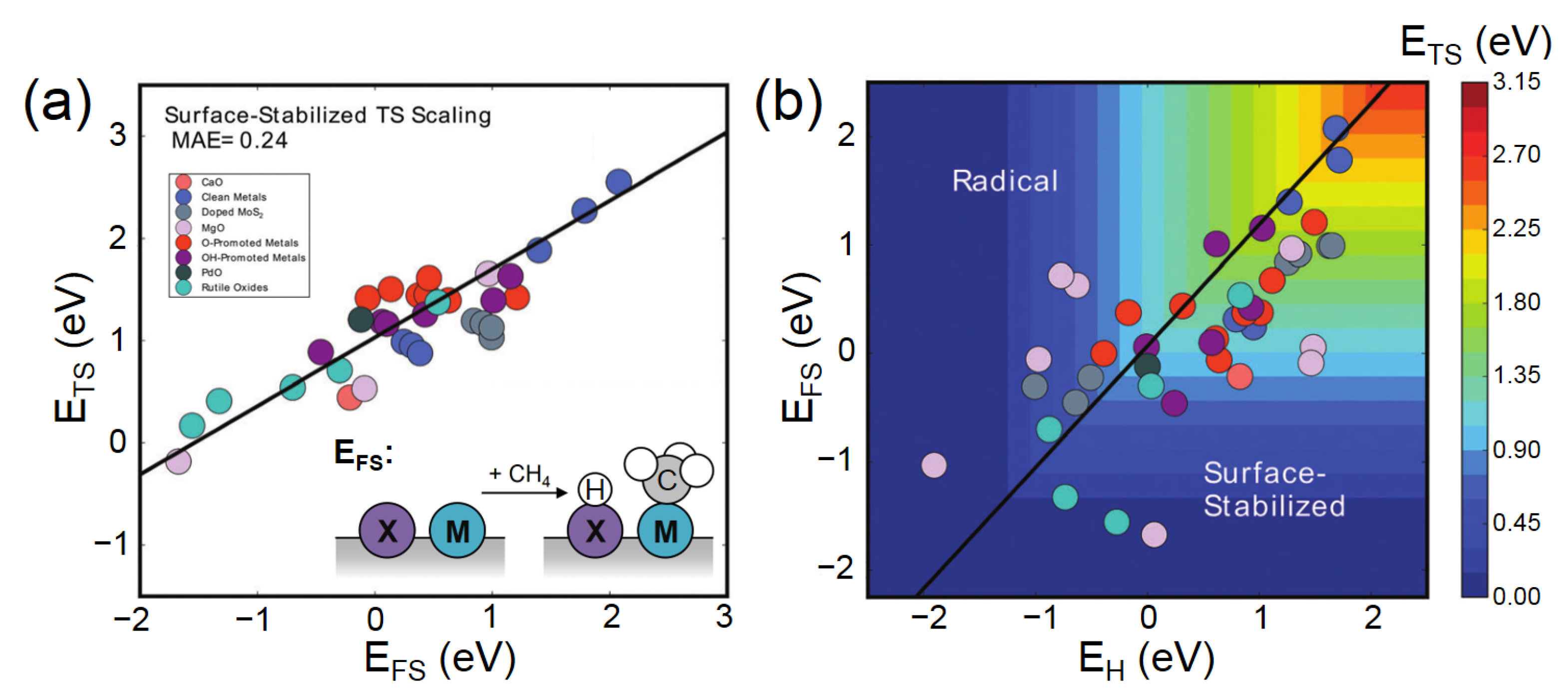
3.1. Activity Descriptors for Homolytic Mechanisms
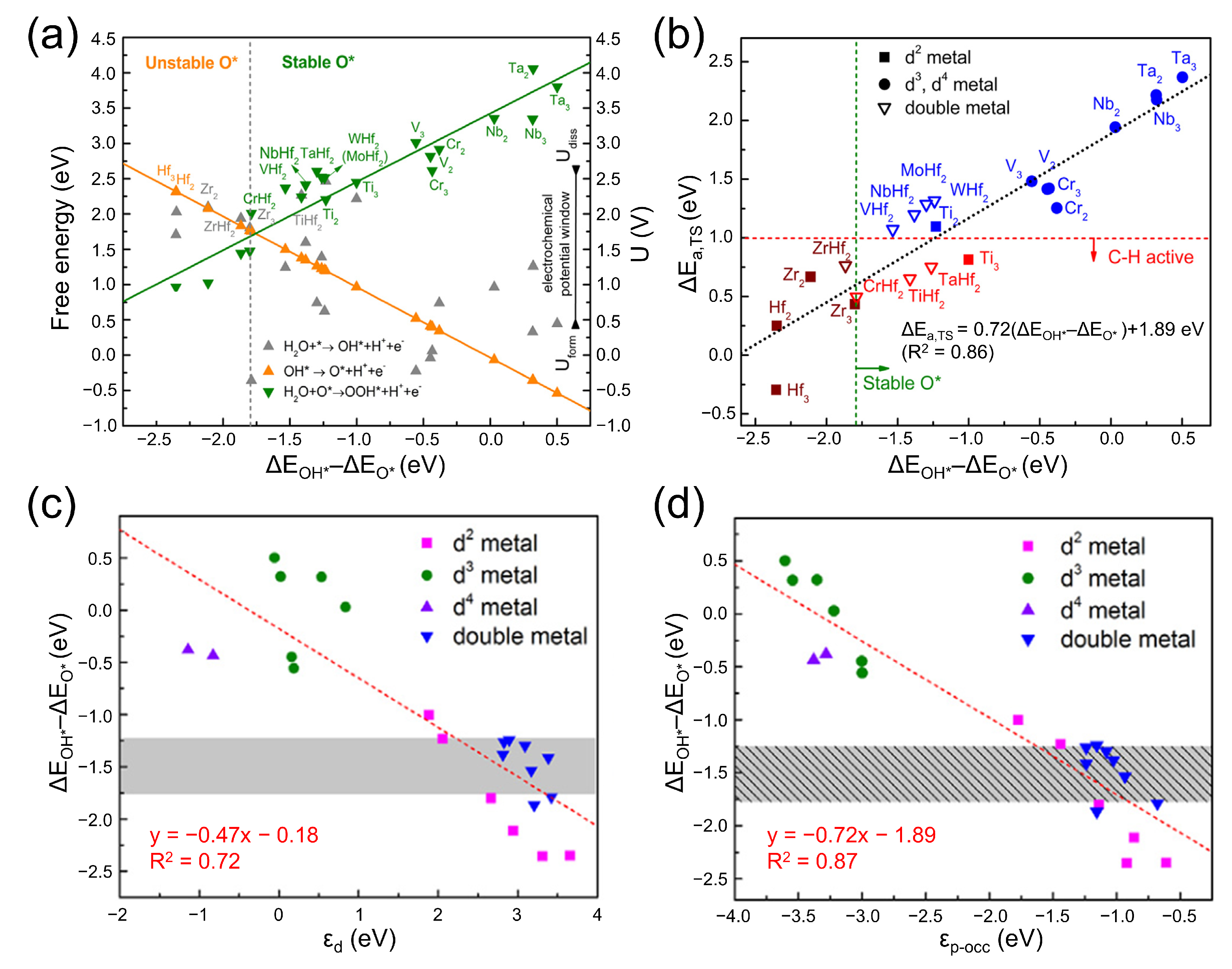
3.2. Activity Descriptors for Heterolytic Mechanisms
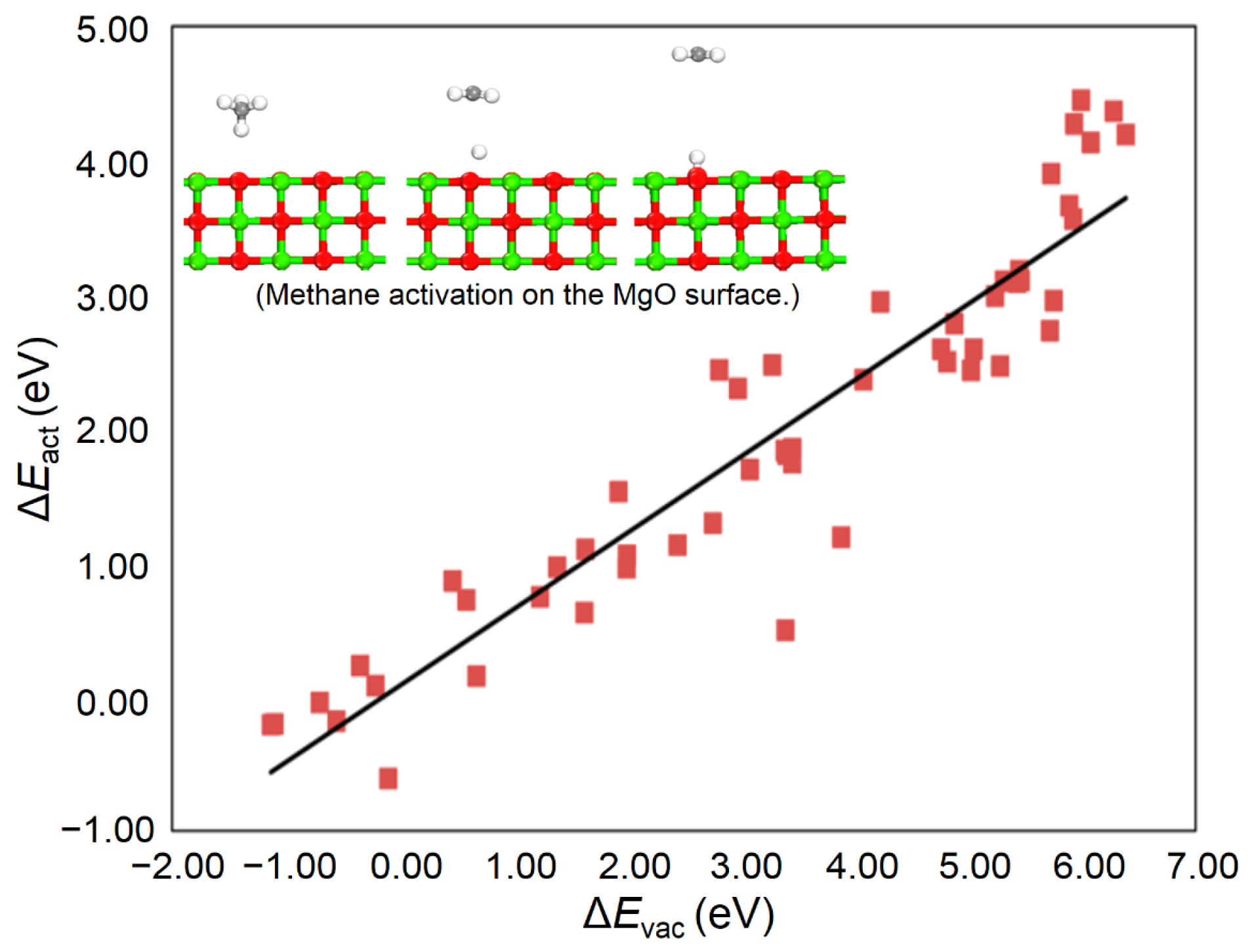
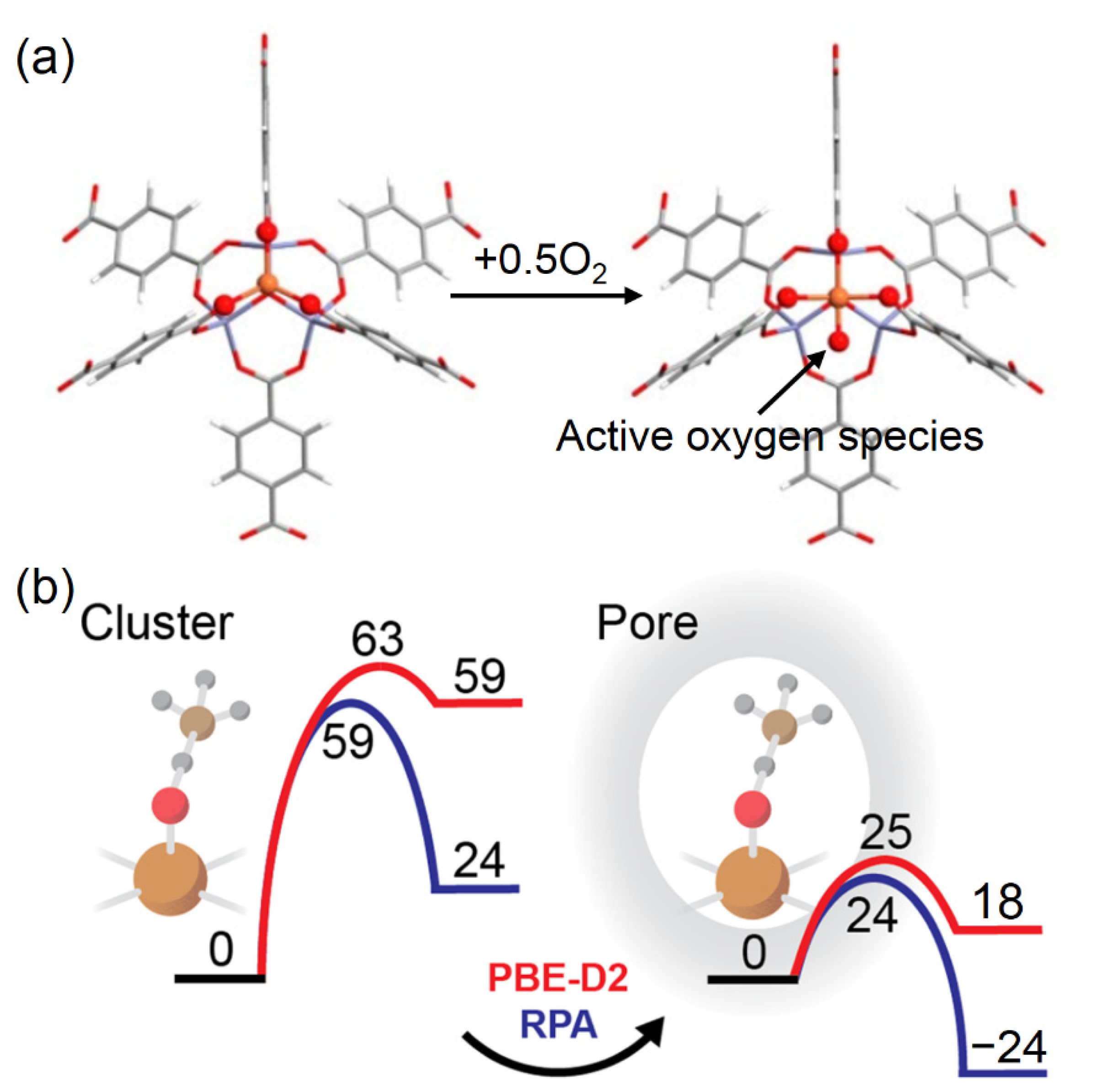
3.3. Additional Electronic and Structural Descriptors
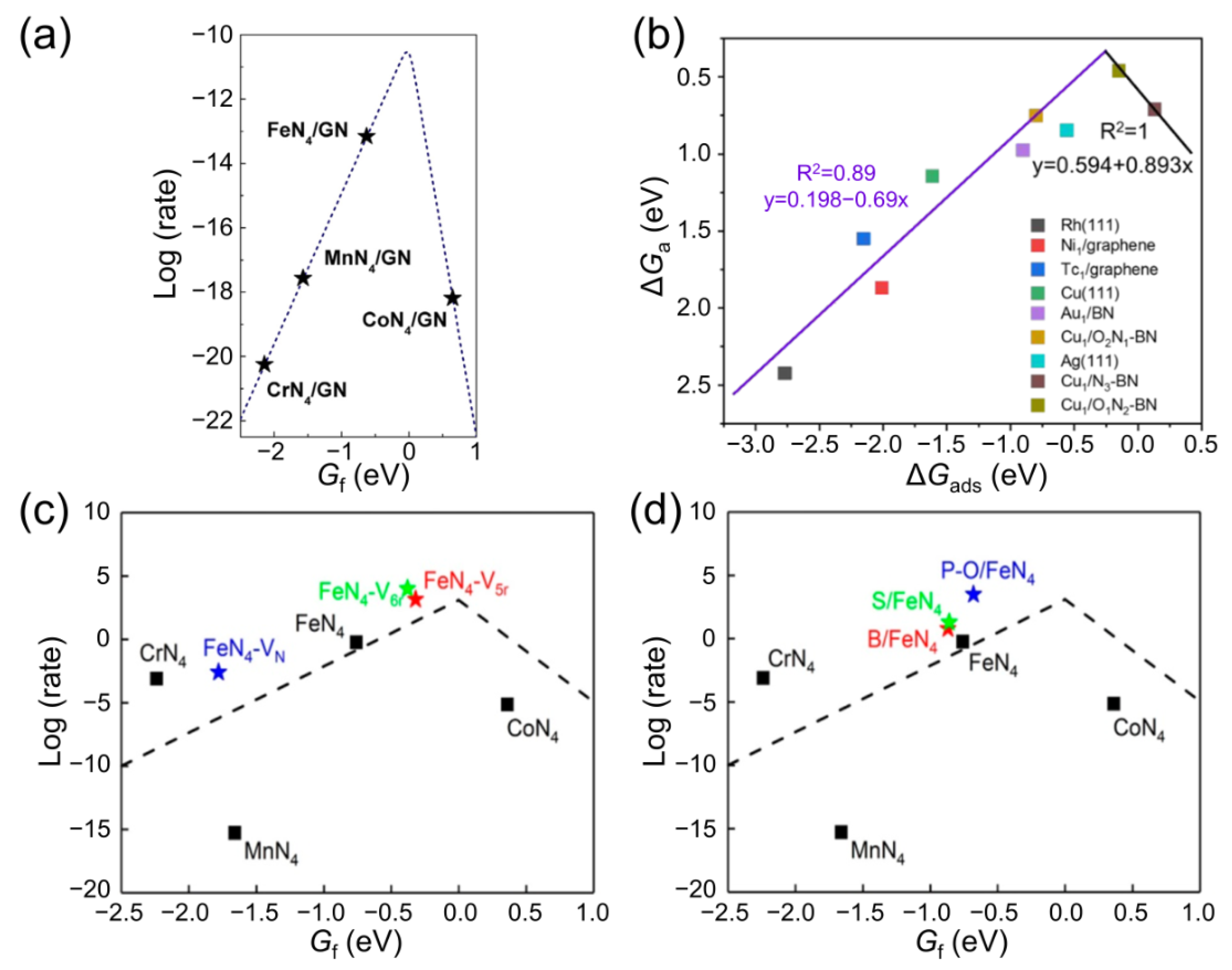
3.4. Moderate Principles Based Descriptors in Catalyst Design
3.5. Applications of Activity Descriptors
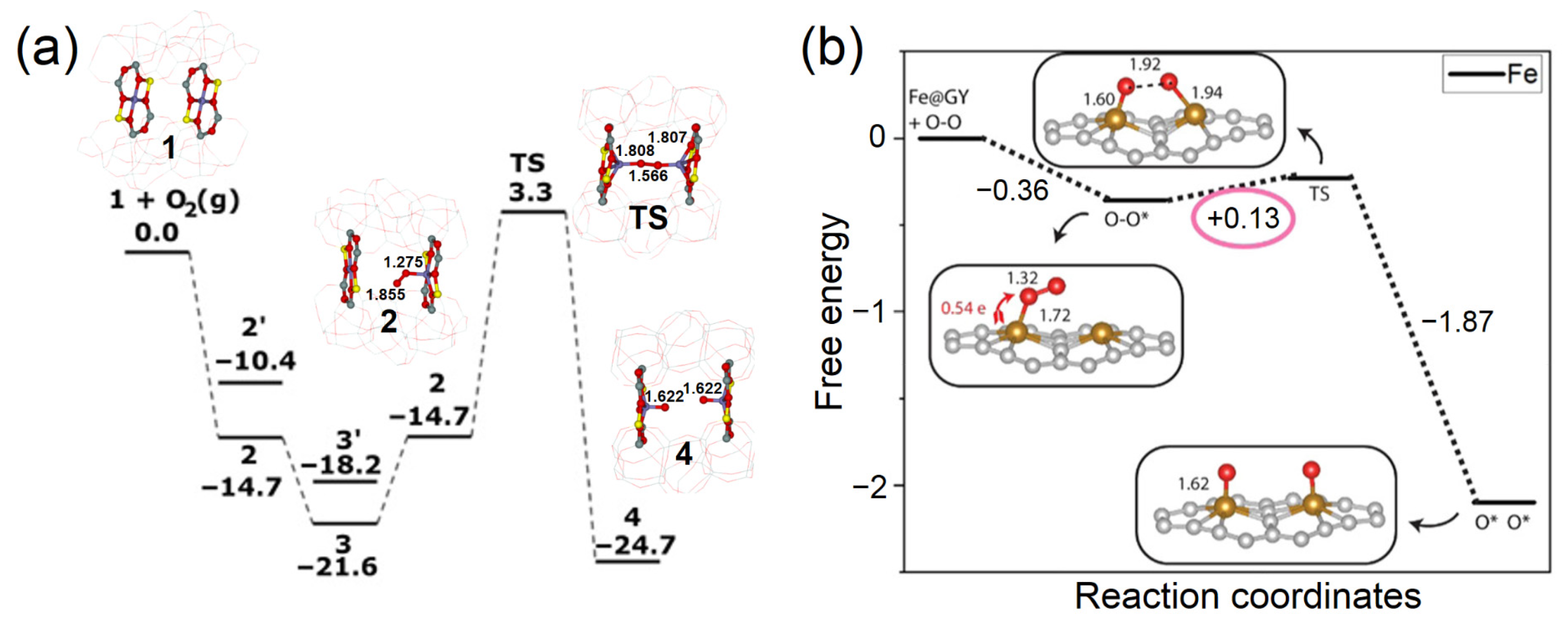
3.6. Breaking the Scaling Relationship of Activity Descriptors
4. Selectivity Descriptors
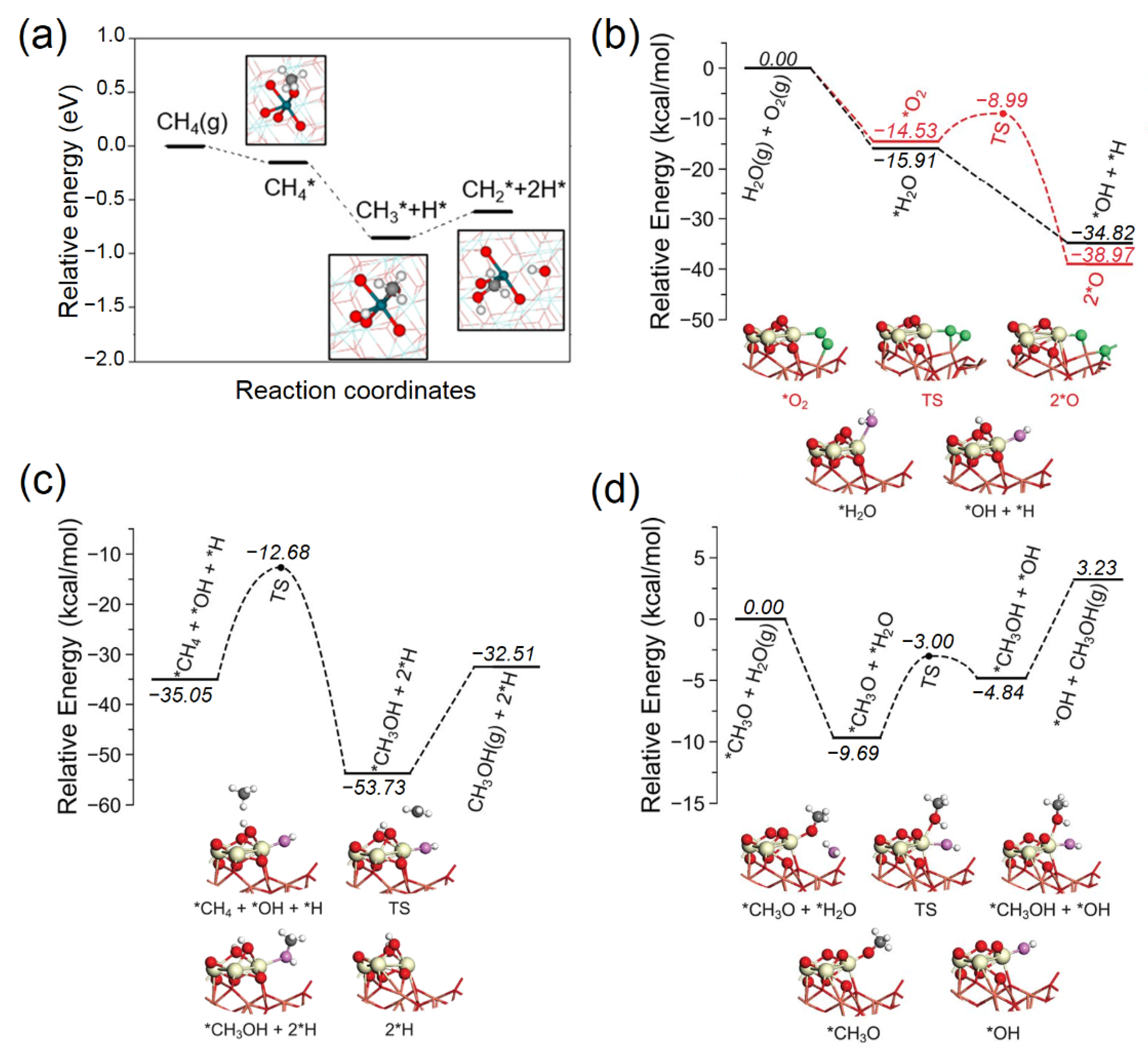
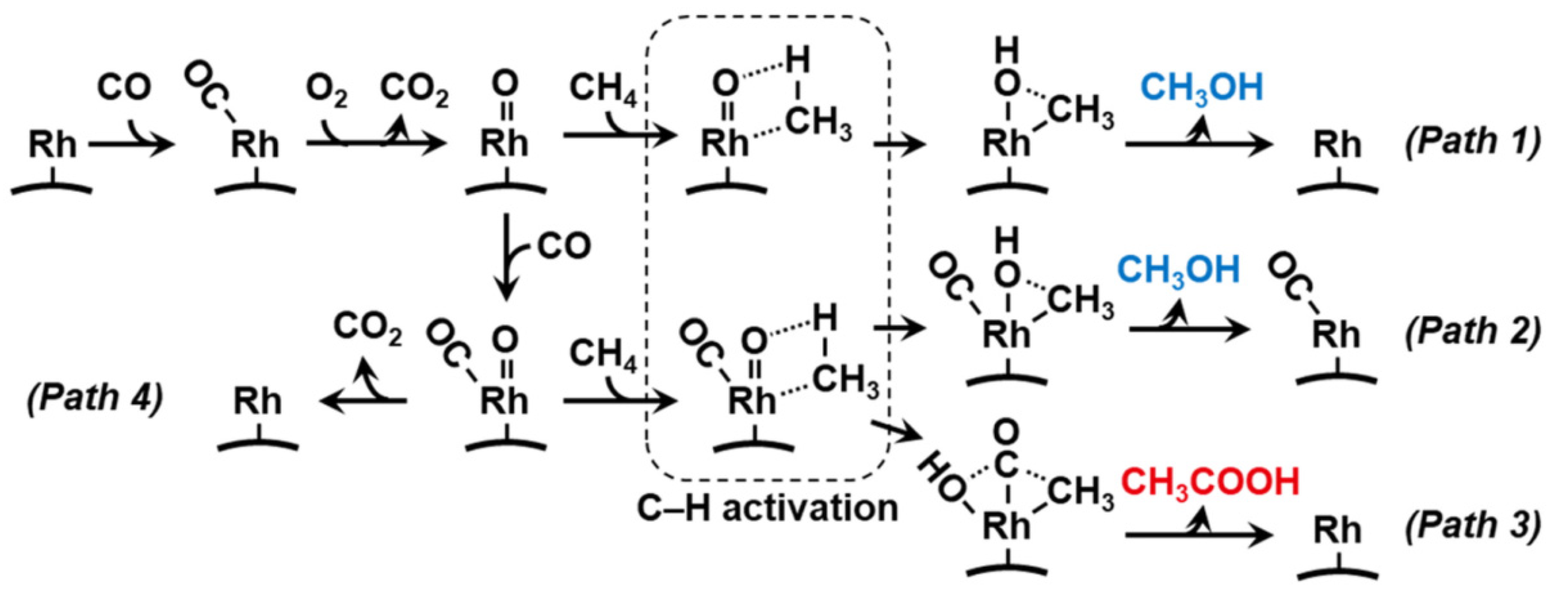
4.1. Conversion–Selectivity Limit in Continuous Methane to Methanol
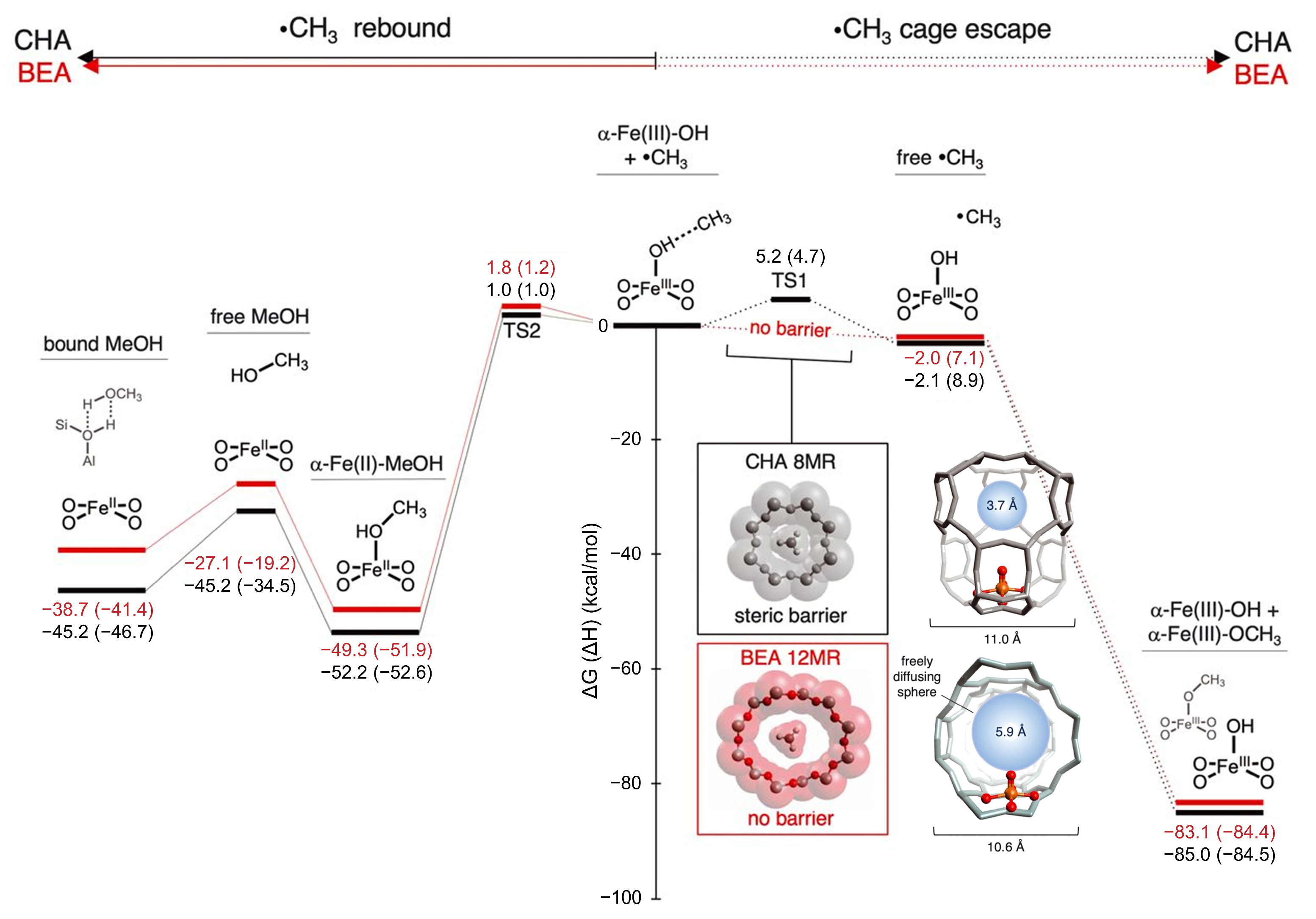
4.2. Protection Strategies for Methanol in Continuous Reaction
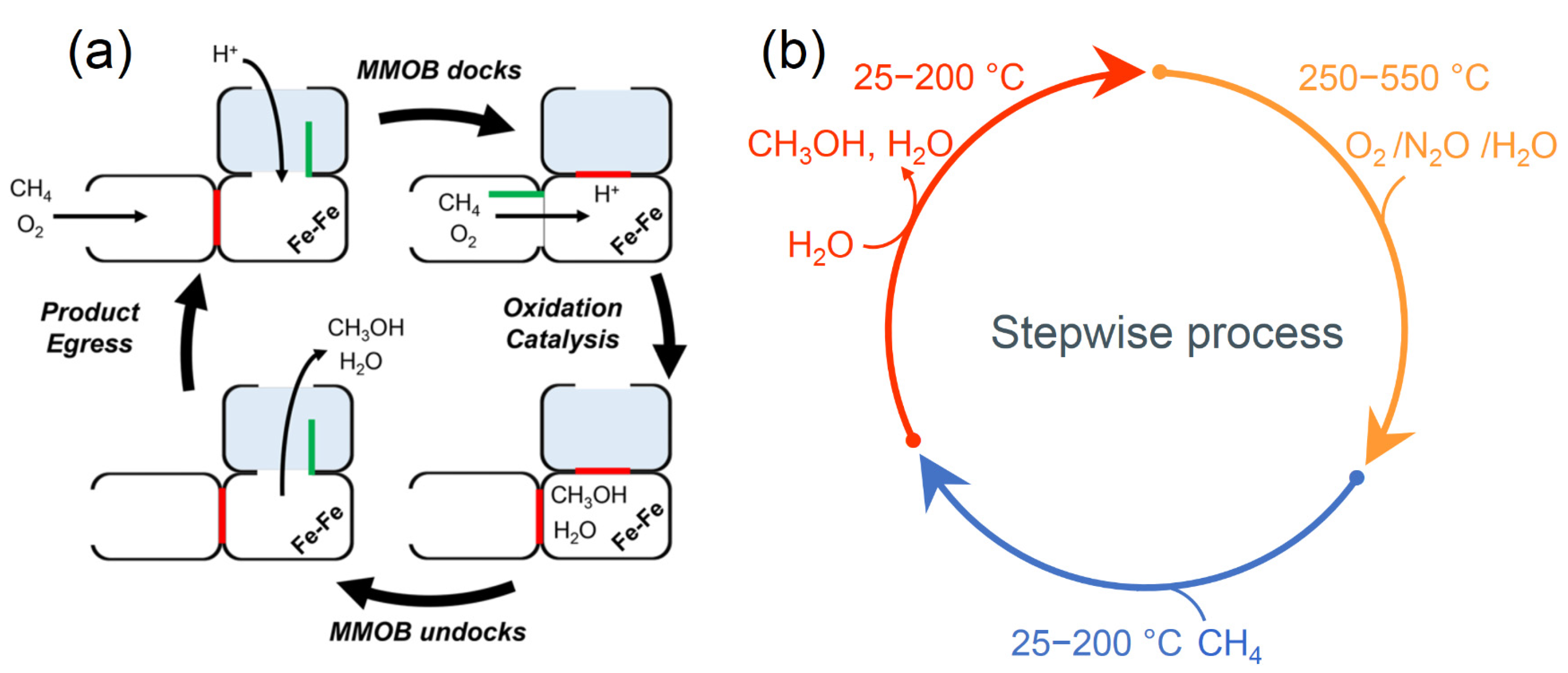
4.3. Stepwise Processes to Protect Methanol
5. Perspectives
5.1. The Synergistic Effect of Multicenter Active Sites
5.2. Active Site Microenvironment
5.3. Co-Catalyst
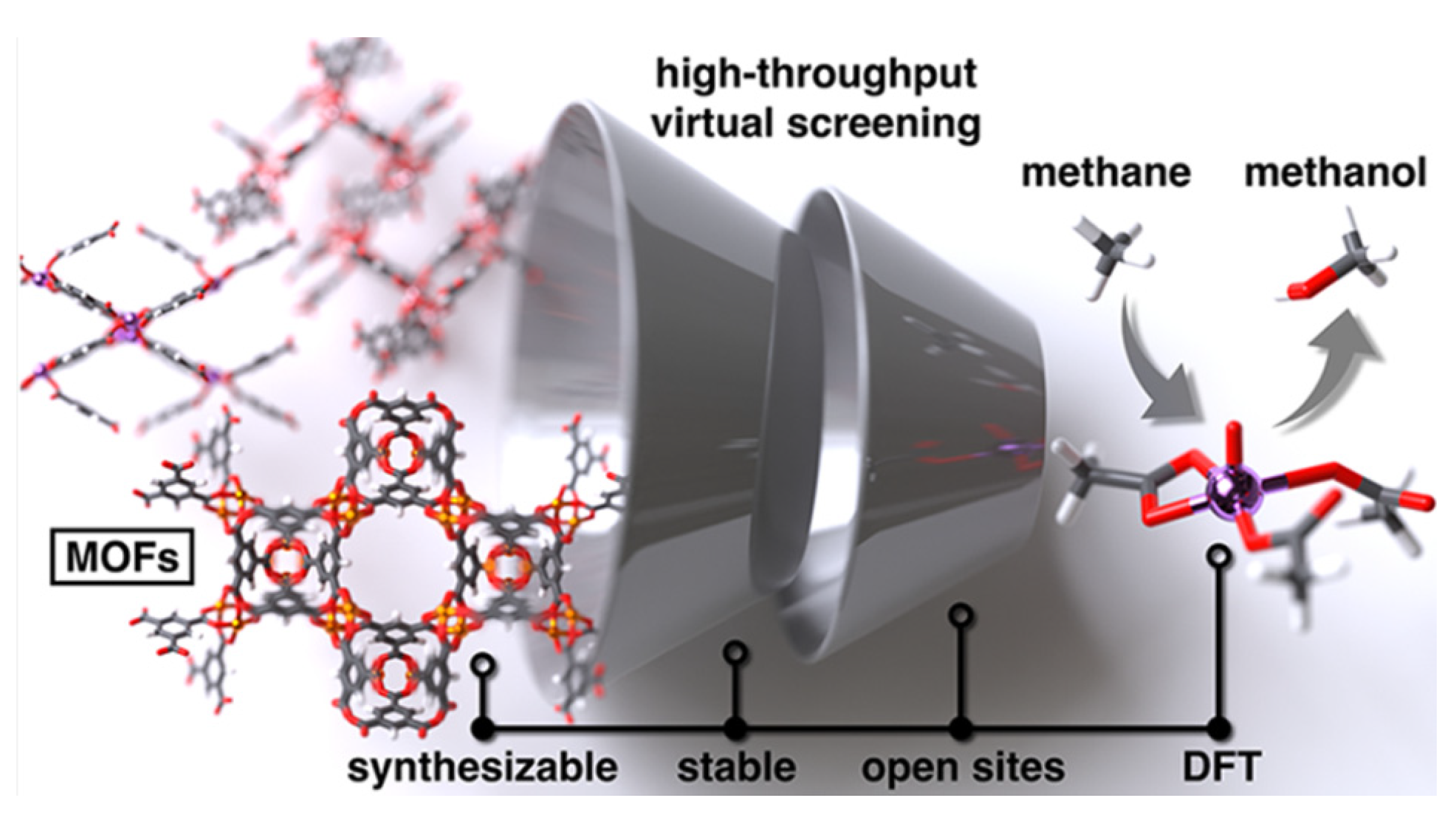
5.4. Machine Learning-Assisted Techniques
5.5. Multicomponent Catalytic Materials
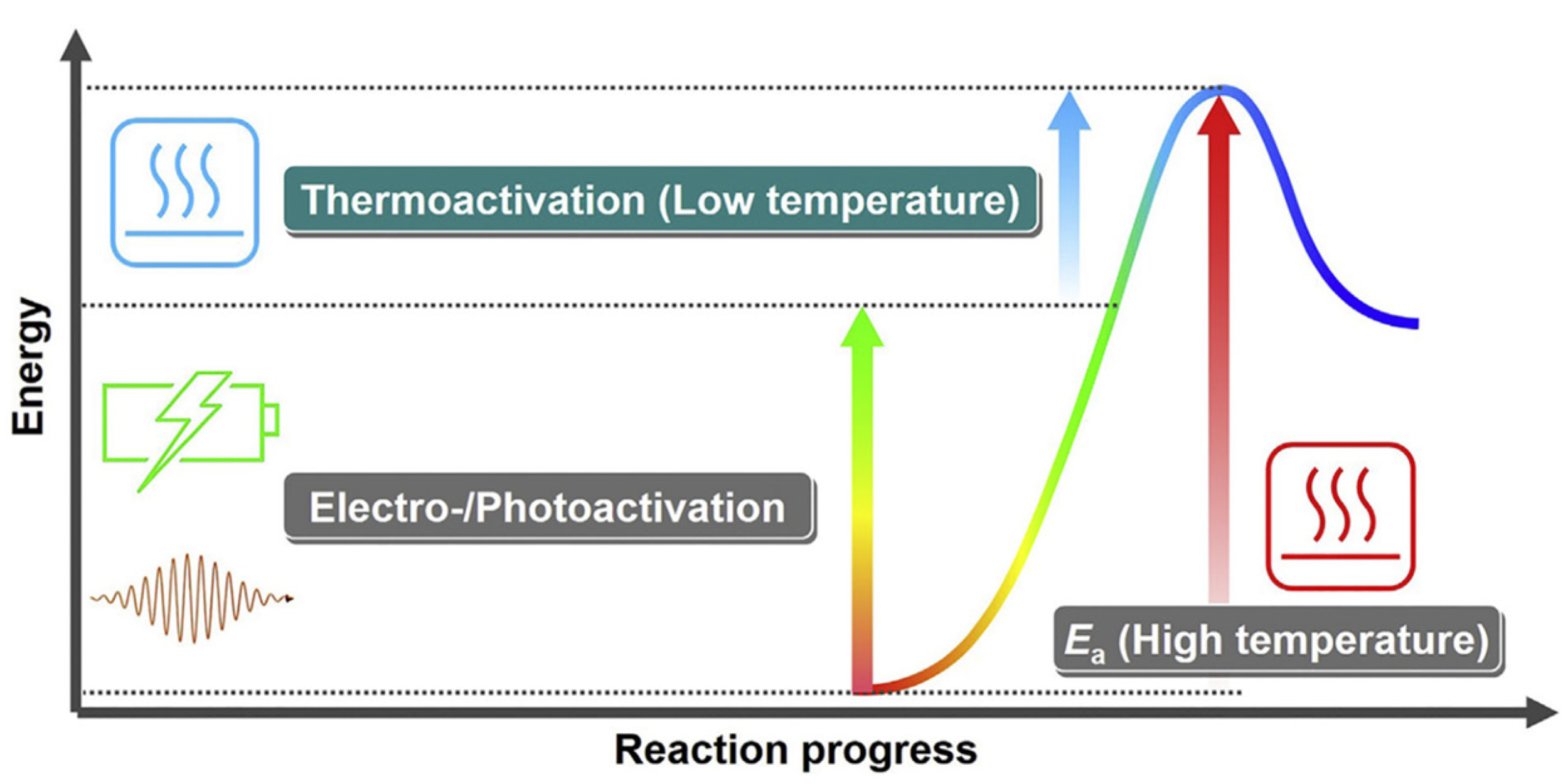
5.6. Additional Energy Source
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nkinahamira, F.; Yang, R.; Zhu, R.; Zhang, J.; Ren, Z.; Sun, S.; Xiong, H.; Zeng, Z. Current Progress on Methods and Technologies for Catalytic Methane Activation at Low Temperatures. Adv. Sci. 2023, 10, 2204566. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and Beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Sarangi, P.K.; Bhatia, L.; Singh, A.K.; Shadangi, K.P. Conversion of Methane to Methanol: Technologies and Future Challenges. Biomass Conv. Bioref. 2022, 12, 1851–1875. [Google Scholar] [CrossRef]
- Freakley, S.J.; Dimitratos, N.; Willock, D.J.; Taylor, S.H.; Kiely, C.J.; Hutchings, G.J. Methane Oxidation to Methanol in Water. Acc. Chem. Res. 2021, 54, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Dummer, N.F.; Willock, D.J.; He, Q.; Howard, M.J.; Lewis, R.J.; Qi, G.; Taylor, S.H.; Xu, J.; Bethell, D.; Kiely, C.J.; et al. Methane Oxidation to Methanol. Chem. Rev. 2023, 123, 6359–6411. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Huang, R.; Deng, D. Catalytic Conversion of C1 Molecules under Mild Conditions. EnergyChem 2021, 3, 100050. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Jones, W.; Liu, Y.; He, Q.; Song, W.; Du, P.; Yang, B.; An, H.; Farmer, D.M.; et al. Identifying Key Mononuclear Fe Species for Low-Temperature Methane Oxidation. Chem. Sci. 2021, 12, 3152–3160. [Google Scholar] [CrossRef]
- Kumar, P.; Al-Attas, T.A.; Hu, J.; Kibria, M.G. Single Atom Catalysts for Selective Methane Oxidation to Oxygenates. ACS Nano 2022, 16, 8557–8618. [Google Scholar] [CrossRef]
- Newton, M.A.; Knorpp, A.J.; Sushkevich, V.L.; Palagin, D.; Van Bokhoven, J.A. Active Sites and Mechanisms in the Direct Conversion of Methane to Methanol Using Cu in Zeolitic Hosts: A Critical Examination. Chem. Soc. Rev. 2020, 49, 1449–1486. [Google Scholar] [CrossRef]
- Wei, Y.-S.; Zhang, M.; Zou, R.; Xu, Q. Metal–Organic Framework-Based Catalysts with Single Metal Sites. Chem. Rev. 2020, 120, 12089–12174. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Zhao, Y.; Wu, P.; Liu, G.; Sun, F.; Ma, J.; Jiang, Z.; Sun, Y.; Zeng, G. Defective C3N4 Frameworks Coordinated Diatomic Copper Catalyst: Towards Mild Oxidation of Methane to C1 Oxygenates. Appl. Catal. B 2021, 299, 120682. [Google Scholar] [CrossRef]
- Fan, J.; Liang, S.; Zhu, K.; Mao, J.; Cui, X.; Ma, C.; Yu, L.; Deng, D. Boosting Room-Temperature Conversion of Methane via Confining Cu Atoms in Ultrathin Ru Nanosheets. Chem. Catal. 2022, 2, 2253–2261. [Google Scholar] [CrossRef]
- Liu, H.; Kang, L.; Wang, H.; Jiang, Q.; Liu, X.Y.; Wang, A. Ru Single-Atom Catalyst Anchored on Sulfated Zirconia for Direct Methane Conversion to Methanol. Chin. J. Catal. 2023, 46, 64–71. [Google Scholar] [CrossRef]
- Bai, S.; Liu, F.; Huang, B.; Li, F.; Lin, H.; Wu, T.; Sun, M.; Wu, J.; Shao, Q.; Xu, Y.; et al. High-Efficiency Direct Methane Conversion to Oxygenates on a Cerium Dioxide Nanowires Supported Rhodium Single-Atom Catalyst. Nat. Commun. 2020, 11, 954. [Google Scholar] [CrossRef]
- Sharma, R.; Poelman, H.; Marin, G.B.; Galvita, V.V. Approaches for Selective Oxidation of Methane to Methanol. Catalysts 2020, 10, 194. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Yoshizawa, K. Methane Selective Oxidation to Methanol by Metal-Exchanged Zeolites: A Review of Active Sites and Their Reactivity. Catal. Sci. Technol. 2019, 9, 1744–1768. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Su, H.-Y.; Sun, K.; Wang, W.-Q.; Zeng, Z.; Calle-Vallejo, F.; Li, W.-X. Establishing and Understanding Adsorption-Energy Scaling Relations with Negative Slopes. J. Phys. Chem. Lett. 2016, 7, 5302–5306. [Google Scholar] [CrossRef]
- Zheng, H.; Li, R.; Zhong, C.; Li, Z.; Kang, Y.; Deng, J.; Song, W.; Zhao, Z. Theoretical Design of Transition Metal-Doped TiO2 for the Selective Catalytic Reduction of NO with NH3 by DFT Calculations. Catal. Sci. Technol. 2022, 12, 1429–1440. [Google Scholar] [CrossRef]
- Zhao, Z.-J.; Liu, S.; Zha, S.; Cheng, D.; Studt, F.; Henkelman, G.; Gong, J. Theory-Guided Design of Catalytic Materials Using Scaling Relationships and Reactivity Descriptors. Nat. Rev. Mater. 2019, 4, 792–804. [Google Scholar] [CrossRef]
- Jia, C.; Wang, Q.; Yang, J.; Ye, K.; Li, X.; Zhong, W.; Shen, H.; Sharman, E.; Luo, Y.; Jiang, J. Toward Rational Design of Dual-Metal-Site Catalysts: Catalytic Descriptor Exploration. ACS Catal. 2022, 12, 3420–3429. [Google Scholar] [CrossRef]
- Seemakurthi, R.R.; Canning, G.; Wu, Z.; Miller, J.T.; Datye, A.K.; Greeley, J. Identification of a Selectivity Descriptor for Propane Dehydrogenation through Density Functional and Microkinetic Analysis on Pure Pd and Pd Alloys. ACS Catal. 2021, 11, 9588–9604. [Google Scholar] [CrossRef]
- Chen, S.; Zaffran, J.; Yang, B. Descriptor Design in the Computational Screening of Ni-Based Catalysts with Balanced Activity and Stability for Dry Reforming of Methane Reaction. ACS Catal. 2020, 10, 3074–3083. [Google Scholar] [CrossRef]
- Xie, Z.; Li, Z.; Tang, P.; Song, Y.; Zhao, Z.; Kong, L.; Fan, X.; Xiao, X. The Effect of Oxygen Vacancies on the Coordinatively Unsaturated Al-O Acid-Base Pairs for Propane Dehydrogenation. J. Catal. 2021, 397, 172–182. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Zheng, H.; Chen, L.; Song, W.; Zhao, Z.; Li, J.; Liu, J. The Nature of Ni-O Pairs for Ethane Activation on NiO(100) and NiO(110) Surfaces. Mol. Catal. 2019, 474, 110417. [Google Scholar] [CrossRef]
- Singh, A.R.; Montoya, J.H.; Rohr, B.A.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. Computational Design of Active Site Structures with Improved Transition-State Scaling for Ammonia Synthesis. ACS Catal. 2018, 8, 4017–4024. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the Computational Design of Solid Catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef]
- Nandy, A.; Zhu, J.; Janet, J.P.; Duan, C.; Getman, R.B.; Kulik, H.J. Machine Learning Accelerates the Discovery of Design Rules and Exceptions in Stable Metal–Oxo Intermediate Formation. ACS Catal. 2019, 9, 8243–8255. [Google Scholar] [CrossRef]
- Gani, T.Z.H.; Kulik, H.J. Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV=O. ACS Catal. 2018, 8, 975–986. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; López, N. Strategies to Break Linear Scaling Relationships. Nat. Catal. 2019, 2, 971–976. [Google Scholar] [CrossRef]
- Doan, H.A.; Wang, X.; Snurr, R.Q. Computational Screening of Supported Metal Oxide Nanoclusters for Methane Activation: Insights into Homolytic versus Heterolytic C–H Bond Dissociation. J. Phys. Chem. Lett. 2023, 14, 5018–5024. [Google Scholar] [CrossRef] [PubMed]
- Olivos-Suarez, A.I.; Szécsényi, À.; Hensen, E.J.M.; Ruiz-Martinez, J.; Pidko, E.A.; Gascon, J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Theoretical Investigation of Methane Hydroxylation over Isoelectronic [FeO]2+- and [MnO]+-Exchanged Zeolites Activated by N2O. Inorg. Chem. 2017, 56, 10370–10380. [Google Scholar] [CrossRef]
- Szécsényi, Á.; Li, G.; Gascon, J.; Pidko, E.A. Unraveling Reaction Networks behind the Catalytic Oxidation of Methane with H2O2 over a Mixed-Metal MIL-53(Al,Fe) MOF Catalyst. Chem. Sci. 2018, 9, 6765–6773. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Böttger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef]
- Zhu, K.; Liang, S.; Cui, X.; Huang, R.; Wan, N.; Hua, L.; Li, H.; Chen, H.; Zhao, Z.; Hou, G.; et al. Highly Efficient Conversion of Methane to Formic Acid under Mild Conditions at ZSM-5-Confined Fe-Sites. Nano Energy 2021, 82, 105718. [Google Scholar] [CrossRef]
- Barona, M.; Snurr, R.Q. Exploring the Tunability of Trimetallic Mof Nodes for Partial Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2020, 12, 28217–28231. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Rodriguez, J.A.; Weaver, J.F. Low Temperature Activation of Methane on Metal-Oxides and Complex Interfaces: Insights from Surface Science. Acc. Chem. Res. 2020, 53, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver, J.F. Low-Temperature Activation of Methane on the IrO2(110) Surface. Science 2017, 356, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Latimer, A.A.; Aljama, H.; Kakekhani, A.; Yoo, J.S.; Kulkarni, A.; Tsai, C.; Garcia-Melchor, M.; Abild-Pedersen, F.; Nørskov, J.K. Mechanistic Insights into Heterogeneous Methane Activation. Phys. Chem. Chem. Phys. 2017, 19, 3575–3581. [Google Scholar] [CrossRef]
- Szécsényi, Á.; Li, G.; Gascon, J.; Pidko, E.A. Mechanistic Complexity of Methane Oxidation with H2 2 by Single-Site Fe/ZSM-5 Catalyst. ACS Catal. 2018, 8, 7961–7972. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Ma, R.; You, R.; Zeng, H.; Rui, Z. Metal–Organic Framework-Derived IrO2/CuO Catalyst for Selective Oxidation of Methane to Methanol. ACS Energy Lett. 2019, 4, 2945–2951. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, L.; Wang, W. Direct Functionalization of Methane into Ethanol over Copper Modified Polymeric Carbon Nitride via Photocatalysis. Nat. Commun. 2019, 10, 506. [Google Scholar] [CrossRef]
- Ravi, M.; Sushkevich, V.L.; Knorpp, A.J.; Newton, M.A.; Palagin, D.; Pinar, A.B.; Ranocchiari, M.; van Bokhoven, J.A. Misconceptions and Challenges in Methane-to-Methanol over Transition-Metal-Exchanged Zeolites. Nat. Catal. 2019, 2, 485–494. [Google Scholar] [CrossRef]
- Yuan, S.; Li, Y.; Peng, J.; Questell-Santiago, Y.M.; Akkiraju, K.; Giordano, L.; Zheng, D.J.; Bagi, S.; Román-Leshkov, Y.; Shao-Horn, Y. Conversion of Methane into Liquid Fuels-Bridging Thermal Catalysis with Electrocatalysis. Adv. Energy Mater. 2020, 10, 2002154. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, P.; Yang, J.; Zhu, Y.-A.; Chen, D. C–H Bond Activation in Light Alkanes: A Theoretical Perspective. Chem. Soc. Rev. 2021, 50, 4299–4358. [Google Scholar] [CrossRef]
- Wang, G.; Huang, L.; Chen, W.; Zhou, J.; Zheng, A. Rationally Designing Mixed Cu–(μ-O)–M (M = Cu, Ag, Zn, Au) Centers over Zeolite Materials with High Catalytic Activity towards Methane Activation. Phys. Chem. Chem. Phys. 2018, 20, 26522–26531. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Wang, Z.; Chen, X.; Song, W.; Zhao, Z.; Wei, Y.; Zhang, X. Hetero-Metallic Active Sites in Omega (MAZ) Zeolite-Catalyzed Methane Partial Oxidation: A DFT Study. Ind. Eng. Chem. Res. 2021, 60, 2400–2409. [Google Scholar] [CrossRef]
- Ma, D.; Cao, X.; Cao, Z. Selective Oxidation of CH4 to CH3OH by Transition-Metal Single-Atom-Embedded N-Doped Graphene Catalysts with Oxidants N2O and O2: Oxygen Adsorption Energy as an Activity Descriptor. J. Phys. Chem. C 2023, 127, 5800–5809. [Google Scholar] [CrossRef]
- Ganai, A.; Ball, B.; Sarkar, P. Modulating the Energetics of C–H Bond Activation in Methane by Utilizing Metalated Porphyrinic Metal–Organic Frameworks. J. Phys. Chem. Lett. 2023, 14, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
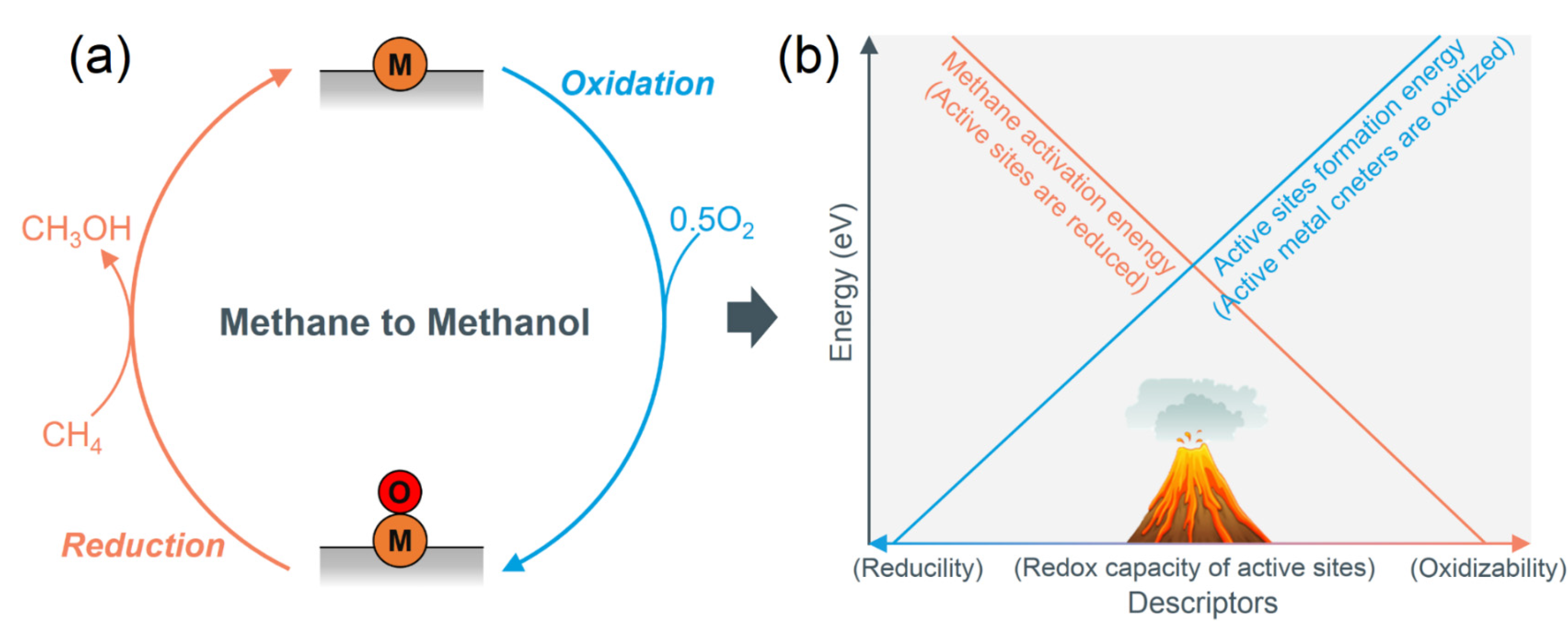
- Bligaard, T.; Nørskov, J.K.; Dahl, S.; Matthiesen, J.; Christensen, C.H.; Sehested, J. The Brønsted–Evans–Polanyi Relation and the Volcano Curve in Heterogeneous Catalysis. J. Catal. 2004, 224, 206–217. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kulkarni, A.R.; Aljama, H.; Montoya, J.H.; Yoo, J.S.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J.K. Understanding Trends in C–H Bond Activation in Heterogeneous Catalysis. Nat. Mater. 2017, 16, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Mahyuddin, M.H.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Theoretical Overview of Methane Hydroxylation by Copper–Oxygen Species in Enzymatic and Zeolitic Catalysts. Acc. Chem. Res. 2018, 51, 2382–2390. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
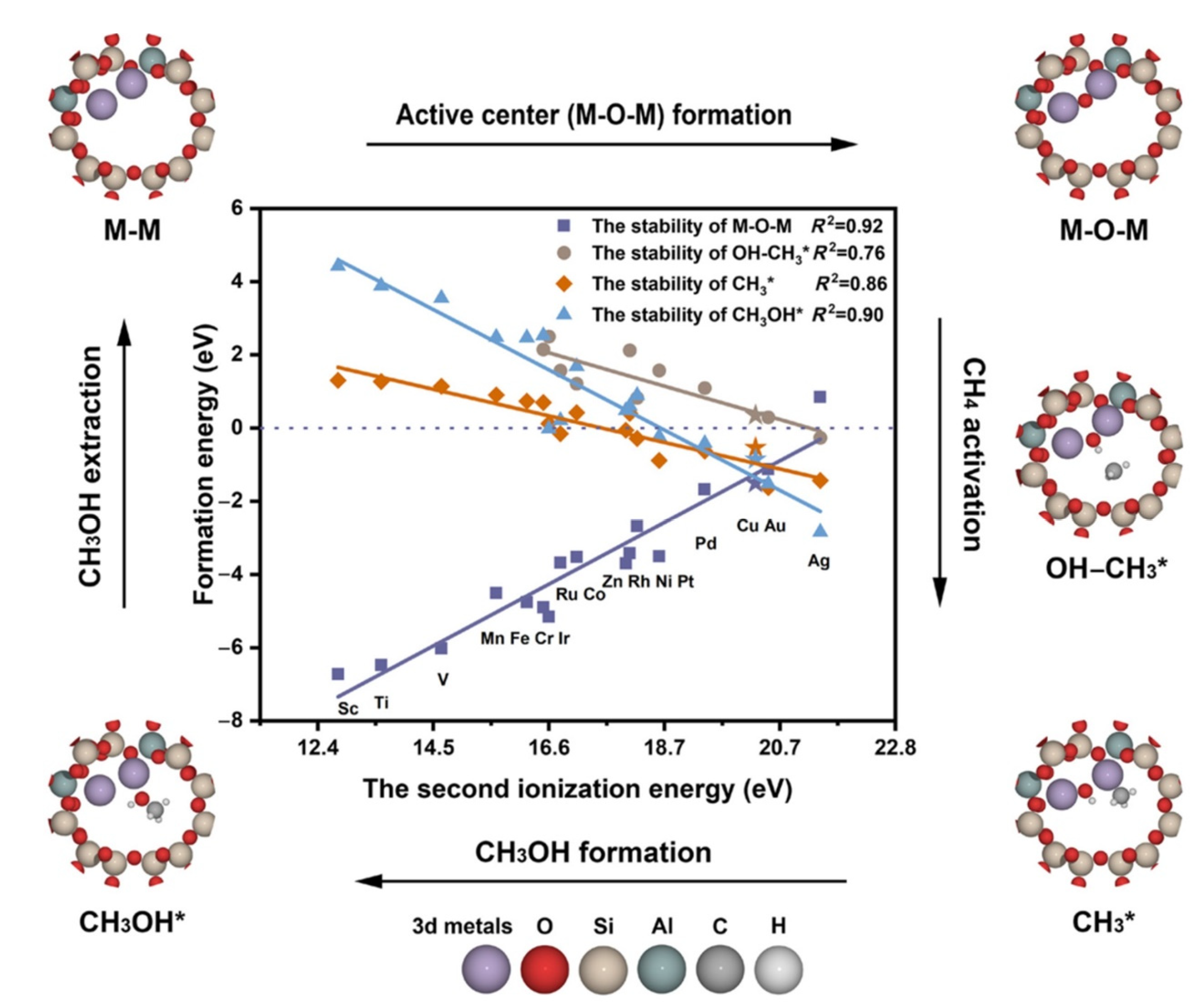
- Kumar, G.; Lau, S.L.J.; Krcha, M.D.; Janik, M.J. Correlation of Methane Activation and Oxide Catalyst Reducibility and Its Implications for Oxidative Coupling. ACS Catal. 2016, 6, 1812–1821. [Google Scholar] [CrossRef]
- Fung, V.; Tao, F.; Jiang, D. Low-Temperature Activation of Methane on Doped Single Atoms: Descriptor and Prediction. Phys. Chem. Chem. Phys. 2018, 20, 22909–22914. [Google Scholar] [CrossRef]
- Liu, C.; Li, G.; Pidko, E.A. Property–Activity Relations for Methane Activation by Dual-metal Cu–Oxo Trimers in ZSM-5 Zeolite. Small Methods 2018, 2, 1800266. [Google Scholar] [CrossRef]
- Zheng, J.; Ye, J.; Ortuño, M.A.; Fulton, J.L.; Gutiérrez, O.Y.; Camaioni, D.M.; Motkuri, R.K.; Li, Z.; Webber, T.E.; Mehdi, B.L.; et al. Selective Methane Oxidation to Methanol on Cu-Oxo Dimers Stabilized by Zirconia Nodes of an NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 9292–9304. [Google Scholar] [CrossRef] [PubMed]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Structure–Activity Relationships That Identify Metal–Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
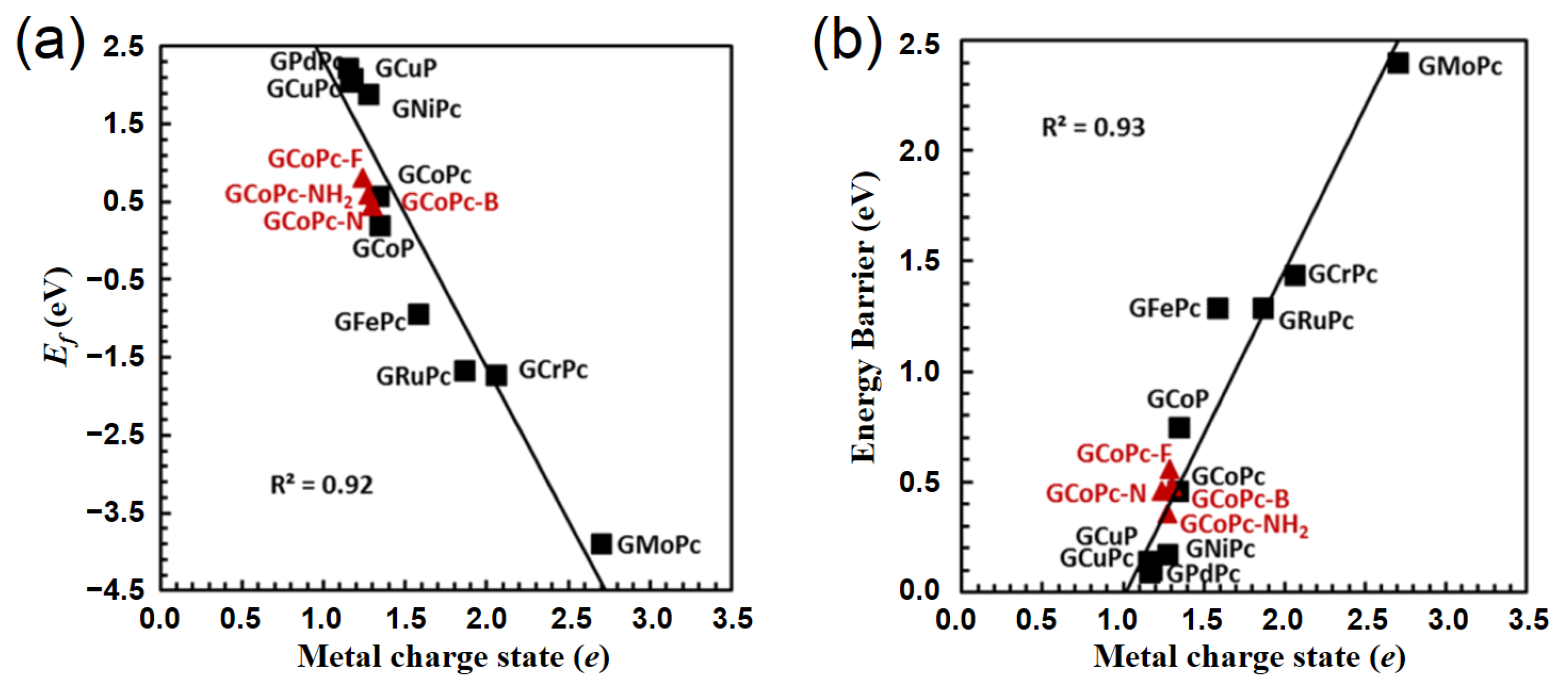
- Filonowich, D.; Luna, M.; Quinn, T.; Choudhury, P. Electronic Descriptor of Single Metal-Oxo Species on Phthalocyanine- and Porphyrin-Functionalized Graphene toward Methane Activation Process. J. Phys. Chem. C 2020, 124, 4502–4510. [Google Scholar] [CrossRef]
- Zha, S.; Zhao, Z.-J.; Chen, S.; Liu, S.; Liu, T.; Studt, F.; Gong, J. Predicting the Catalytic Activity of Surface Oxidation Reactions by Ionization Energies. CCS Chem. 2020, 2, 262–270. [Google Scholar] [CrossRef]
- Xu, J.; Cao, X.-M.; Hu, P. Improved Prediction for the Methane Activation Mechanism on Rutile Metal Oxides by a Machine Learning Model with Geometrical Descriptors. J. Phys. Chem. C 2019, 123, 28802–28810. [Google Scholar] [CrossRef]
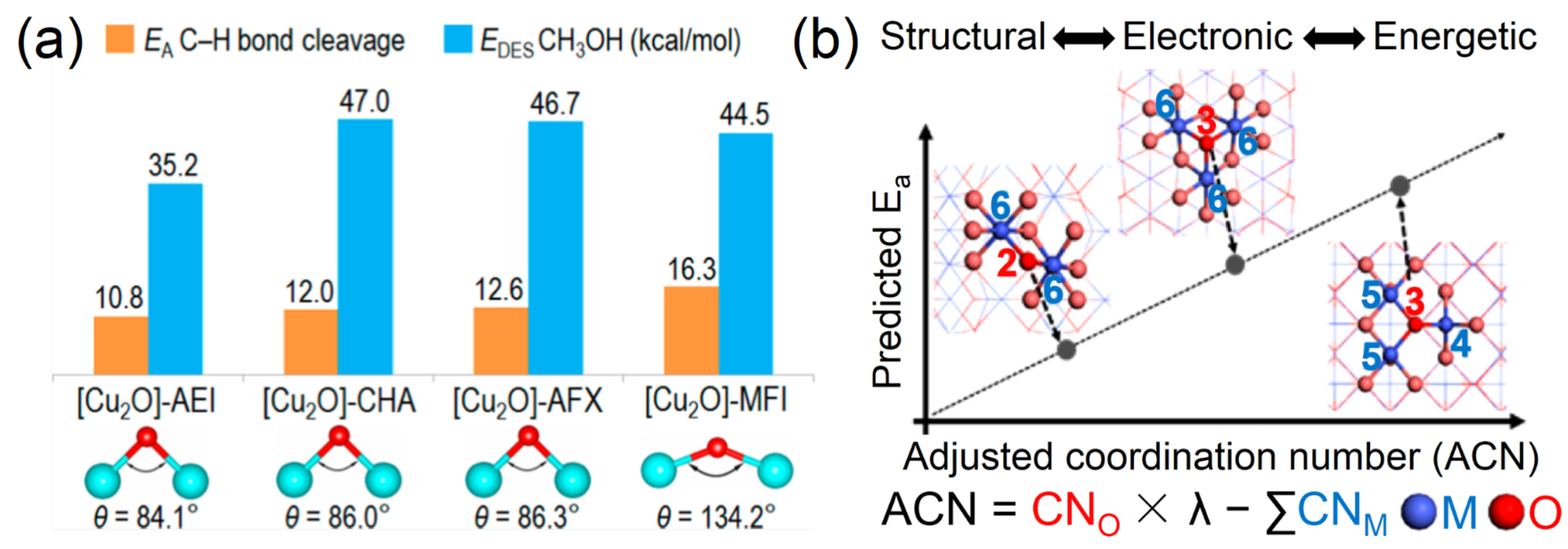
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Miyanishi, M.; Yoshizawa, K. Roles of Zeolite Confinement and Cu–O–Cu Angle on the Direct Conversion of Methane to Methanol by [Cu2(μ-O)]2+-Exchanged Aei, Cha, Afx, and Mfi Zeolites. ACS Catal. 2017, 7, 3741–3751. [Google Scholar] [CrossRef]
- Fung, V.; Tao, F.F.; Jiang, D. General Structure–Reactivity Relationship for Oxygen on Transition-Metal Oxides. J. Phys. Chem. Lett. 2017, 8, 2206–2211. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Jiao, S.; Fu, X.; Huang, H. Descriptors for the Evaluation of Electrocatalytic Reactions: D-Band Theory and Beyond. Adv. Funct. Mater. 2022, 32, 2107651. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why Gold Is the Noblest of All the Metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier Principle to a Predictive Theory of Transition-Metal Heterogeneous Catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Choi, C.; Yoon, S.; Jung, Y. Shifting the Scaling Relations of Single-Atom Catalysts for Facile Methane Activation by Tuning the Coordination Number. Chem. Sci. 2021, 12, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-Temperature Methane Conversion by Graphene-Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef]
- Tan, X.; Tahini, H.A.; Smith, S.C. Defect Engineering in Graphene-Confined Single-Atom Iron Catalysts for Room-Temperature Methane Conversion. J. Phys. Chem. C 2021, 125, 12628–12635. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.-J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Cation-Exchanged Zeolites for the Selective Oxidation of Methane to Methanol. Catal. Sci. Technol. 2018, 8, 114–123. [Google Scholar] [CrossRef]
- Wang, S.; Xin, Y.; Yuan, J.; Wang, L.; Zhang, W. Direct Conversion of Methane to Methanol on Boron Nitride-Supported Copper Single Atoms. Nanoscale 2022, 14, 5447–5453. [Google Scholar] [CrossRef]
- Kang, Y.; Li, Z.; Lv, X.; Song, W.; Wei, Y.; Zhang, X.; Liu, J.; Zhao, Z. Active Oxygen Promoted Electrochemical Conversion of Methane on Two-Dimensional Carbide (MXenes): From Stability, Reactivity and Selectivity. J. Catal. 2021, 393, 20–29. [Google Scholar] [CrossRef]
- Szécsényi, Á.; Khramenkova, E.; Chernyshov, I.Y.; Li, G.; Gascon, J.; Pidko, E.A. Breaking Linear Scaling Relationships with Secondary Interactions in Confined Space: A Case Study of Methane Oxidation by Fe/ZSM-5 Zeolite. ACS Catal. 2019, 9, 9276–9284. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, S.; Lee, H. Heterogeneous Atomic Catalysts Overcoming the Limitations of Single-Atom Catalysts. ACS Nano 2020, 14, 14355–14374. [Google Scholar] [CrossRef]
- Brozek, C.K.; Dincă, M. Ti3+-, V2+/3+-, Cr2+/3+-, Mn2+-, and Fe2+-Substituted MOF-5 and Redox Reactivity in Cr- and Fe-MOF-5. J. Am. Chem. Soc. 2013, 135, 12886–12891. [Google Scholar] [CrossRef] [PubMed]
- Göltl, F.; Michel, C.; Andrikopoulos, P.C.; Love, A.M.; Hafner, J.; Hermans, I.; Sautet, P. Computationally Exploring Confinement Effects in the Methane-to-Methanol Conversion Over Iron-Oxo Centers in Zeolites. ACS Catal. 2016, 6, 8404–8409. [Google Scholar] [CrossRef]
- Valvekens, P.; Vermoortele, F.; De Vos, D. Metal–Organic Frameworks as Catalysts: The Role of Metal Active Sites. Catal. Sci. Technol. 2013, 3, 1435. [Google Scholar] [CrossRef]
- Vitillo, J.G.; Lu, C.C.; Cramer, C.J.; Bhan, A.; Gagliardi, L. Influence of First and Second Coordination Environment on Structural Fe(II) Sites in MIL-101 for C–H Bond Activation in Methane. ACS Catal. 2021, 11, 579–589. [Google Scholar] [CrossRef]
- Wang, V.C.-C.; Maji, S.; Chen, P.P.-Y.; Lee, H.K.; Yu, S.S.-F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef]
- Bagherzadeh Mostaghimi, A.H.; Al-Attas, T.A.; Kibria, M.G.; Siahrostami, S. A Review on Electrocatalytic Oxidation of Methane to Oxygenates. J. Mater. Chem. A 2020, 8, 15575–15590. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Kim, R.S.; Oh, S.; Surendranath, Y. Catalytic Methane Monofunctionalization by an Electrogenerated High-Valent Pd Intermediate. ACS Cent. Sci. 2017, 3, 1174–1179. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by Using Copper-Promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Antil, N.; Chauhan, M.; Akhtar, N.; Newar, R.; Begum, W.; Malik, J.; Manna, K. Metal–Organic Framework-Encaged Monomeric Cobalt(III) Hydroperoxides Enable Chemoselective Methane Oxidation to Methanol. ACS Catal. 2022, 12, 11159–11168. [Google Scholar] [CrossRef]
- Tang, X.; Wang, L.; Yang, B.; Fei, C.; Yao, T.; Liu, W.; Lou, Y.; Dai, Q.; Cai, Y.; Cao, X.-M.; et al. Direct Oxidation of Methane to Oxygenates on Supported Single Cu Atom Catalyst. Appl. Catal. B 2021, 285, 119827. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, Y.; Jiang, Y.; Zhang, X.; Long, C.; An, P.; Zhu, Y.; Shao, S.; Yan, Z.; Li, G.; et al. Fe-O Clusters Anchored on Nodes of Metal–Organic Frameworks for Direct Methane Oxidation. Angew. Chem. Int. Ed. 2021, 60, 5811–5815. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Hu, J.; Tian, L.; Liang, J.; Lin, J.; Li, L.; Zhu, C.; Wang, X. Zirconium-oxo Nodes of Mofs with Tunable Electronic Properties Provide Effective •OH Species for Enhanced Methane Hydroxylation. Angew. Chem. Int. Ed. 2022, 61, e202205077. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Wei, F.; Lin, J.; Zhou, Y.; Sun, L.; Shang, X.; Lin, S.; Wang, X. Retrofitting Zr-Oxo Nodes of UiO-66 by Ru Single Atoms to Boost Methane Hydroxylation with Nearly Total Selectivity. J. Am. Chem. Soc. 2023, 145, 13169–13180. [Google Scholar] [CrossRef]
- Xiao, P.; Wang, Y.; Lu, Y.; De Baerdemaeker, T.; Parvulescu, A.-N.; Müller, U.; De Vos, D.; Meng, X.; Xiao, F.-S.; Zhang, W.; et al. Effects of Al Distribution in the Cu-Exchanged AEI Zeolites on the Reaction Performance of Continuous Direct Conversion of Methane to Methanol. Appl. Catal. B 2023, 325, 122395. [Google Scholar] [CrossRef]
- Rungtaweevoranit, B.; Abdel-Mageed, A.M.; Khemthong, P.; Eaimsumang, S.; Chakarawet, K.; Butburee, T.; Kunkel, B.; Wohlrab, S.; Chainok, K.; Phanthasri, J.; et al. Structural Evolution of Iron-Loaded Metal–Organic Framework Catalysts for Continuous Gas-Phase Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2023, 15, 26700–26709. [Google Scholar] [CrossRef]
- Koishybay, A.; Shantz, D.F. Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13. J. Am. Chem. Soc. 2020, 142, 11962–11966. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic Zeolite Modification for in Situ Peroxide Formation in Methane Oxidation to Methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Li, W.; Li, Z.; Zhang, H.; Liu, P.; Xie, Z.; Song, W.; Liu, B.; Zhao, Z. Efficient Catalysts of Surface Hydrophobic Cu-BTC with Coordinatively Unsaturated Cu(I) Sites for the Direct Oxidation of Methane. Proc. Natl. Acad. Sci. USA 2023, 120, e2206619120. [Google Scholar] [CrossRef]
- Kye, S.-H.; Park, H.S.; Zhang, R.; Yang, H.J.; Lee, K.H.; Suh, H.; Kim, J.-G.; Kim, M.G.; Hwang, G.S.; Hur, N.H. Partial Oxidation of Methane to Methanol by Isolated Pt Catalyst Supported on a CeO2 Nanoparticle. J. Chem. Phys. 2020, 152, 054715. [Google Scholar] [CrossRef]
- Lustemberg, P.G.; Palomino, R.M.; Gutiérrez, R.A.; Grinter, D.C.; Vorokhta, M.; Liu, Z.; Ramírez, P.J.; Matolín, V.; Ganduglia-Pirovano, M.V.; Senanayake, S.D.; et al. Direct Conversion of Methane to Methanol on Ni-Ceria Surfaces: Metal–Support Interactions and Water-Enabled Catalytic Conversion by Site Blocking. J. Am. Chem. Soc. 2018, 140, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective Activation of Methane on Single-Atom Catalyst of Rhodium Dispersed on Zirconia for Direct Conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, E.; Orozco, I.; Liao, W.; Palomino, R.M.; Rui, N.; Duchoň, T.; Nemšák, S.; Grinter, D.C.; Mahapatra, M.; et al. Water-Promoted Interfacial Pathways in Methane Oxidation to Methanol on a CeO2-Cu2O Catalyst. Science 2020, 368, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Rui, N.; Rosales, R.; Kang, J.; Nemšák, S.; Senanayake, S.D.; Rodriguez, J.A.; Liu, P. Highly Selective Methane to Methanol Conversion on Inverse SnO2/Cu2O/Cu(111) Catalysts: Unique Properties of Sno2 Nanostructures and the Inhibition of the Direct Oxidative Combustion of Methane. ACS Catal. 2022, 12, 11253–11262. [Google Scholar] [CrossRef]
- Huang, E.; Orozco, I.; Ramírez, P.J.; Liu, Z.; Zhang, F.; Mahapatra, M.; Nemšák, S.; Senanayake, S.D.; Rodriguez, J.A.; Liu, P. Selective Methane Oxidation to Methanol on Zno/Cu2O/Cu(111) Catalysts: Multiple Site-Dependent Behaviors. J. Am. Chem. Soc. 2021, 143, 19018–19032. [Google Scholar] [CrossRef]
- Ross, M.O.; Rosenzweig, A.C. A Tale of Two Methane Monooxygenases. J. Biol. Inorg. Chem. 2017, 22, 307–319. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Viewpoint on the Partial Oxidation of Methane to Methanol Using Cu- and Fe-Exchanged Zeolites. ACS Catal. 2018, 8, 8306–8313. [Google Scholar] [CrossRef]
- Simons, M.C.; Prinslow, S.D.; Babucci, M.; Hoffman, A.S.; Hong, J.; Vitillo, J.G.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; et al. Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes. J. Am. Chem. Soc. 2021, 143, 12165–12174. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective Anaerobic Oxidation of Methane Enables Direct Synthesis of Methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, K.H.; Cho, S.J.; Park, E.D. Partial Oxidation of Methane with Hydrogen Peroxide over Fe-ZSM-5 Catalyst. Catal. Today 2021, 376, 113–118. [Google Scholar] [CrossRef]
- Lange, J.-P.; Sushkevich, V.L.; Knorpp, A.J.; Van Bokhoven, J.A. Methane-to-Methanol via Chemical Looping: Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Tabor, E.; Dedecek, J.; Mlekodaj, K.; Sobalik, Z.; Andrikopoulos, P.C.; Sklenak, S. Dioxygen Dissociation over Man-Made System at Room Temperature to Form the Active α-Oxygen for Methane Oxidation. Sci. Adv. 2020, 6, eaaz9776. [Google Scholar] [CrossRef] [PubMed]
- Dedecek, J.; Tabor, E.; Andrikopoulos, P.C.; Sklenak, S. Splitting Dioxygen over Distant Binuclear Transition Metal Cationic Sites in Zeolites. Effect of the Transition Metal Cation. Int. J. Quantum Chem. 2021, 121, e26611. [Google Scholar] [CrossRef]
- Mlekodaj, K.; Lemishka, M.; Sklenak, S.; Dedecek, J.; Tabor, E. Dioxygen Splitting at Room Temperature over Distant Binuclear Transition Metal Centers in Zeolites for Direct Oxidation of Methane to Methanol. Chem. Commun. 2021, 57, 3472–3475. [Google Scholar] [CrossRef]
- Tabor, E.; Lemishka, M.; Sobalik, Z.; Mlekodaj, K.; Andrikopoulos, P.C.; Dedecek, J.; Sklenak, S. Low-Temperature Selective Oxidation of Methane over Distant Binuclear Cationic Centers in Zeolites. Commun. Chem. 2019, 2, 71. [Google Scholar] [CrossRef]
- Jasin Arachchige, L.; Dong, A.; Wang, T.; Li, H.; Zhang, X.L.; Wang, F.; Su, H.; Sun, C. Mechanistic Insights into Direct Methane Oxidation to Methanol on Single-Atom Transition-Metal-Modified Graphyne. ACS Appl. Nano Mater. 2021, 4, 12006–12016. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Second-Sphere Effects on Methane Hydroxylation in Cu-Zeolites. J. Am. Chem. Soc. 2018, 140, 9236–9243. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bols, M.L.; Rhoda, H.M.; Plessers, D.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Cage Effects Control the Mechanism of Methane Hydroxylation in Zeolites. Science 2021, 373, 327–331. [Google Scholar] [CrossRef]
- Wu, L.; Fan, W.; Wang, X.; Lin, H.; Tao, J.; Liu, Y.; Deng, J.; Jing, L.; Dai, H. Methane Oxidation over the Zeolites-Based Catalysts. Catalysts 2023, 13, 604. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.; Fung, V.; Jiang, D.; Huang, W.; Zhang, S.; Iwasawa, Y.; Sakata, T.; Nguyen, L.; Zhang, X.; et al. Single Rhodium Atoms Anchored in Micropores for Efficient Transformation of Methane under Mild Conditions. Nat. Commun. 2018, 9, 1231. [Google Scholar] [CrossRef]
- Shan, J.; Li, M.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild Oxidation of Methane to Methanol or Acetic Acid on Supported Isolated Rhodium Catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Moteki, T.; Tominaga, N.; Ogura, M. Mechanism Investigation and Product Selectivity Control on CO-Assisted Direct Conversion of Methane into C1 and C2 Oxygenates Catalyzed by Zeolite-Supported Rh. Appl. Catal. B 2022, 300, 120742. [Google Scholar] [CrossRef]
- Li, H.; Fei, M.; Troiano, J.L.; Ma, L.; Yan, X.; Tieu, P.; Yuan, Y.; Zhang, Y.; Liu, T.; Pan, X.; et al. Selective Methane Oxidation by Heterogenized Iridium Catalysts. J. Am. Chem. Soc. 2023, 145, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, W.; Tang, Y.; Cao, W.; Docherty, S.R.; Wu, F.; Cheng, K.; Zhang, Q.; Copéret, C.; Wang, Y. Selective Oxidation of Methane to Methanol over Au/H-MOR. J. Am. Chem. Soc. 2023, 145, 12928–12934. [Google Scholar] [CrossRef]
- Yu, H.; Jiang, J. Accelerating Catalysts Design by Machine Learning. Sci. Bull. 2020, 65, 1593–1594. [Google Scholar] [CrossRef]
- Ishikawa, A.; Tateyama, Y. A First-Principles Microkinetics for Homogeneous–Heterogeneous Reactions: Application to Oxidative Coupling of Methane Catalyzed by Magnesium Oxide. ACS Catal. 2021, 11, 2691–2700. [Google Scholar] [CrossRef]
- Adamji, H.; Nandy, A.; Kevlishvili, I.; Román-Leshkov, Y.; Kulik, H.J. Computational Discovery of Stable Metal–Organic Frameworks for Methane-to-Methanol Catalysis. J. Am. Chem. Soc. 2023, 145, 14365–14378. [Google Scholar] [CrossRef]
- Andrade, L.S.; Lima, H.H.L.B.; Silva, C.T.P.; Amorim, W.L.N.; Poço, J.G.R.; López-Castillo, A.; Kirillova, M.V.; Carvalho, W.A.; Kirillov, A.M.; Mandelli, D. Metal–Organic Frameworks as Catalysts and Biocatalysts for Methane Oxidation: The Current State of the Art. Coord. Chem. Rev. 2023, 481, 215042. [Google Scholar] [CrossRef]
- Hall, J.N.; Li, M.; Bollini, P. Light Alkane Oxidation over Well-Defined Active Sites in Metal–Organic Framework Materials. Catal. Sci. Technol. 2022, 12, 418–435. [Google Scholar] [CrossRef]
- Imyen, T.; Znoutine, E.; Suttipat, D.; Iadrat, P.; Kidkhunthod, P.; Bureekaew, S.; Wattanakit, C. Methane Utilization to Methanol by a Hybrid Zeolite@metal–Organic Framework. ACS Appl. Mater. Interfaces 2020, 12, 23812–23821. [Google Scholar] [CrossRef]
- Li, S.; Ahmed, R.; Yi, Y.; Bogaerts, A. Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2021, 11, 590. [Google Scholar] [CrossRef]
- Sher Shah, M.S.A.; Oh, C.; Park, H.; Hwang, Y.J.; Ma, M.; Park, J.H. Catalytic Oxidation of Methane to Oxygenated Products: Recent Advancements and Prospects for Electrocatalytic and Photocatalytic Conversion at Low Temperatures. Adv. Sci. 2020, 7, 2001946. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yang, J.; Moon, J.H. Solar Cell-Powered Electrochemical Methane-to-Methanol Conversion with CuO/CeO2 Catalysts. ACS Energy Lett. 2021, 6, 893–899. [Google Scholar] [CrossRef]
- Chawdhury, P.; Wang, Y.; Ray, D.; Mathieu, S.; Wang, N.; Harding, J.; Bin, F.; Tu, X.; Subrahmanyam, C. A Promising Plasma-Catalytic Approach towards Single-Step Methane Conversion to Oxygenates at Room Temperature. Appl. Catal. B 2021, 284, 119735. [Google Scholar] [CrossRef]
- Zhu, S.; Li, X.; Pan, Z.; Jiao, X.; Zheng, K.; Li, L.; Shao, W.; Zu, X.; Hu, J.; Zhu, J.; et al. Efficient Photooxidation of Methane to Liquid Oxygenates over ZnO Nanosheets at Atmospheric Pressure and Near Room Temperature. Nano Lett. 2021, 21, 4122–4128. [Google Scholar] [CrossRef]























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Chen, Y.; Xie, Z.; Song, W.; Liu, B.; Zhao, Z. Rational Design of the Catalysts for the Direct Conversion of Methane to Methanol Based on a Descriptor Approach. Catalysts 2023, 13, 1226. https://doi.org/10.3390/catal13081226
Li Z, Chen Y, Xie Z, Song W, Liu B, Zhao Z. Rational Design of the Catalysts for the Direct Conversion of Methane to Methanol Based on a Descriptor Approach. Catalysts. 2023; 13(8):1226. https://doi.org/10.3390/catal13081226
Chicago/Turabian StyleLi, Zhi, Yanjun Chen, Zean Xie, Weiyu Song, Baijun Liu, and Zhen Zhao. 2023. "Rational Design of the Catalysts for the Direct Conversion of Methane to Methanol Based on a Descriptor Approach" Catalysts 13, no. 8: 1226. https://doi.org/10.3390/catal13081226
APA StyleLi, Z., Chen, Y., Xie, Z., Song, W., Liu, B., & Zhao, Z. (2023). Rational Design of the Catalysts for the Direct Conversion of Methane to Methanol Based on a Descriptor Approach. Catalysts, 13(8), 1226. https://doi.org/10.3390/catal13081226