1. Introduction
Ziegler-Natta catalysts and metallocene catalysts can catalyze the copolymerization of olefins efficiently [
1,
2,
3,
4]. However, the coordination copolymerization of ethylene with polar comonomer is a challenge because the nonbonded electron pair of the heteroatom from the polar comonomer tends to form a complex with the center metal, resulting in deactivation of the transition metal catalyst. Up to now, copolymerization of ethylene with polar monomers has been performed by a conventional radical process. Polymers prepared via a radical process have poor stereo-controlling [
5]. In recent years, the design of non-metallocene catalysts has been given much attention because non-metallocene catalysts possess broad “tolerance” to electron-donating atoms such as N, O, P
etc. Thus, using non-metallocene catalysts to prepare functional polyolefins is a preferred approach. Changing the ligand structure of the catalyst can make the center metal less oxophilic and enhance the stereo-controlling ability for the polymer. Marks [
6] adopted CGCTiMe
2/Ph
3CB
+(C
6F
5)
4− to catalyze the copolymerization of ethylene with 5-hexenylsilane. A functional group was introduced to the copolymer. The result showed that 5-hexenylsilane readily underwent insertion into the polymer chain as well as affecting intramolecular chain termination. Palladium complexes with bulky substituted α-diimine ligands can catalyze the copolymerization of ethylene and α-olefins with methyl acrylate to give high-molar-mass polymers [
7]. Nozaki
et al. [
8] reported that phosphine-sulfonate methyl palladium complex can catalyze copolymerization of ethylene with acrylonitrile. It was demonstrated that acrylonitrile units were inserted into the linear polyethylene chain. Furthermore, acrylonitrile units were shown at the terminating end of the copolymer chain and in the backbone. In the following years, Nozaki
et al., prepared copolymers of ethylene with various polar monomers such as allyl monomers [
9], vinyl acetate [
10]
etc. by use of palladium/phosphine-sulfonate catalyst. Furthermore, they utilized palladium/alkylphosphine-sulfonate catalysts to synthesize copolymers of ethylene with polar monomers featured with higher weight average molecular weight of 1.77 × 10
5 g/mol by tuning the substituent groups of the phosphorus atom constituent of the ligand [
11]. Fujita [
12] demonstrated the molecular tailoring of polymer can be realized by changing the ligand structure. He prepared Al-PEs by a bis(phenoxy-imine) zirconium complex with methylalumoxane, which confirmed that the stereo-controlling ability of the non-metallocene catalysts can be enhanced. Chen
et al. [
13] reported chemoselective, stereospecific, and living polymerization of polar divinyl monomers by chiral zirconium catalysts. Li
et al. [
14] obtained the new functional iPPs with high molecular weights and abundant polar groups by hafnium catalyst tandem with click chemistry. Hu
et al. [
15] performed copolymerizations of ethylene and polar comonomers with bis(phenoxyketimine) group IV complexes. The results revealed that early transition metal complexes are favorable for the preparation of high molecular weight functionalized polyolefins containing a high content of polar groups. Before the report, the copolymers of ethylene with polar monomers had low weight average molecular weight of about 1 × 10
4 g/mol. Guan
et al. [
16] and Ye
et al. [
17] adopted Pd-diimine catalysts to catalyze the copolymerization of ethylene and acrylic monomers. The copolymers were highly branched and contained double bonds; and the comonomer insertion rate was 2.0 mol % and 3.6 mol %. Nevertheless, the catalytic activities were relatively low, about 1.6 × 10
2 g/(mol Pd h) and 9.2 × 10
2 g/(mol Pd h), respectively. In our previous work, non-metallocene catalysts with [N, N, O] [
18], [N, N, N] [
19] and [N, N, O, O] [
20,
21] ligands were synthesized and carried out for homopolymerization of ethylene and copolymerization of ethylene with α-olefins or polar monomers such as acrylonitrile.
Herein, we report the copolymers of ethylene with N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester prepared by non-metallocene catalysts with [N, Si, N, P]-type ligands based on diisopropyl phosphorus-phenylamine and their derivatives.
2. Results and Discussion
The obtained [N, Si, N, P] non-metallocene catalysts (Cat.
1–
6) were used for ethylene copolymerization, with MAO used as cocatalyst. The effects of centre metal atoms (Ti and Zr) and the structure of catalyst ligands on ethylene copolymerization were investigated, and the data are compiled in
Table 1.
Table 1.
Effects of the metal atoms and the structure of catalyst ligands on ethylene copolymerization.
Table 1.
Effects of the metal atoms and the structure of catalyst ligands on ethylene copolymerization.
| Run | Catalysts | A a (×104) | N-cont b (mol %) | Mw c (×105) | Mw/Mn c | Tm d (°C) |
|---|
| 1 | Cat.1 (Ti, L1) | 127 | - | 7.56 | 1.87 | 135.6 |
| 2 e | 3.68 | 1.31 | 1.58 | 2.59 | 137.0 |
| 3 | Cat.2 (Zr, L1) | 95.2 | - | 7.28 | 1.77 | 135.8 |
| 4 e | 1.61 | 0.41 | 1.35 | 2.91 | 135.9 |
| 5 | Cat.3 (Ti, L2) | 121 | - | 7.58 | 1.75 | 135.1 |
| 6 e | 5.76 | 1.78 | 1.48 | 2.78 | 137.8 |
| 7 | Cat.4 (Zr, L2) | 109 | - | 7.26 | 1.81 | 135.2 |
| 8 e | 2.06 | 0.56 | 1.42 | 2.76 | 136.1 |
| 9 | Cat.5 (Ti, L3) | 168 | - | 8.26 | 1.79 | 135.6 |
| 10 e | 6.32 | 2.42 | 1.73 | 2.89 | 138.7 |
| 11 | Cat.6 (Zr, L3) | 156 | - | 6.42 | 1.92 | 134.7 |
| 12 e | 2.45 | 0.91 | 1.53 | 2.66 | 136.5 |
From
Table 1, the results indicated that these catalysts were favorable for both homopolymerization of ethylene and copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester. The highest catalytic activity for homopolymerization of ethylene was up to 1.68 × 10
6 g PE (mol Ti)
−1 h
−1 and that for copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester was up to 6.32 × 10
4 g P (mol Ti)
−1 h
−1 catalyzed by Cat.
5. Titanium complex showed a higher catalytic activity for ethylene copolymerization than zirconium complex. We also noticed that the structure of the ligands and the center metal atoms of these catalysts influenced the ethylene copolymerization behavior. Cat.
1 to Cat.
6 were used for both homopolymerization of ethylene and copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester. The MWs were reduced from 8.26 × 10
5 g/mol to 6.42 × 10
5 g/mol for polyethylene, and from 1.73 × 10
5 g/mol to 1.35 × 10
5 g/mol for copolymer of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester. The MWDs were slightly broadened from 1.75 to 1.87 for polyethylene and from 2.59 to 2.91 for the copolymers. This result implied that the chain transfer took place to some extent when the comonomer was added into the polymerization system.
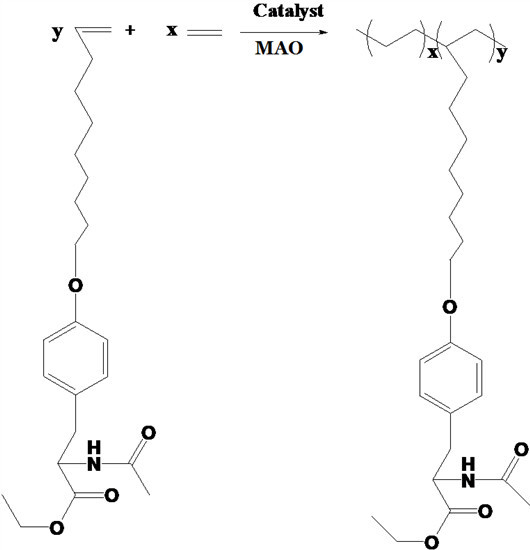
Scheme 1.
Copolymerization of ethylene and N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester.
Scheme 1.
Copolymerization of ethylene and N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester.
It is obvious that Cat.
5 and Cat.
6 with L
3 exhibited the highest catalytic activity (Runs 9 to 12 in
Table 1), compared with other catalysts with the same transition metals. This depends on the electrical and steric effect of the ligands. Compared with L
3, L
2 with methyl on the benzene ring had a large steric block. Methyl on the para-position of L
2 has a strong electron donating effect that leads to high electron density around the active center. It hinders the forming monomer from approaching the metal atom.
As shown in
Table 1, the catalyst with a fluorine atom on the para-position of aniline showed higher activity than that with a methyl group as it decreases the cloud density of the metal center, attracting ethylene more to the active center. The catalysts include a large five-membered ring, ensuring the stability of the active centers.
The effects of polymerization conditions on copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester catalyzed by Cat.
5 were investigated: temperature, catalyst concentration, Al/Ti ratio, and concentration of comonomer. The results are listed in
Table 2.
From
Table 2, we noticed that Cat.
5 exhibited the highest catalytic activity of 6.56 × 10
4 g P (mol
−1 Ti
−1 h
−1) (run 14 in
Table 2) at 50 °C for the copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester and then decreased. As shown in
Table 2, the catalytic activity decreased when the temperature was lower or higher (runs 13 and 15 in
Table 2). It might be due to the fact that temperature not only influences the chain propagation rate but also the stability of active species [
19]. With an increase of temperature from 25 °C to 70 °C, the MWs of the polymers became low and the MWDs became slightly broader. It is possible that the chain transfer rate increasing with temperature influences the MWs and MWDs of the copolymers. The incorporation ratio of
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester changed slightly at different temperatures.
Table 2.
Effects of polymerization conditions on copolymerization of ethylene with N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester catalyzed by Cat.5.
Table 2.
Effects of polymerization conditions on copolymerization of ethylene with N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester catalyzed by Cat.5.
| Run | T a (°C) | C b (×10−4) | Common c (g/L) | Al/M d | A e (×104) | Mw f (×105) | Mw/Mn f | Tm g (°C) | N-cont h (mol %) |
|---|
| 13 | 25 | 2.0 | 10 | 600 | 4.71 | 3.66 | 2.71 | 137.7 | 1.67 |
| 14 | 50 | 2.0 | 10 | 600 | 6.49 | 2.73 | 2.91 | 138.6 | 2.39 |
| 15 | 70 | 2.0 | 10 | 600 | 5.24 | 2.31 | 3.09 | 138.5 | 2.26 |
| 16 | 50 | 1.0 | 10 | 600 | 3.96 | 2.81 | 3.11 | 136.3 | 0.81 |
| 17 | 50 | 3.0 | 10 | 600 | 6.12 | 2.63 | 3.32 | 138.2 | 2.14 |
| 18 | 50 | 4.0 | 10 | 600 | 5.66 | 2.43 | 3.22 | 138.0 | 1.89 |
| 19 | 50 | 5.0 | 10 | 600 | 5.20 | 2.31 | 3.21 | 137.6 | 1.47 |
| 20 | 50 | 2.0 | 5 | 600 | 6.63 | 2.77 | 2.91 | 137.3 | 1.35 |
| 21 | 50 | 2.0 | 20 | 600 | 5.35 | 2.87 | 2.93 | 138.9 | 2.56 |
| 22 | 50 | 2.0 | 10 | 300 | 2.44 | 1.89 | 2.93 | 137.9 | 1.86 |
| 23 | 50 | 2.0 | 10 | 1000 | 2.61 | 0.87 | 3.64 | 138.1 | 2.08 |
As shown in
Table 2, when the catalyst concentration was 2.0 × 10
−4 g/L, the catalytic activity exhibited the highest value of 6.49 × 10
4 g PE (mol
−1 Ti
−1 h
−1). Because the active centers of the catalysts are easily deactivated by impurity, low catalyst concentration cannot catalyze copolymerization efficiently. However, too high a catalyst concentration leads to fast polymer chain aggregation within the same polymerization time, it is more difficult for the monomers to approach the active centers. So either low or high catalyst concentration decreased the catalytic activity (runs 16–19 in
Table 2).
The concentration of
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester in the feed influenced the incorporation content of
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester. For example, when the concentration of
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester was 5 g/L in the reaction system, the resulted insertion content of the comonomer was 1.35 mol %; while when the concentration increased to 10 g/L, the insertion content of the comonomer increased to 2.39 mol %. However, on continuing to increase the concentration to 20 g/L, the insertion content of the comonomer slightly increased to 2.56 mol % (runs 14, 20 and 21 in
Table 2). The results showed that there was a limit to the amount of the comonomer. Too much comonomer does not work efficiently for polymerization.
The catalytic activity of the copolymerization of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester reached 6.63 × 10
4 g P/(mol Ti h) when the Al/Ti ratio in mol was 600. The catalytic activity of the catalysts for the copolymerization decreased when the Al/Ti ratio in mol was low or high. Less MAO readily promoted active center transfer to β-H, resulting in lower activity, lower MWs, and broader MWDs. Excess MAO propagated chain transfer to MAO, resulting in lower MWs and broader MWDs. Chain transfer to MAO has been identified as the major chain transfer pathway for a titanium or zirconium catalyst with the hetero atoms of the polar ligand in ethylene copolymerization [
12,
22]. So the proper amount of MAO was important for the stablity of the active center and for controlling the ability of chain transfer to β-H and MAO. The reaction system purification and chain transfer reaction to MAO consumed MAO. However, high MAO can reduce catalytic activity for polymerization, and also make chain transfer easy (run 23 in
Table 2).
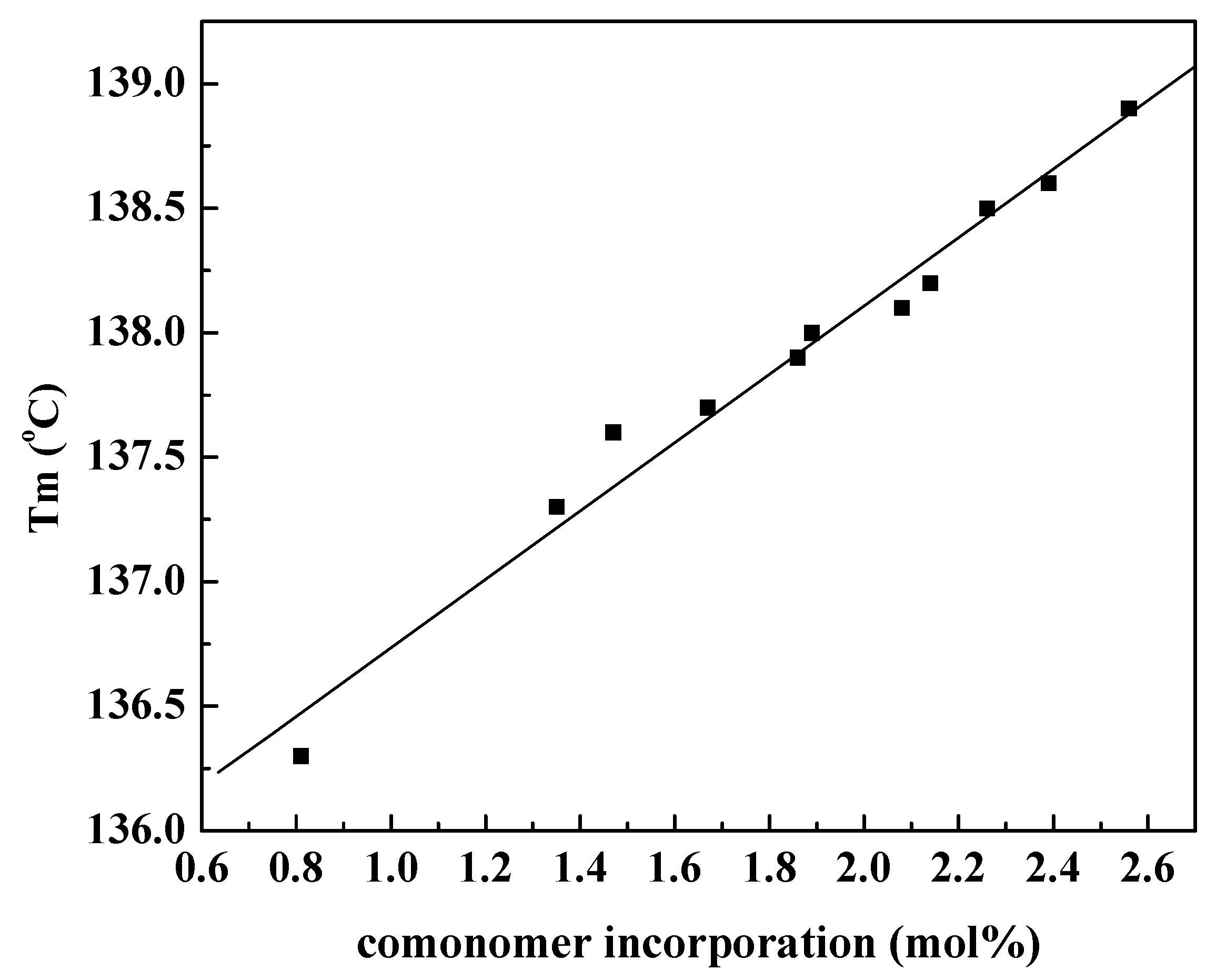
Figure 1 (the data from
Table 2) showed that the melting temperature of the resultant polymers gradually increased with increasing the
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester contents of the copolymers, which can be contributed to the hydrogen bonding interaction between the branch chains. Furthermore, each polymer has only one melting temperature, implying that the copolymer possessed uniform
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester incorporation.
Figure 1.
Variation of melting temperature of copolymers versus N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester incorporation.
Figure 1.
Variation of melting temperature of copolymers versus N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester incorporation.
The GPC curves of ethylene homopolymer and copolymers (
Figure 2) were monomodal and symmetric in shape. GPC results exhibited that the MWs of ethylene homopolymer (
Figure 2A) and copolymers (
Figure 2B–D) were 8.26 × 10
5 g/mol, 2.81 × 10
5 g/mol, 2.73×10
5 g/mol, 2.77 × 10
5 g/mol, respectively. MWs of the copolymers were lower than that of the homopolymer, however, MWDs of the copolymers were broader, which indicated that the chain transfer took place during the polymerization when
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester was added into the polymerization system. The MWD of the homopolyethylene was about three, suggesting that the polymerization behavior was restricted to the single mechanism.
The CP-MAS
13C-NMR spectrum of the polyethylene prepared by Cat.
5 (run 14 in
Table 2) was performed (
Figure 3B). Only methylene peaks showed in the spectra. The signals at δ = 33.5, 32.4, and 31.2 ppm were attributed to methylene carbon atoms from monoclinic crystalline, orthorhombic crystalline, and amorphous fraction, respectively [
23]. Other signals did not appear, which corresponds with linear polyethylene without branching.
Figure 2.
GPC curves of ethylene homopolymer (A, run 9 in
Table 1) and copolymers of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester (B, run 16; C, run 14; D, run 20 in
Table 2).
Figure 2.
GPC curves of ethylene homopolymer (A, run 9 in
Table 1) and copolymers of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester (B, run 16; C, run 14; D, run 20 in
Table 2).
Figure 3.
CP-MAS
13C-NMR spectra of polyethylene (A, run 9 in
Table 1) and copolymer of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester (B, run 14 in
Table 2).
Figure 3.
CP-MAS
13C-NMR spectra of polyethylene (A, run 9 in
Table 1) and copolymer of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester (B, run 14 in
Table 2).
The CP-MAS
13C-NMR spectrum of the copolymer of ethylene with
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester (run 14 in
Table 2) is presented in
Figure 3B. One broad signal at δ = 21.43–34.27 ppm represents methylene in the backbone and polymer chains and signals at δ = 154.30, 131.23, 127.52 and 114.33 ppm represent the carbon atoms on the benzene ring in tyrosine ethyl ester. This indicated that the comonomer had inserted into the polyethylene chain. The signals at δ = 173.62 and 170.06 ppm are the signals of amide group and ester group, respectively. Signals at δ = 97.43, −35.26 ppm are sideband signals of methylene. The area integral ratio of the resonance signals of methylene, methane (δ = 21.43–34.27 ppm) to tyrosine ethyl ester group (δ= 114–154 ppm) is 83.6:1. In other words,
x/
y = (83.6 – 2)/2 = 81.6/2 = 40.8/1. The content of comonomer inserted into copolymer was
y/(
x +
y) = 1/(40.8 + 1) = 2.39 mol %, Where
x was defined as the number of ethylene units and
y defined as the number of comonomer units in the copolymer chain.
FT-IR spectra of the obtained copolymers were performed (
Figure 4). Bands at 2917 cm
−1, 2849 cm
−1, 1471 cm
−1, and 718 cm
−1 are characters of linear polyethylene. Because of the rocking vibration of methylene, ((CH
2)
n,
n > 3) showed bending vibration absorptions at 718 cm
−1, 2917 cm
−1, 2849 cm
−1 and 1471 cm
−1 which were assigned to the bands of C–H stretching vibration absorption and bending vibration absorption. There was a band evident at 1740 cm
−1 (
Figure 4B) which is the C=O vibration absorption of the saturated ester from the branched group. Compared to the C=O vibration absorption of
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester at 1760 cm
−1 [
24], the bands of C=O of the copolymer B shifted towards the red area and the vibration absorption wave number became 1740 cm
−1, which contributed to the forming of hydrogen bonds among inter- and intra- of the copolymer chains [
25]. In addition, the bands at 3332 cm
−1 and 3050 cm
−1 corresponded to N–H vibration absorptions, indicating molecular association took place between inter- or intra- of the copolymer chains. The band at 1250 cm
−1 corresponded to C–O vibration absorption. Stretching vibration absorption and bending vibration absorption of C–O on the ester group were shown at 1096 cm
−1and 1022 cm
−1. The band at 1656 cm
−1 corresponded to C=O vibration absorption of the amide group. The band at 802 cm
−1 represented the vibration absorption of the substituted benzene. The results indicated that the comonomer
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester was incorporated into the copolymer chains.
Figure 4.
FT-IR spectra of ethylene homopolymer (A, run 9 in
Table 1) and ethylene/
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester copolymer (B, run 14 in
Table 2).
Figure 4.
FT-IR spectra of ethylene homopolymer (A, run 9 in
Table 1) and ethylene/
N-acetyl-
O-(dec-9-enyl)-
l-tyrosine ethyl ester copolymer (B, run 14 in
Table 2).
3. Experimental Section
3.1. General Remarks
Aniline (99.8%), N-acetyl-l-tyrosine ethyl ester (99%) and 10-bromo-1-decene (97%) were purchased from J & K in Beijing, China. 2,4,6-trimethylaniline (99%), dichlorodimethylsilane (99.8%), diphenyldichlorosilane (97%), 2,4,6-trifluoroaniline (97%), methylmagnesium chloride (CH3MgCl) of 22 wt. % in THF and methylaluminoxane (MAO) of 10 wt. % in toluene were purchased from Acros Organics Agent in Shanghai, China. Chlorodiisopropylphosphine (99%) was purchased from Strem Chemicals in Beijing, China. Toluene, n-hexane, acetone, and diethyl ether came from Beijing Chemicals Company in Beijing, China. Toluene and n-hexane were further purified by refluxing over sodium under normal pressure for 48 h prior to use. Acetone and diethyl ether were dried over activated Davison 5 Å molecular sieves prior to use.
3.2. Characterization
1H-NMR spectra were recorded on a Varian INOVA 600 MHz spectrometer (Karlsruhe, Germany) in deuterated chloroform (CDCl3) solution at 25 °C and tetramethylsilane (TMS) was used as reference. Elemental analyses were performed on a Perkin Elmer 2400 microanalyzer (Taunton, MA, USA), using a combustion method with quantitative oxygen with a thermal conductivity detector. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet 5DXC FT-IR spectrograph (Madison, WI, USA). The spectra were obtained at 40 cm−1 resolution, and average data were obtained from at least 32 scans in the standard wavenumber range from 500 to 4000 cm−1. Mass spectra were recorded by Esquire-LC mass spectroscopy (Bruker, Karlsruhe, Germany), acetone as dissolvent. 13C-NMR spectra were obtained on a Varian INOVA 500 MHz (125 MHz for 13C-NMR) instrument. The conditions used for quantitative 13C-NMR were a copolymer content up to 15 wt. % in solution, using ortho-dichlorobezene (d4) as the solvent at 125 °C, tetramethylsilane (TMS) as internal reference. The molecular weight and molecular weight distribution (MWD) were measured with a PL-GPC 200 instrument (Varian, Palo Alto, CA, USA), using standard polyethylene (PE) as reference and 1,2,4-trichlorobenzene as solvent at 150 °C. DSC thermograms were recorded with a PA5000-DSC instrument (Perkin-Elmer, Munich, Germany) at a rate of 10 K min−1.
3.3. Polymerization Procedure
All polymerization manipulations were carried out in flamed 300 mL Schlenk-type glassware on a dual-manifold Schlenk line. Freshly distilled toluene (80 mL), the desired amount of catalysts (Cat.1–Cat.6) and MAO were introduced into the glassware. The mixture was stirred for 15 min for preactivation. A desired amount of N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester dissolved in toluene was treated with an equivalent of AlEt3 as a protecting reagent for the desired time before it was injected into the Schlenk flask. The pressure of ethylene in the feed was 0.2 MPa and the reactor was rapidly heated to the desired temperature. The polymerization was maintained for 1 h. The reaction was terminated with 10 wt. % HCl in alcohol. The obtained product was filtered, washed, and then dried to constant weight in a vacuum oven at 80 °C. The product was weighed and the catalytic activity calculated.
3.4. Synthesis of N-acetyl-O-(dec-9-enyl)-l-tyrosine Ethyl Ester
N-acetyl-l-tyrosine ethyl ester (5 g, 18.6 mmol) in acetone (80 mL), K2CO3 (3.7 g, 26.8 mmol) and 10-bromo-1-decene (4.07 mL, 18.6 mmol) were added to Schlenk-type glassware in that order. The mixture was stirred at reflux for 2 days. Then the solvent was removed in vacuo, and the remaining viscous oil was dissolved in a mixture of aqueous 5% NaOH/Et2O (200 mL). The organic layer was collected, dried with MgSO4 and concentrated. The resultant viscous oil was washed with 200 mL of hexane, 50 mL at a time, and purified by vacuum. A white solid powder was obtained with a yield of 45.2%. 1H-NMR (600 MHz, CDCl3): δ 6.99 (d, 2H, benzene), δ 6.81 (d, 2H, benzene), δ 5.88 (m, 1H, CH=CH2), δ 5.82 (d, 1H, NH–CH), δ 4.94–4.98 (d, 2H, –CH2–CH=CH2), δ 4.81 (q, 1H, CH–NH), δ 4.17 (q, 2H, CH2–CH3), δ 3.93 (t, 3H, CH2–O), δ 3.06 (d, 2H, CH2=CH), δ 2.05 (m, 2H, CH2–CH), δ 1.99 (s, 3H, CH3–C),δ 1.43–1.34 (m, 10H, CH2–CH2), δ 1.27 (t, 3H, CH3–CH2); FT-IR (cm−1, KBr): 3323 (N–H), 1730 (C=O), 1647 (C–N), 1550 (N–H). Anal. calcd. (%) for C23H35NO4 (389.3 g/mol): C, 70.90; H, 8.99; N, 3.60; found: C, 70.91; H, 9.00; N, 3.58. ESI-MS m/z calculated for [M + H]+. C19H26NO4: 390.30; found 390.36.
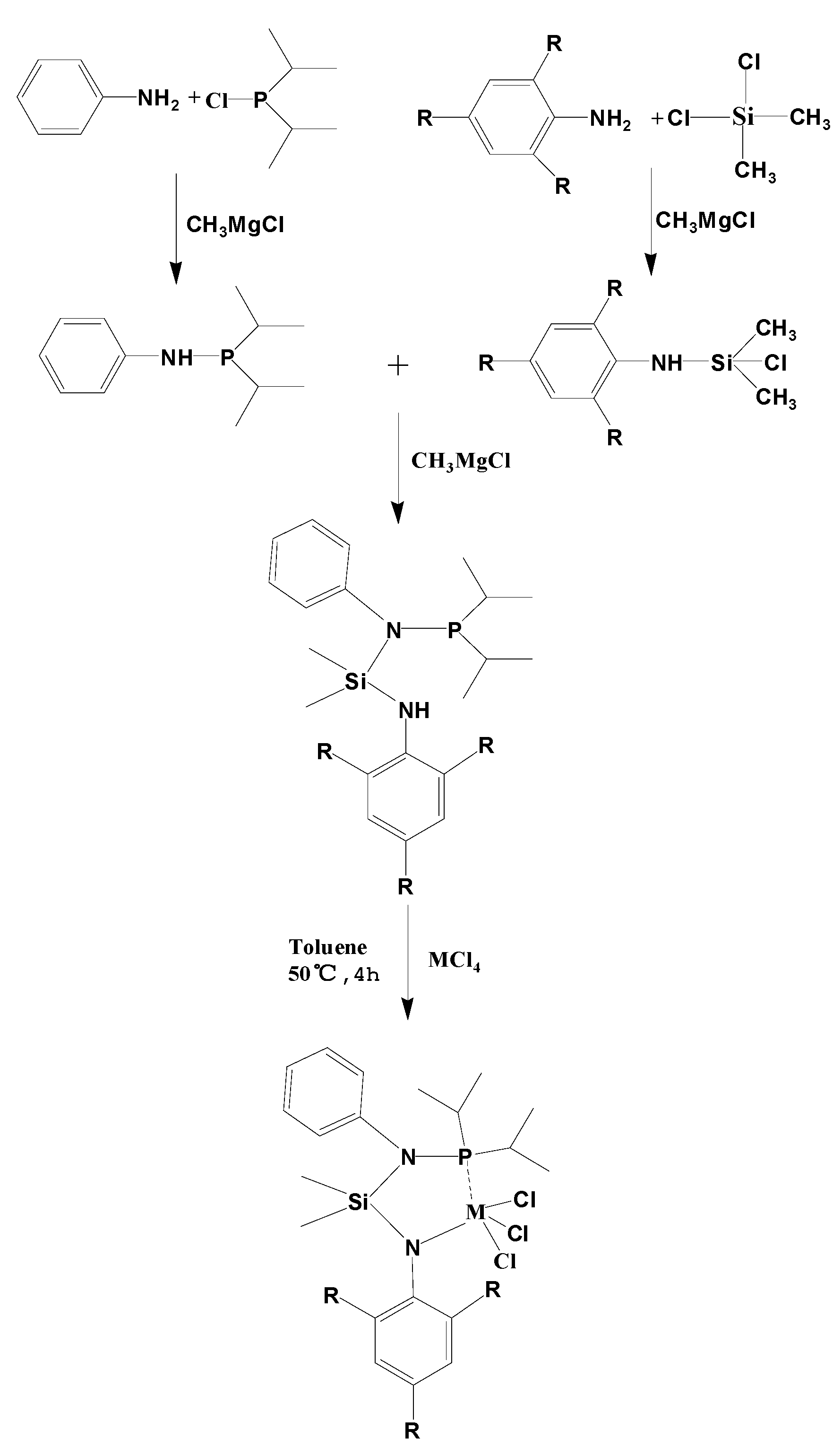
3.5. Synthesis of Catalyst Precursors
Aniline (1.5 mL, 16.2 mmol) was treated with an equiv. of CH
3MgCl for 3 h in toluene, then dichlorodimethylsilane (2 mL, 16.2 mmol) was injected. The mixture was stirred for 5 h at room temperature. Subsequently, the solvent was removed under vacuum and the residue was dissolved in hexane. The solution was concentrated. A colorless and clear liquid was obtained. Diisopropylphosphone-aniline (3.1 mL, 11.4 mmol) was treated with an equiv. of CH
3MgCl for 3 h in toluene, then (2.8 mL, 11.4 mmol) chlorodimethyisilane-aniline was added. The mixture was stirred for 5 h at room temperature. Subsequently, the solvent was removed
in vacuo and the residual was dissolved in hexane. The solution was concentrated. A colorless and clear liquid (Ligand
1 (L
1)) was obtained (1.03 g, 18.8%;
Scheme 2).
1H-NMR (400MHz, DMSO-
d6): δ 6.75–7.24 (m, 10H, benzene): δ 3.72 (s, 1H, –N
H–C), δ 1.78 (m, 2H, C
H–(CH
3)
2), δ 1.07 (d, 12H, CH–(C
H3)
2), δ 0.25 (s, 6H, Si–(C
H3)
2);
31P-NMR (400MHz, DMSO-
d6): δ 49.81. Element analysis calculated (%): C, 63.88; H, 5.87; N, 6.81; found: C, 63.89; H, 5.88; N, 6.82. FT-IR (cm
−1, KBr): 3276 (N–H). ESI-MS
m/
z calculated for [M + H]
+. C
20H
32N
2SiP: 359.02 found 359.05.
L
1 Ti complex (Cat.
1) was prepared by the treatment of L
1 (0.334 g, 1.0 mmol) with TiCl
4 (0.11 mL, 1.0 mmol) in toluene 50 mL at 0 °C. The reaction was held for 6 h at 40 °C. The mixture was filtered. After washing four times and then drying, a brown yellow powder (Cat.
1) was obtained (0.60 g, 84.8%;
Scheme 2) and further characterized. Cat.
1 was confirmed by
1H (
13C) NMR, ESI-MS
m/
z and microanalysis (
Table 1). The ligand
2 (L
2) derived from 2, 4, 6-trimethylaniline and the ligand
3 (L
3) from 2,4,6-trifluoroaniline.
Scheme 2.
Synthesis of the non-metallocene catalysts. L1: R = H; L2: R = CH3; L3: R = F; M = Ti and Zr.
Scheme 2.
Synthesis of the non-metallocene catalysts. L1: R = H; L2: R = CH3; L3: R = F; M = Ti and Zr.
L2 (C23H37N2SiP, Fw = 400.2): 1H-NMR (400 MHz, DMSO-d6): δ 6.42–7.10 (m, 5H, benzene), δ 6.38 (s, 2H, benzene), δ 4.26 (s, 1H, N–H), δ 2.35 (s, 9H, methyl on benzene), δ 1.79 (m, 2H, CH–(CH3)2), δ 1.06 (d, 12H, CH–(CH3)2), δ 0.14 (s, 6H, methyl). 31P-NMR (400 MHz, DMSO-d6): δ 49.45. Element analysis calculated (%): C, 64.98; H, 5.00; N, 5.83; found: C, 65.01; H, 4.98; N, 5.85. FT-IR (cm−1, KBr): 3287 (N–H). ESI-MS m/z calculated for [M + H]+. C23H38N2SiP: 401.20, found 401.25.
L
3 (C
20H
28N
2SiPF
3,
Fw = 412):
1H-NMR (400 MHz, DMSO-
d6): δ 6.45–7.03 (m, 5H, benzene), 6.21 (s, 2H, benzene), δ 4.1 (s, 1H, N
H–), δ 1.77 (m, 2H, C
H–(CH
3)
2), δ 1.02 (d, 12H, CH–(C
H3)
2), δ 0.14 (s, 6H, methyl).
31P-NMR (400 MHz, DMSO-
d6): δ 49.76. Element analysis calculated (%): C, 64.98; H, 5.00; N, 5.83; found: C, 65.01; H, 4.98; N, 5.85. FT-IR (cm
−1, KBr): 3279 (N–H). ESI-MS
m/
z calculated for [M + H]
+. C
20H
29N
2SiPF
3: 413.31 found 413.29. Cat.
2–
6 were synthesized according to the method mentioned above.
1H (
13C) NMR, ESI-MS
m/
z and microanalysis data of Cat.
1–
6 are compiled in
Table 1. From the
13C-NMR results in
Table 3, the complexes from the same ligand (L
1, L
2 or L
3) complexed with different transition metals (Ti and Zr) showed almost identical chemical shifts, which indicated that the transition metals coordinated with the heteroatoms, and not with the carbon atoms of these ligands. A lot of work was done to obtain single crystals, unfortunately, there was not one good enough for X-ray analysis.
Table 3.
The parameters for catalysts 1–6.
Table 3.
The parameters for catalysts 1–6.
| Catalyst | Met | Ligand | Microanalysis | 1H-NMR | ESI-MS m/z | 13C-NMR | 31P-NMR |
|---|
| 1 | Ti | L1 | C20H30N2SiPTiCl3 (405): calcd. C 53.89, H 4.49, N 4.84; found C 53.87, H 4.51, N 4.82 | DMSO: δ 6.46–7.24 (m, 10H, benzene), δ 1.78 (m, 2H, CH–(CH3)2), δ 1.07 (d, 12H, CH–(CH3)2), δ 0.25 (s, 6H, methyl) | calculated for [M + H]+. C20H31N2SiPTiCl3: 406.12 found 406.17 | DMSO: δ 0.30 (Si(CH3)2), δ 16.41 (C–(CH3)2), δ 21.2 (C–(CH3)2), δ 115.1 (Ar–C), δ 118.5 (Ar–C), δ 129.3(Ar–C), δ 146.7 (C–N) | DMSO: δ 49.79 |
| 2 | Zr | L1 | C20H30N2SiPZrCl3 (448): calcd. C 53.33, H 4.48, N 4.89; found C 53.34, H 4.45, N 4.90 | DMSO: δ 6.44–7.24 (m, 10H, benzene), δ 1.75 (m, 2H,CH–(CH3)2), δ 1.06 (d, 12H, CH–(CH3)2), δ 0.27 (s, 6H, methyl) | calculated for [M + H]+. C20H31N2SiPZrCl3: 449.31 found 449.35 | DMSO: δ 0.26 (Si(CH3)2), δ 16.40 (C–(CH3)2), δ 21.6 (C–(CH3)2), δ 115.3 (Ar–C), δ 118.2 (Ar–C), δ 129.1 (Ar–C), δ 146.6 (C–N) | DMSO: δ 49.77 |
| 3 | Ti | L2 | C23H36N2SiPTiCl3 (447): calcd. C 55.35, H 4.94, N 4.61; found C 55.39, H 4.91, N 4.63 | DMSO: δ 6.46–7.08 (m, 5H, benzene); δ 6.38 (s, 2H, benzene),δ 2.36 (s, 9H, methyl on benzene),δ 1.79 (m, 2H, CH–(CH3)2), δ 1.06 (d, 12H, CH–(CH3)2), δ 0.15 (s, 6H, methyl) | calculated for [M + H]+. C23H37N2SiPTiCl3: 448.23 found 448.28 | DMSO: δ 0.3 (Si(CH3)2), δ 0.4 (Si(CH3)2), δ 12.4 (C(CH3)), δ 16.41 (C–(CH3)2), δ 21.2 (C–(CH3)2), δ 115.1 (Ar–C), δ 118.6 (Ar–C), δ 124.1 (C(CH3)), δ 127.5 (C(CH3)), δ 127.8 (Ar–C), δ 129.4 (Ar–C), δ 145.2 (C–N), δ 146.5 (C–N) | DMSO: δ 49.47 |
| 4 | Zr | L2 | C23H36N2SiPZrCl3 (490): calcd. C 54.90, H 4.90, N 4.58; found C 54.87, H 4.89, N 4.61 | DMSO: δ 6.46–7.10 (m, 5H, benzene); δ 6.40 (s, 2H, benzene),δ 2.26(s, 9H, methyl on benzene),δ 1.77 (m, 2H, CH–(CH3)2), δ 1.08 (d, 12H, CH–(CH3)2), δ 0.16 (s, 6H, methyl) | calculated for [M + H]+. C23H37N2SiPZrCl3: 491.27 found 491.32 | DMSO: δ 0.3 (Si(CH3)2), δ 0.4 (Si(CH3)2), δ 12.3 (C(CH3)), δ 16.40 (C–(CH3)2), δ 21.2 (C–(CH3)2), δ 115.1 (Ar–C), δ 118.8 (Ar–C), δ 124.1 (C(CH3)), δ 127.5 (C(CH3)), δ 127.8 (Ar–C), δ 129.3 (Ar–C), δ 145.2 (C–N), δ 146.5 (C–N) | DMSO: δ 49.44 |
| 5 | Ti | L3 | C20H27N2SiPF3TiCl3 (459): calcd. C 49.21, H 3.63, N 4.42; found C 49.19, H 3.61, N 4.45 | DMSO: δ 6.41–7.02 (m, 5H, benzene); 6.21 (s, 2H, benzene); δ 1.77 (m, 2H, CH–(CH3)2), δ 1.04 (d, 12H, CH–(CH3)2), δ 0.14 (s, 6H, methyl) | calculated for [M + H]+. C20H28N2SiPTiCl3: 460.31 found 460.34 | DMSO: δ 0.3 (Si(CH3)2), δ 0.4 (Si(CH3)2), δ 16.43 (C–(CH3)2), δ 21.6 (C–(CH3)2), δ 98.8 (Ar–C), δ 115.1 (Ar–C), δ 116.3 (C–N),δ 118.5 (Ar–C), δ 129.3 (Ar–C), δ 146.7 (C–N), δ 151.9 (C–F), δ 155.3 (C–F) | DMSO: δ 49.71 |
| 6 | Zr | L3 | C20H27N2SiPF3ZrCl3 (502): calcd. C 48.83, H 3.60, N 4.38; found C 48.81,H 3.64, N 3.57 | DMSO: δ 6.42–7.06 (m, 5H, benzene); 6.24 (s, 2H, benzene); δ 1.76 (m, 2H, CH–(CH3)2), δ 1.06 (d, 12H, CH–(CH3)2), δ 0.15 (s, 6H, methyl) | calculated for [M + H]+. C20H28N2SiPZrCl3: 503.66 found 503.65 | DMSO: δ 0.3 (Si(CH3)2), δ 0.4 (Si(CH3)2), δ 16.42(C–(CH3)2), δ 21.4 (C–(CH3)2), δ 98.7 (Ar–C),δ 115.3 (Ar–C), δ 116.1 (C–N), δ 118.3 (Ar–C), δ 129.3 (Ar–C), δ 146.7 (C–N), δ 151.6 (C–F), δ 155.2 (C–F) | DMSO: δ 49.74 |


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
