
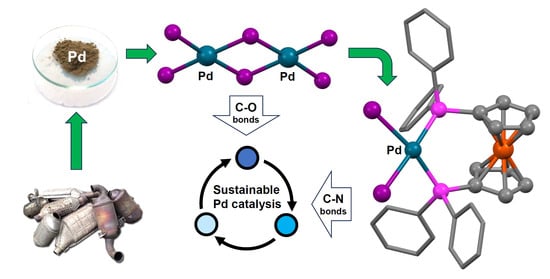
Palladium Complexes Derived from Waste as Catalysts for C-H Functionalisation and C-N Bond Formation

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Palladium Complexes
2.2. Oxidative C-H Functionalisation
2.3. Amination Reactions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johansson Seechurn, C.C.; Kitching, M.O.; Colacot, T.J.; Snieckus, V. Palladium-catalysed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012, 51, 5062–5085. [Google Scholar] [CrossRef] [PubMed]
- Gensch, T.; Hopkinson, M.N.; Glorius, F.; Wencel-Delord, J. Mild metal-catalysed C–H activation: Examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalisation reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Muci, A.R.; Buchwald, S.L. Practical Palladium Catalysts for C-N and C-O Bond Formation. In Cross-Coupling Reactions; A Practical Guide (2002); Miyaura, N., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 219, pp. 131–209. [Google Scholar]
- Gazvoda, M.; Dhanjee, H.H.; Rodriguez, J.; Brown, J.S.; Farquhar, C.E.; Truex, N.L.; Loas, A.; Buchwald, S.L.; Pentelute, B.L. Palladium-mediated incorporation of carboranes into small molecules, peptides, and proteins. J. Am. Chem. Soc. 2022, 144, 7852–7860. [Google Scholar] [CrossRef] [PubMed]
- Reichert, E.C.; Feng, K.; Sather, A.C.; Buchwald, S.L. Pd-Catalyzed Amination of Base-Sensitive Five-Membered Heteroaryl Halides with Aliphatic Amines. J. Am. Chem. Soc. 2023, 145, 3323–3329. [Google Scholar] [CrossRef] [PubMed]
- ACS Green Chemistry Institute: Endangered Elements. 2020. Available online: https://www.acs.org/content/acs/en/greenchemistry/research-innovation/endangered-elements.html (accessed on 22 April 2024).
- Johnson Matthey. PGM Market Report. 2019, pp. 1–48. Available online: http://www.platinum.matthey.com/services/market-research/pgm-market-reports (accessed on 22 April 2024).
- Nuss, P.; Eckelman, M.J. Life cycle assessment of metals: A scientific synthesis. PLoS ONE 2014, 9, e101298. [Google Scholar] [CrossRef] [PubMed]
- Michałek, T.; Hessel, V.; Wojnicki, M. Production, recycling and economy of palladium: A critical review. Materials 2023, 17, 45. [Google Scholar] [CrossRef]
- Macklin, M.G.; Thomas, C.J.; Mudbhatkal, A.; Brewer, P.A.; Hudson-Edwards, K.A.; Lewin, J.; Scussolini, P.; Eilander, D.; Lechner, A.; Owen, J.; et al. Impacts of metal mining on river systems: A global assessment. Science 2023, 381, 1345–1350. [Google Scholar] [CrossRef]
- Glaister, B.J.; Mudd, G.M. The environmental costs of platinum–PGM mining and sustainability: Is the glass half-full or half-empty? Miner. Eng. 2010, 23, 438–450. [Google Scholar] [CrossRef]
- Yakoumis, I.; Panou, M.; Moschovi, A.M.; Panias, D. Recovery of platinum group metals from spent automotive catalysts: A review. Cleaner Eng. Technol. 2021, 3, 100112. [Google Scholar] [CrossRef]
- Paiva, A.P.; Piedras, F.V.; Rodrigues, P.G.; Nogueira, C.A. Hydrometallurgical recovery of platinum-group metals from spent auto-catalysts—Focus on leaching and solvent extraction. Sep. Purif. Technol. 2022, 286, 120474. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Hu, Z.; Yao, M.; Li, Y. A review on the Pd-based three-way catalyst. Catal. Rev. 2015, 57, 79–144. [Google Scholar] [CrossRef]
- McCarthy, S.; Braddock, D.C.; Wilton-Ely, J.D.E.T. Strategies for sustainable palladium catalysis. Coord. Chem. Rev. 2021, 442, 213925. [Google Scholar] [CrossRef]
- Hagelüken, B.C. Recycling the platinum group metals: A European perspective. Platinum Met. Rev. 2012, 56, 29–35. [Google Scholar] [CrossRef]
- Dong, H.; Zhao, J.; Chen, J.; Wu, Y.; Li, B. Recovery of platinum group metals from spent catalysts: A review. Int. J. Miner. Process. 2015, 145, 108–113. [Google Scholar] [CrossRef]
- Serpe, A.; Bigoli, F.; Cabras, M.C.; Fornasiero, P.; Graziani, M.; Mercuri, M.L.; Montini, T.; Pilia, L.; Trogu, E.F.; Deplano, P. Pd-dissolution through a mild and effective one-step reaction and its application for Pd-recovery from spent catalytic converters. Chem. Commun. 2005, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Jantan, K.A.; Chan, K.W.; Melis, L.; White, A.J.P.; Marchiò, L.; Deplano, P.; Serpe, A.; Wilton-Ely, J.D.E.T. From recovered palladium to molecular and nanoscale catalysts. ACS Sustain. Chem. Eng. 2019, 7, 12389–12398. [Google Scholar] [CrossRef]
- Jantan, K.A.; Kwok, C.Y.; Chan, K.W.; Marchiò, L.; White, A.J.P.; Deplano, P.; Serpe, A.; Wilton-Ely, J.D.E.T. From recovered metal waste to high-performance palladium catalysts. Green Chem. 2017, 19, 5846–5853. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A highly selective catalytic method for the oxidative functionalisation of C-H bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef]
- McCarthy, S.; Desaunay, O.; Jie, A.L.; Hassatzky, M.; White, A.J.P.; Deplano, P.; Braddock, D.C.; Serpe, A.; Wilton-Ely, J.D.E.T. Homogeneous gold catalysis using complexes recovered from waste electronic equipment. ACS Sustain. Chem. Eng. 2022, 10, 15726–15734. [Google Scholar] [CrossRef]
- Cuscusa, M.; Rigoldi, A.; Artizzu, F.; Cammi, R.; Fornasiero, P.; Deplano, P.; Marchiò, L.; Serpe, A. Ionic couple-driven palladium leaching by organic triiodide solutions. ACS Sustain. Chem. Eng. 2017, 5, 4359–4370. [Google Scholar] [CrossRef]
- McCarthy, S.; Braddock, D.C.; Wilton-Ely, J.D.E.T. From waste to green applications: The use of recovered gold and palladium in catalysis. Molecules 2021, 26, 5217. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Ely, J.D.E.T. The use of recovered metal complexes in catalysis. Johns. Matthey Technol. Rev. 2023, 67, 300–302. [Google Scholar]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An improved catalyst system for aromatic carbon−nitrogen bond formation: The possible involvement of bis(phosphine) palladium complexes as key intermediates. J. Am. Chem. Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Driver, M.S.; Hartwig, J.F. A second-generation catalyst for aryl halide amination: Mixed secondary amines from aryl halides and primary amines catalysed by (DPPF)PdCl2. J. Am. Chem. Soc. 1996, 118, 7217–7218. [Google Scholar] [CrossRef]
- Colacot, T.J.; Qian, H.; Cea-Olivares, R.; Hernandez-Ortega, S. Synthesis, X-ray, spectroscopic and a preliminary Suzuki coupling screening studies of a complete series of dppfMX2 (M = Pt, Pd; X = Cl, Br, I). J. Organomet. Chem. 2001, 637, 691–697. [Google Scholar] [CrossRef]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C-H) bond activation at Pd IV: A Frontier in C-H functionalisation catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Powers, D.C.; Ritter, T. Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation. Nat. Chem. 2009, 1, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Westermann, E. Phosphane-free palladium-catalysed coupling reactions: The decisive role of Pd nanoparticles. Angew. Chem. Int. Ed. 2000, 39, 165–168. [Google Scholar] [CrossRef]
- Baumann, C.G.; De Ornellas, S.; Reeds, J.P.; Storr, T.E.; Williams, T.J.; Fairlamb, I.J.S. Formation and propagation of well-defined Pd nanoparticles (PdNPs) during C-H bond functionalisation of heteroarenes: Are nanoparticles a moribund form of Pd or an active catalytic species? Tetrahedron 2014, 70, 6174–6187. [Google Scholar] [CrossRef]
- Reay, A.J.; Fairlamb, I.J.S. Catalytic C–H bond functionalisation chemistry: The case for quasi-heterogeneous catalysis. Chem. Commun. 2015, 51, 16289–16307. [Google Scholar] [CrossRef]
- Guram, A.S.; Rennels, R.A.; Buchwald, S.L. A simple catalytic method for the conversion of aryl bromides to arylamines. Angew. Chem. Int. Ed. 1995, 34, 1348–1350. [Google Scholar] [CrossRef]
- Wagaw, S.; Rennels, R.A.; Buchwald, S.L. Palladium-catalyzed coupling of optically active amines with aryl bromides. J. Am. Chem. Soc. 1997, 119, 8451–8458. [Google Scholar] [CrossRef]
- Ali, M.H.; Buchwald, S.L. An improved method for the palladium-catalysed amination of aryl iodides. J. Org. Chem. 2001, 66, 2560–2565. [Google Scholar] [CrossRef] [PubMed]
- Campeau, L.C.; Parisien, M.; Jean, A.; Fagnou, K. Catalytic direct arylation with aryl chlorides, bromides, and iodides: Intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc. 2006, 128, 581–590. [Google Scholar] [CrossRef]
- Forero-Cortés, P.A.; Haydl, A.M. The 25th anniversary of the Buchwald–Hartwig amination: Development, applications, and outlook. Org. Process Res. Dev. 2019, 23, 1478–1483. [Google Scholar] [CrossRef]
- Azzena, U.; Carraro, M.; Pisano, L.; Monticelli, S.; Bartolotta, R.; Pace, V. Cyclopentyl methyl ether: An elective ecofriendly ethereal solvent in classical and modern organic chemistry. ChemSusChem 2019, 12, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical-and less classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- SHELXTL v5.1; Bruker AXS: Madison, WI, USA, 1998.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. (2003, 2009) PLATON, A Multipurpose Crystallographic Tool. Acta. Cryst. 2015, C71, 9–18. [Google Scholar]
- Old, D.W.; Wolfe, J.P.; Buchwald, S.L. A Highly Active Catalyst for Palladium-Catalyzed Cross-Coupling Reactions: Room-Temperature Suzuki Couplings and Amination of Unactivated Aryl Chlorides. J. Am. Chem. Soc. 1998, 120, 9722–9723. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Tomori, H.; Sadighi, J.P.; Yin, J.; Buchwald, S.J. Simple, Efficient Catalyst System for the Palladium-Catalyzed Amination of Aryl Chlorides, Bromides, and Triflates. J. Org. Chem. 2000, 65, 1158–1174. [Google Scholar] [CrossRef]
- Semeniuchenko, V.; Braje, W.M.; Organ, M.G. Sodium Butylated Hydroxytoluene: A Functional Group Tolerant, Eco-Friendly Base for Solvent-Free, Pd-Catalysed Amination. Chem. Eur. J. 2021, 27, 12535–12539. [Google Scholar] [CrossRef] [PubMed]
- Hajra, A.; Wei, Y.; Yoshikai, N. Palladium-Catalyzed Aerobic Dehydrogenative Aromatization of Cyclohexanone Imines to Arylamines. Org. Lett. 2012, 14, 5488–5491. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Run | Conversion to organic product (%) | Recovery yield of 1 (%) |
| 1 | 97 | 75 |
| 2 | 98 | 72 |

| Solvent | Conversion (%) with PdCl2(dppf) | Conversion (%) with [NnBu4]2[Pd2I6] (1) | Conversion (%) with PdI2(dppf) (2) |
|---|---|---|---|
| THF | 90 ± 1 | 55 ± 1 | 81 ± 1 |
| CPME | 98 ± 1 | 92 ± 2 | 90 ± 1 |
| Toluene | 98 ± 1 | 80 ± 1 | 96 ± 2 |
| Pd loading (mol%) | Conversion (%) with PdCl2(dppf) | Conversion (%) with [NnBu4]2[Pd2I6] (1) | Conversion (%) with PdI2(dppf) (2) |
|---|---|---|---|
| 5 | 71 ± 3 | 59 ± 1 | 65 ± 3 |
| 2 | 80 ± 1 | 72 ± 1 | 57 ± 2 |
| Conversion to the Product Shown (%) | |||||
|---|---|---|---|---|---|
| Catalyst 2 | Catalyst 1 | Catalyst 2 | Catalyst 1 | Catalyst 2 | |
| 1 mol% [Pd] 3 mol% [dppf] | 1 mol% [Pd] 3 mol% [dppf] | 2 mol% [Pd] 6 mol% [dppf] | 2 mol% [Pd] 6 mol% [dppf] | 2 mol% [Pd] 15 mol% [dppf] | |
 | 85 ± 2 | 83 ± 1 | 99 ± 1 | 99 ± 1 | 99 ± 1 |
 | 0 | 0 | 45 ± 2 | 60 ± 1 | 98 ± 2 |
 | 19 ± 3 | 26 ± 2 | 80 ± 1 | 96 ± 2 | 85 ± 2 |
 | 17 ± 2 | 35 ± 3 | 17 ± 2 | 21 ± 4 | 45 ± 1 |
 | 62 ± 4 | 59 ± 1 | 81 ± 2 | 97 ± 1 | 87 ± 1 |
 | 15 ± 1 | - | 99 ± 1 | 86 ± 3 | 91 ± 1 |
 | 9 ± 2 | - | 82 ± 2 | 82 ± 2 | 95 ± 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jantan, K.A.; Ekart, G.; McCarthy, S.; White, A.J.P.; Braddock, D.C.; Serpe, A.; Wilton-Ely, J.D.E.T. Palladium Complexes Derived from Waste as Catalysts for C-H Functionalisation and C-N Bond Formation. Catalysts 2024, 14, 295. https://doi.org/10.3390/catal14050295
Jantan KA, Ekart G, McCarthy S, White AJP, Braddock DC, Serpe A, Wilton-Ely JDET. Palladium Complexes Derived from Waste as Catalysts for C-H Functionalisation and C-N Bond Formation. Catalysts. 2024; 14(5):295. https://doi.org/10.3390/catal14050295
Chicago/Turabian StyleJantan, Khairil A., Gregor Ekart, Sean McCarthy, Andrew J. P. White, D. Christopher Braddock, Angela Serpe, and James D. E. T. Wilton-Ely. 2024. "Palladium Complexes Derived from Waste as Catalysts for C-H Functionalisation and C-N Bond Formation" Catalysts 14, no. 5: 295. https://doi.org/10.3390/catal14050295
APA StyleJantan, K. A., Ekart, G., McCarthy, S., White, A. J. P., Braddock, D. C., Serpe, A., & Wilton-Ely, J. D. E. T. (2024). Palladium Complexes Derived from Waste as Catalysts for C-H Functionalisation and C-N Bond Formation. Catalysts, 14(5), 295. https://doi.org/10.3390/catal14050295