On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights


Abstract
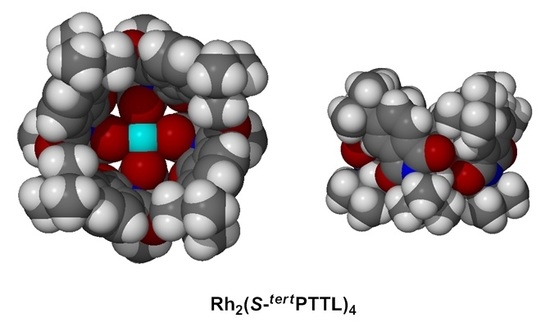
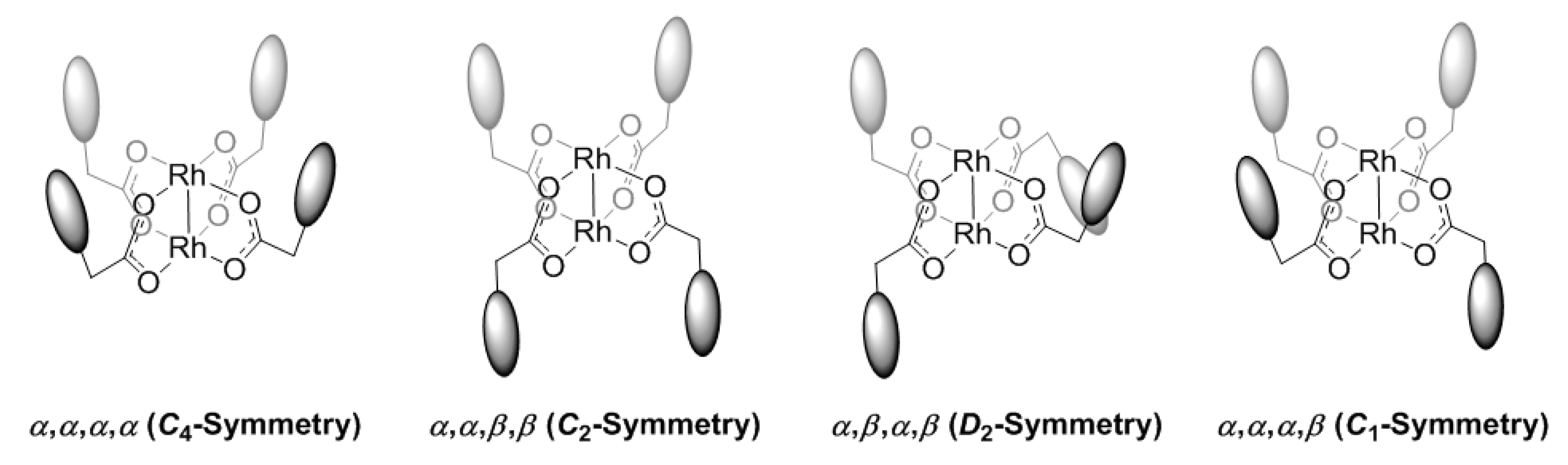
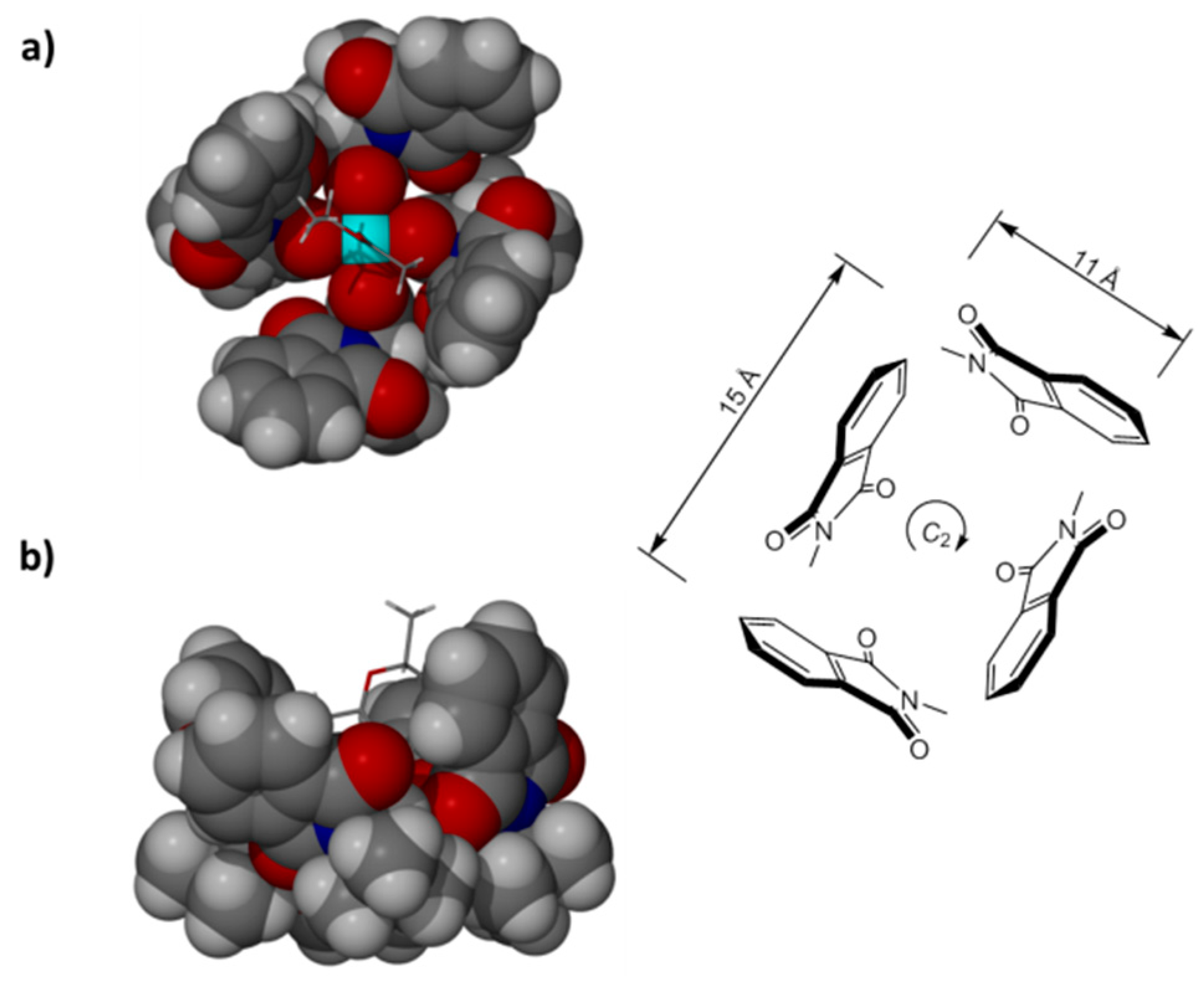
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
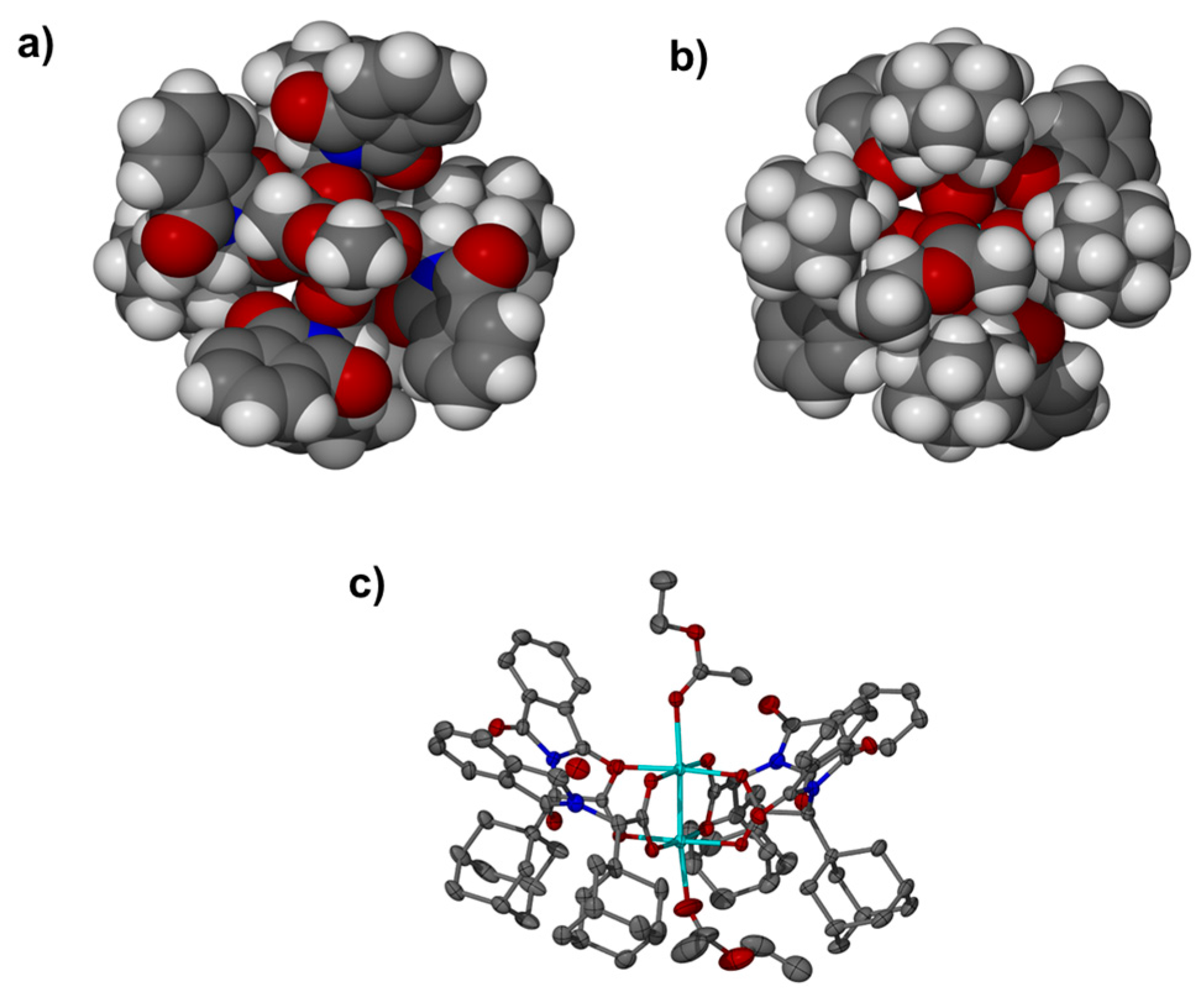{kind=link}
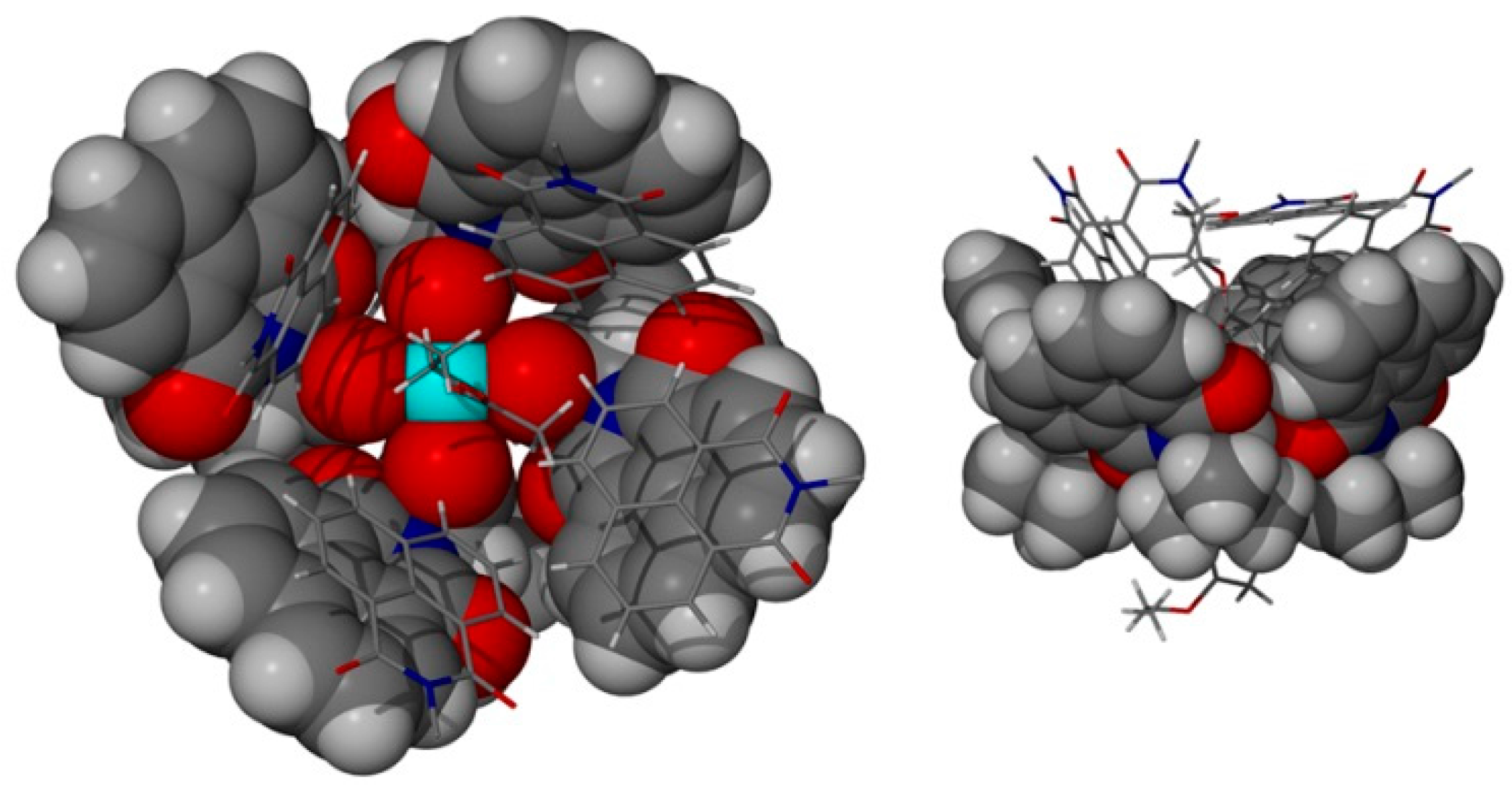{kind=link}
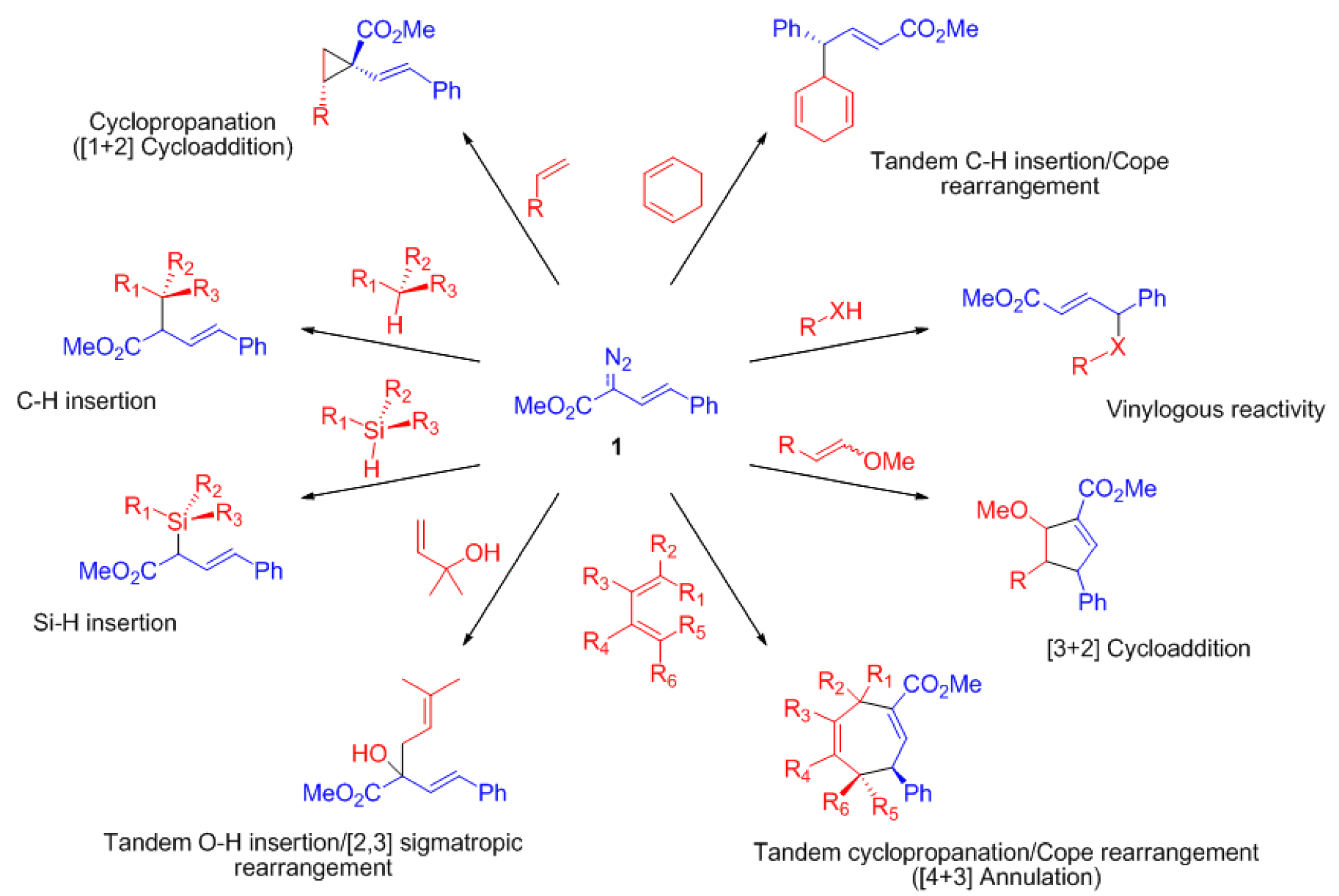{kind=link}
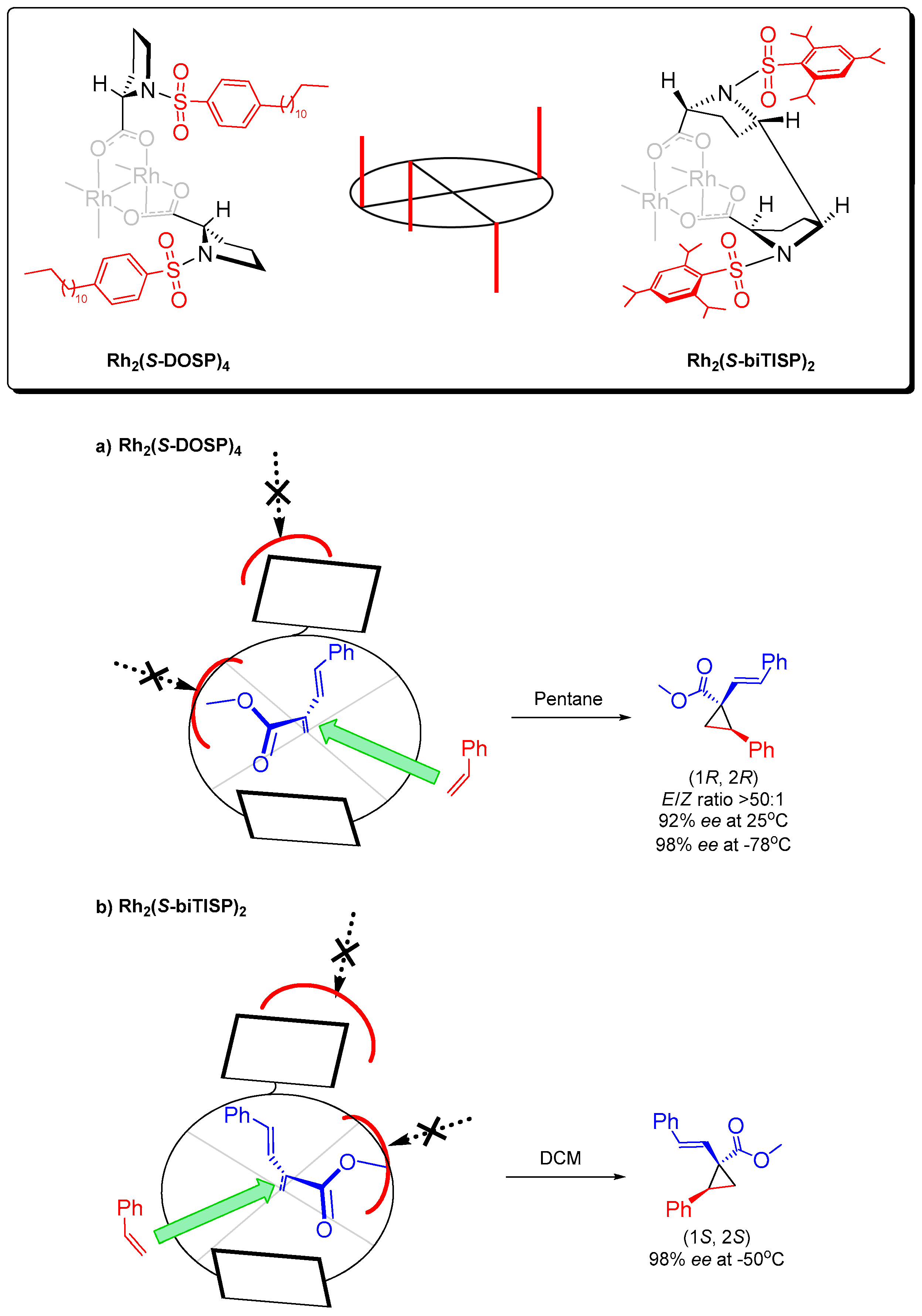{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ligand Blocking Groups Arrangements
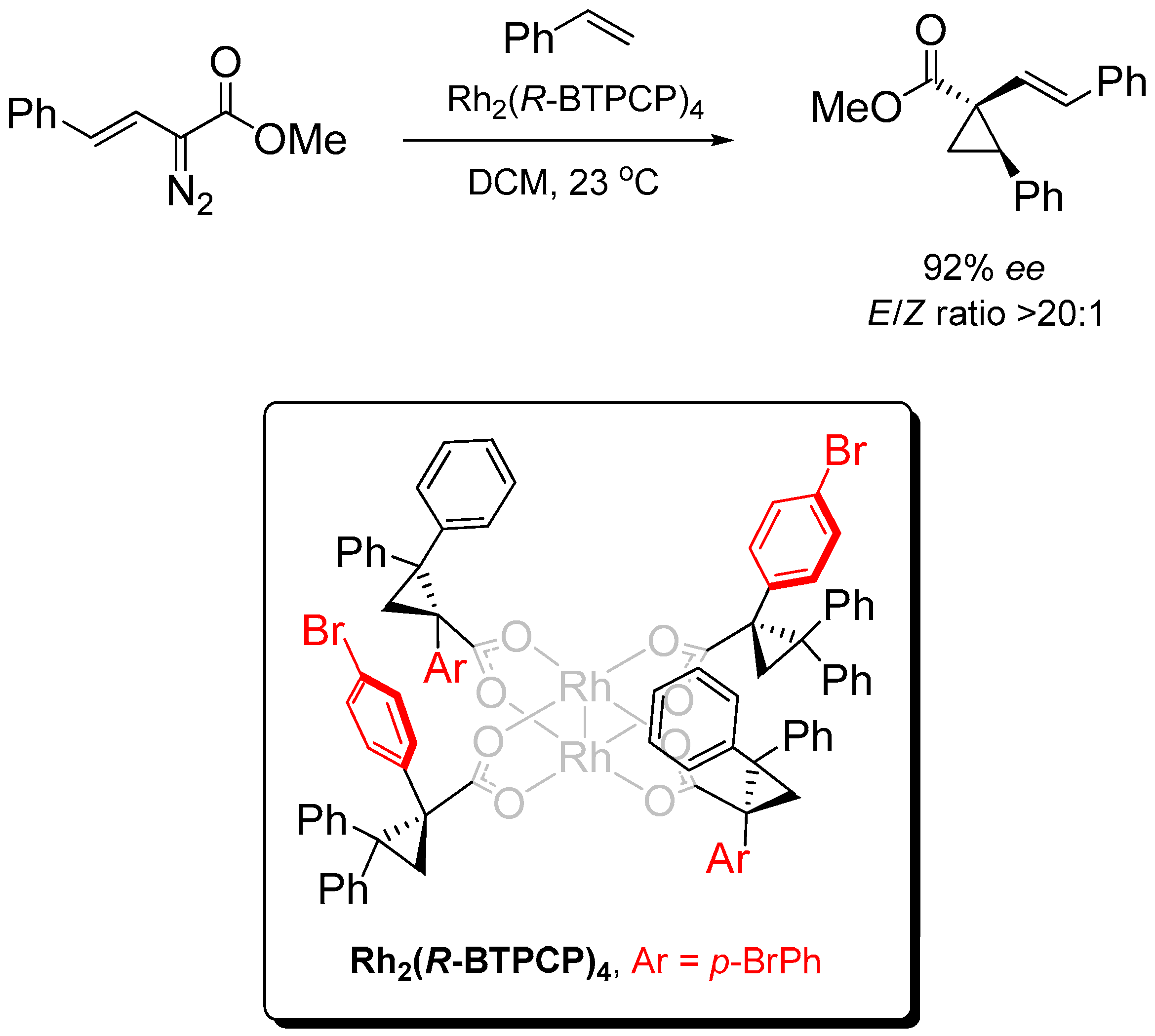
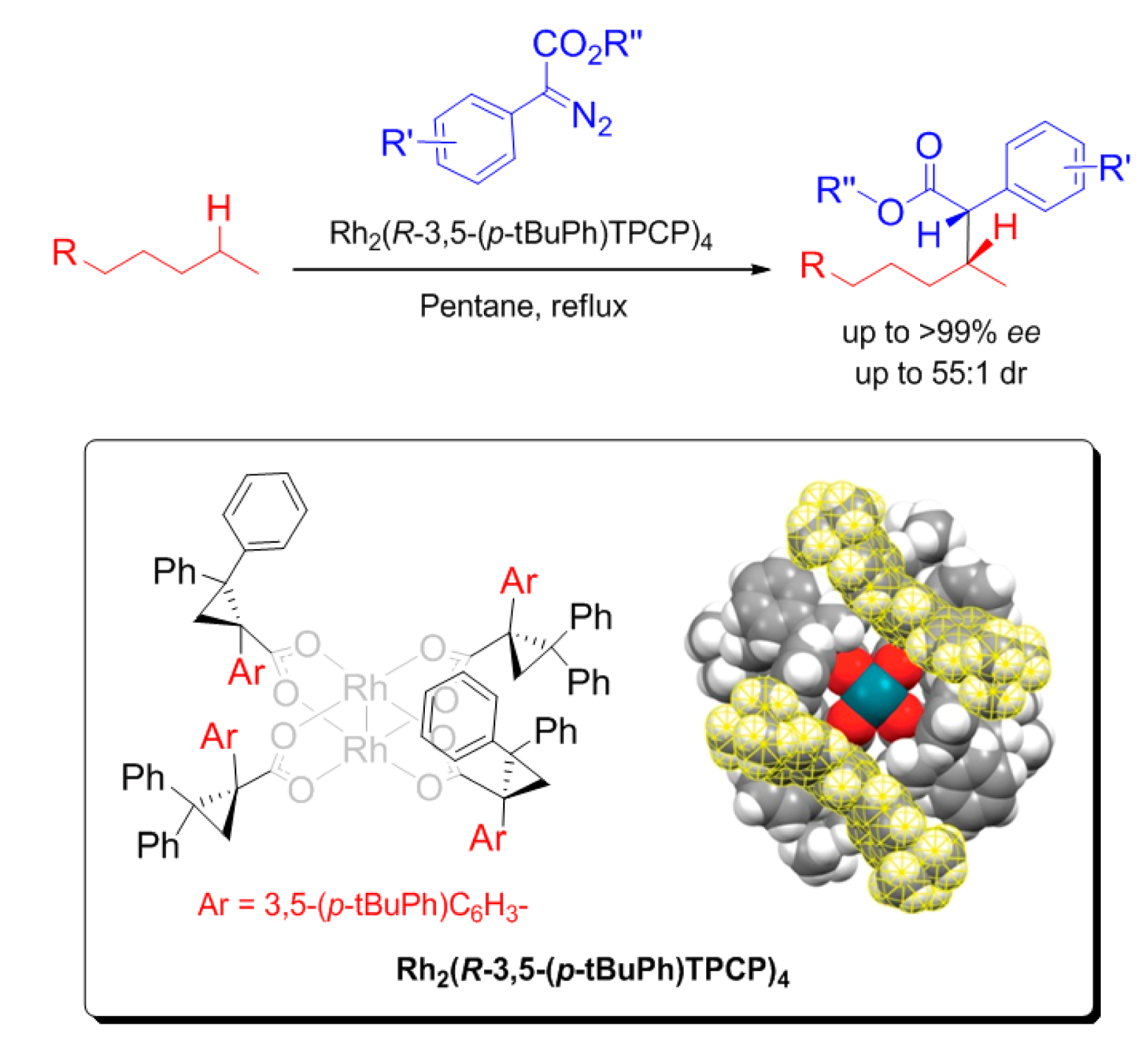
2.1. α,β,α,β-Conformation
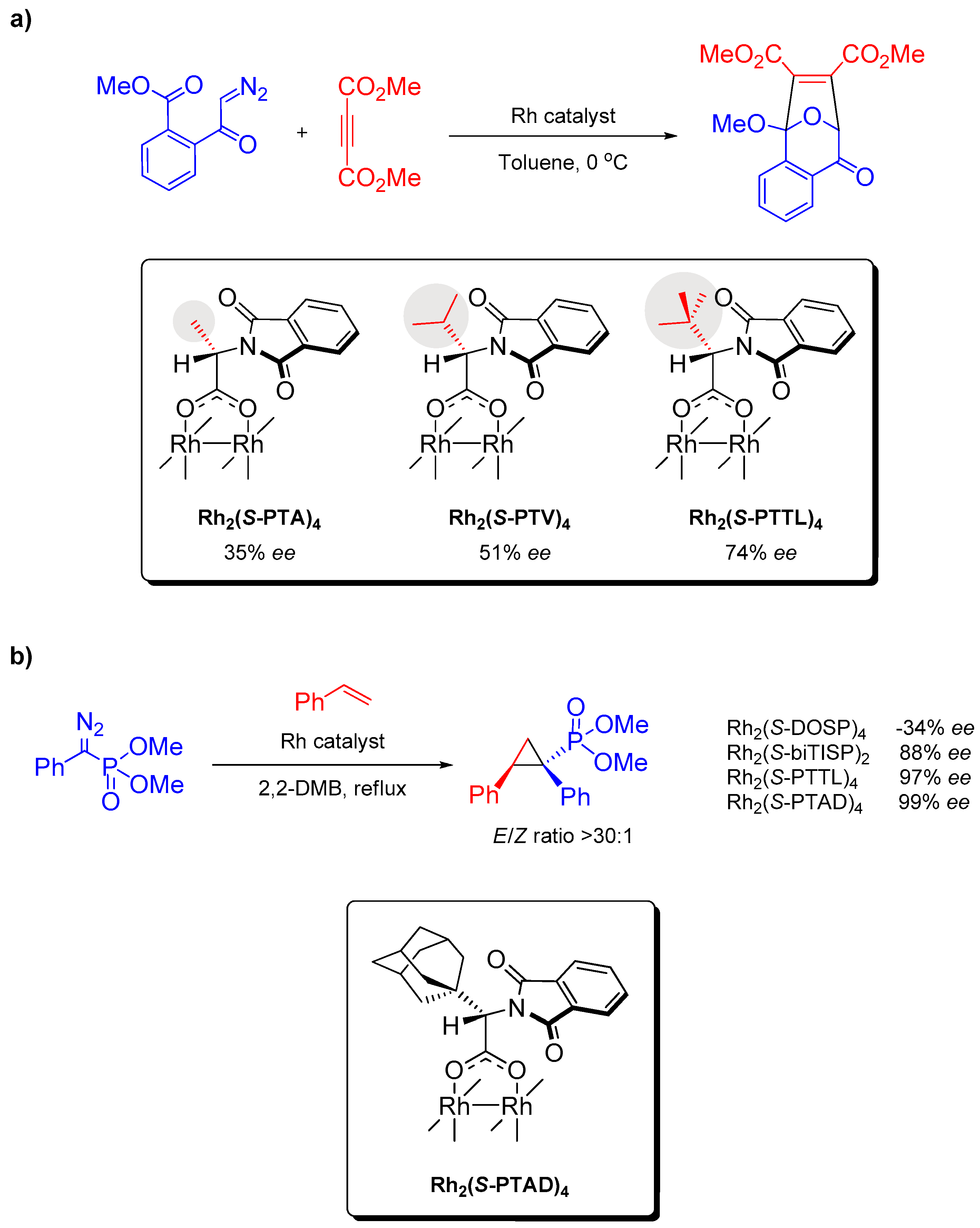
2.2. α,α,β,β- and α,α,α,α-Conformations
2.2.1. Global Catalyst Symmetry
2.2.2. Local Ligand Symmetry
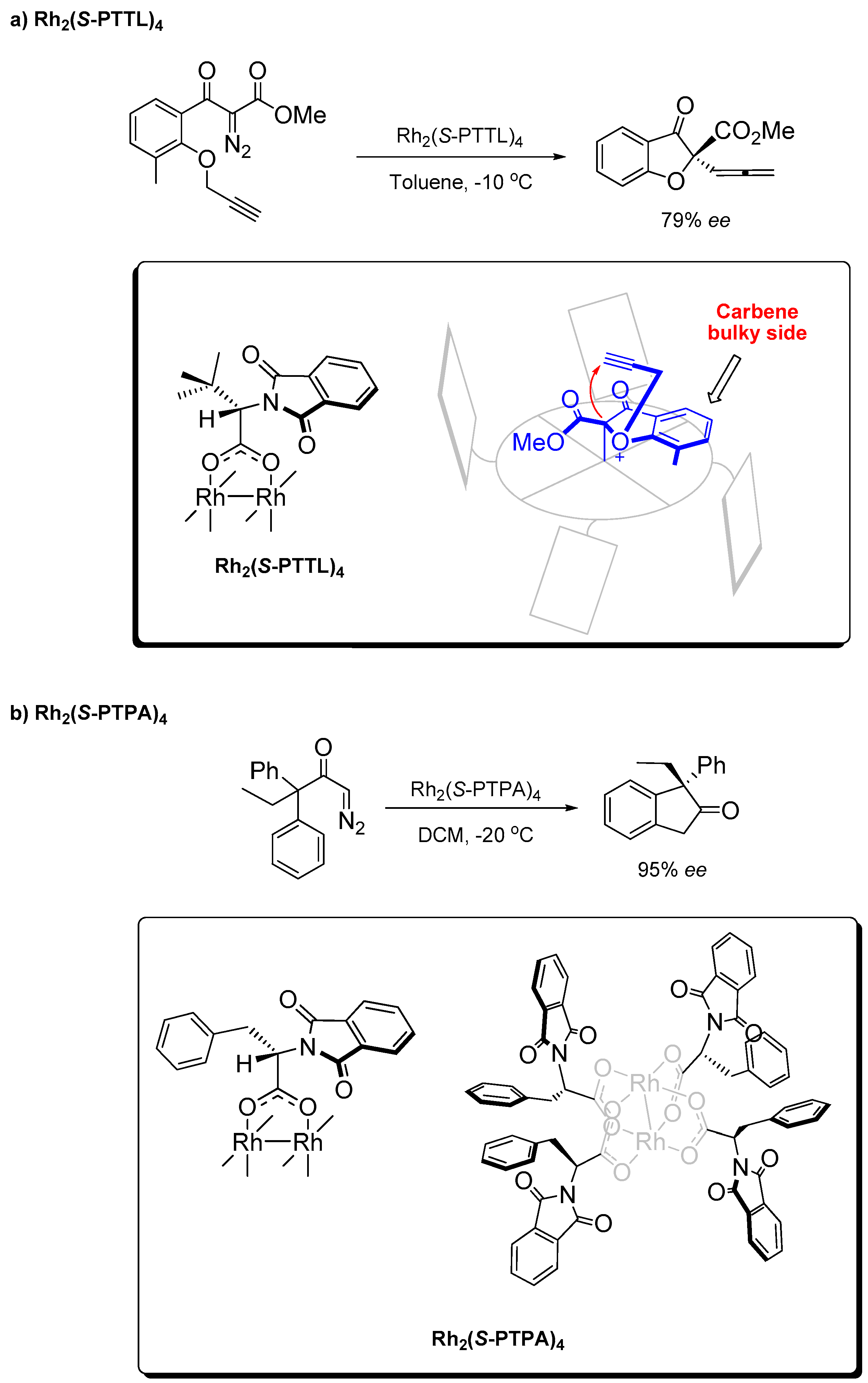
2.3. α,α,α,β-Conformation
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Ghanem, A. Enantiomerically Pure Compounds by Enantioselective Synthetic Chiral Metal Complexes. In Asymmetric Synthesis of Drugs and Natural Products; Nag, A., Ed.; CRC Press: Raton, FL, USA, 2018; in press. [Google Scholar]
- Deng, Y.; Qiu, H.; Srinivas, H.D.; Doyle, M.P. Chiral Dirhodium(II) Catalysts for Selective Metal Carbene Reactions. Curr. Org. Chem. 2016, 20, 61–81. [Google Scholar] [CrossRef]
- Adly, F.G.; Ghanem, A. Chiral Dirhodium (II) Carboxylates and Carboxamidates as Effective Chemzymes in Asymmetric Synthesis of Three-Membered Carbocycles. Chirality 2014, 26, 692–711. [Google Scholar] [CrossRef] [PubMed]
- El-Deftar, M.; Adly, F.G.; Gardiner, M.G.; Ghanem, A. Chiral Dirhodium Catalysts: A New Era for Asymmetric Catalysis. Curr. Org. Chem. 2012, 16, 1808–1836. [Google Scholar] [CrossRef]
- Trindade, A.F.; Coelho, J.A.S.; Afonso, C.A.M.; Veiros, L.F.; Gois, P.M.P. Fine Tuning of Dirhodium(II): Expolring the Axial Modification. ACS Catal. 2012, 2, 370–383. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bois, J.D.; Yu, J.-Q. C-H Functionalization in organic synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic Carbene Insertion into C-H Bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Hedley, S.J. Intermolecular reactions of electron-rich heterocycles with copper and rhodium carbenoids. Chem. Soc. Rev. 2007, 36, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Merlic, C.A.; Zechman, A.L. Selectivity in Rhodium(II) Catalyzed Reactions of Diazo Compounds: Effects of Catalyst ElectoPhilicity, Diazo Substitution and Substrate Substitution. From Chemoselectivity to Enantioselectivity. Synthesis 2003, 34, 1137–1156. [Google Scholar] [CrossRef]
- Colacot, T.J. An overview on the application of “Doyle catalysts” in asymmetric cyclopropanation, cyclopropenation and C-H insertion reactions. Proc. Ind. Acad. Sci. (J. Chem. Sci.) 2000, 11, 197–207. [Google Scholar] [CrossRef]
- Davies, H.M.L. Dirhodium tetra(N-arylsulfonylprolinates) as chiral catalysts for asymmetric transformations of vinyl and aryldiazoacetates. Eur. J. Org. Chem. 1999, 30, 2459–2469. [Google Scholar] [CrossRef]
- Davies, H.M.L. Rhodium-stabilized vinylcarbenoid intermediates in organic synthesis. Curr. Org. Chem. 1998, 2, 463–488. [Google Scholar]
- Collet, F.; Lescot, C.; Liang, C.; Dauban, P. Studies in catalytic C-H amination involving nitrene C-H insertion. Dalton Trans. 2010, 39, 10401–10413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Yan, M.; Huang, D. Catalyzed addition of diazoacetoacetates to imines: Synthesis of highly functionalized aziridines. Org. Biomol. Chem. 2009, 7, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, M.; Tanaka, M.; Abe, T.; Anada, M.; Hashimoto, S. Catalytic enantioselective aziridination of alkenes using chiral dirhodium(II) carboxylates. Heterocycles 2007, 72, 709–721. [Google Scholar] [CrossRef]
- Catino, A.J.; Nichols, J.M.; Forslund, R.E.; Doyle, M.P. Efficient Aziridination of Olefins Catalyzed by Mixed-Valent Dirhodium(II,III) Caprolactamate. Org. Lett. 2005, 7, 2787–2790. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Denton, J.R. Application of donor/acceptor-carbenoids to the synthesis of natural products. Chem. Soc. Rev. 2009, 38, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, R.W.; Mighion, J.D.; Wilkerson-Hill, S.M.; Alford, J.S.; Yoshidomi, T.; Davies, H.M. Enantioselective Intermolecular C-H Functionalization of Allylic and Benzylic sp3 C-H Bonds Using N-Sulfonyl-1, 2, 3-triazoles. Org. Lett. 2016, 18, 3118–3121. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P. Perspective on Dirhodium Carboxamidates as Catalysts. J. Org. Chem. 2006, 71, 9253–9260. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Hodgson, D.M.; Stupple, P.A.; Pierard, F.Y.T.M.; Labande, A.H.; Johnstone, C. Development of dirhodium(II)-catalyzed generation and enantioselective 1,3-dipolar cycloaddition of carbonyl ylides. Chem.-Eur. J. 2001, 7, 4465–4476. [Google Scholar] [CrossRef]
- Doyle, M.P.; Forbes, D.C.; Vasbinder, M.M.; Peterson, C.S. Enantiocontrol in the Generation and Diastereoselective Reactions of Catalytically Generated Oxonium and Iodonium Ylides. Metal-Stabilized Ylides as Reaction Intermediates. J. Am. Chem. Soc. 1998, 120, 7653–7654. [Google Scholar] [CrossRef]
- Doyle, M.P.; Hu, W. Macrocycle formation from catalytic metal carbene transformations. Synlett 2001, 2001, 1364–1370. [Google Scholar] [CrossRef]
- Doyle, M.P.; Phillips, I.M.; Hu, W. A New Class of Chiral Lewis Acid Catalysts for Highly Enantioselective Hetero-Diels-Alder Reactions: Exceptionally High Turnover Numbers from Dirhodium(II) Carboxamidates. J. Am. Chem. Soc. 2001, 123, 5366–5367. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Valenzuela, M.; Huang, P. Asymmetric hetero-Diels-Alder reaction catalyzed by dirhodium(II) carboxamidates. Proc. Natl. Acad. Sci. USA 2004, 101, 5391–5395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wolf, J.; Zavalij, P.; Doyle, M.P. Cationic chiral dirhodium carboxamidates are activated for lewis acid catalysis. Angew. Chem. Int. Ed. 2008, 47, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Shimada, N.; Anada, M.; Hashimoto, S. Enantio- and diastereoselective hetero-Diels-Alder reactions between 4-methyl-substituted Rawal’s diene and aldhydes catalyzed by chiral dirhodium(II) carboxamidates: Catalytic asymmetric synthesis of (−)-cis-aerrangis lactone. Tetrahedron Asymmetry 2014, 25, 63–73. [Google Scholar] [CrossRef]
- Anada, M.; Washio, T.; Shimada, N.; Kitagaki, S.; Nakajima, M.; Shiro, M.; Hashimoto, S. A new dirhodium(II) carboxamidate complex as a chiral Lewis acid catalyst for enantioselective heteo-Diels-Alder reactions. Angew. Chem. Int. Ed. 2004, 43, 2665–2668. [Google Scholar] [CrossRef]
- Hansen, J.H.; Parr, B.T.; Pelphrey, P.; Jin, Q.; Autschbach, J.; Davies, H.M.L. Rhodium(II)-Catalyzed Cross-Coupling of Diazo Compounds. Angew. Chem., Int. Ed. 2011, 50, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Protopopova, M.N. New aspects of catalytic asymmetric cyclopropanation. Tetrahedron 1998, 54, 7919–7946. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Antoulinakis, E.G. Intermolecular Metal-Catalyzed Carbenoid Cyclopropanations. In Organic Reactions; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2001; Volume 57, pp. 1–326. [Google Scholar]
- Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A.B. Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J. Modern Rhodium-Catalyzed Organic Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Davies, H.M.L.; Bruzinski, P.R.; Lake, D.H.; Kong, N.; Fall, M.J. Asymmetric Cyclopropanations by Rhodium(II) N-(Arylsulfonyl)prolinate-Catalyzed Decomposition of Vinyldiazomethanes in the presence of Alkenes. Practical Enantioselective Synthesis of the four Stereoisomers of 2-Phenylcyclopropan-1-amino Acid. J. Am. Chem. Soc. 1996, 118, 6897–6907. [Google Scholar] [CrossRef]
- Reddy, R.P.; Lee, G.H.; Davies, H.M.L. Dirhodium Tetracarboxylate Derived from Adamantylglycine as Chiral Catalyst for Carbenoid Reactions. Org. Lett. 2006, 8, 3437–3440. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Venkataramani, C. Dirhodium Tetraprolinate-Catalyzed Asymmetric Cyclopropanations with High Turnover Numbers. Org. Lett. 2003, 5, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Beckwith, E.J. Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion. Chem. Rev. 2003, 103, 2861–2903. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Hansen, T. Asymmetric Intermolecular Carbenoid C−H Insertions Catalyzed by Rhodium (II)(S)-N-(p-Dodecylphenyl) sulfonylprolinate. J. Am. Chem. Soc. 1997, 119, 9075–9076. [Google Scholar] [CrossRef]
- Davies, H.M.; Hansen, T.; Rutberg, J.; Bruzinski, P.R. Rhodium(II)(S)-N-(arylsulfonyl) prolinate catalyzed asymmetric insertions of vinyl-and phenylcarbenoids into the Si-H bond. Tetrahedron Lett. 1997, 38, 1741–1744. [Google Scholar] [CrossRef]
- Davies, H.M.; Stafford, D.G.; Doan, B.D.; Houser, J.H. Tandem asymmetric cyclopropanation/cope rearrangement. A highly diastereoselective and enantioselective method for the construction of 1,4-cycloheptadienes. J. Am. Chem. Soc. 1998, 120, 3326–3331. [Google Scholar] [CrossRef]
- Davies, H.M.L. [3 + 4] annulations between Rhodium-stabilized vinylcarbenoids and dienes. In Advances in Cycloaddition, Volume 5; Harmata, M., Ed.; JAI Press INC: Stamford, CT, USA, 1999. [Google Scholar]
- Li, Z.; Davies, H.M. Enantioselective C-C Bond Formation by Rhodium-Catalyzed Tandem Ylide Formation/[2,3]-Sigmatropic Rearrangement between Donor/Acceptor Carbenoids and Allylic Alcohols. J. Am. Chem. Soc. 2009, 132, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Wang, Y.; Hu, W. Regioselectivity in Lewis acids catalyzed X–H (O, S, N) insertions of methyl styryldiazoacetate with benzyl alcohol, benzyl thiol and aniline. Tetrahedron Lett. 2007, 48, 3975–3977. [Google Scholar] [CrossRef]
- Hansen, J.H.; Davies, H.M. Vinylogous reactivity of silver(I) vinylcarbenoids. Chem. Sci. 2011, 2, 457–461. [Google Scholar] [CrossRef]
- Hansen, J.H.; Gregg, T.M.; Ovalles, S.R.; Lian, Y.; Autschbach, J.; Davies, H.M. On the mechanism and selectivity of the combined C-H activation/Cope rearrangement. J. Am. Chem. Soc. 2011, 133, 5076–5085. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Jin, Q. Highly Diastereoselective and Enantioselective C-H Functionalization of 1, 2-Dihydronaphthalenes: A Combined C-H Activation/Cope Rearrangement Followed by a Retro-Cope Rearrangement. J. Am. Chem. Soc. 2004, 126, 10862–10863. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Hu, B. Ring expansion of tert-butyl 1-vinylcyclopropane-1-carboxylates to alpha-ethylidenebutyrolactones. J. Org. Chem. 1992, 57, 4309–4312. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Liu, H.; Morehead, A.T. Rhodium Chemzymes: Michaelis-Menten Kinetics in dirhodium(II) carboxylate-catalyzed carbenoid reactions. J. Am. Chem. Soc. 2002, 124, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Yoshikai, N.; Yamanaka, M. Mechanism of C-H Bond Activation/C-C Bond Formation Reaction between Diazo Compound and Alkane Catalyzed by Dirhodium Tetracarboxylate. J. Am. Chem. Soc. 2002, 124, 7181–7192. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Negretti, S.; Musaev, D.G.; Bacsa, J.; Davies, H.M.L. Site-selective and stereoselective functionalization of inactivated C-H bonds. Nature 2016, 533, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Davies, H.M.L. High Symmetry Dirhodium(II) Paddlewheel Complexes as Chiral Catalysts. Coord. Chem. Rev. 2008, 252, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Lin, W.; Charette, A.B. Experimental Evidence for the All-Up Reactive Conformation of Chiral Rhodium(II) Carboxylate Catalysts: Enantioselective Synthesis of cis-Cyclopropane α-Amino Acids. J. Am. Chem. Soc. 2009, 131, 16383–16385. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bruzinski, P.R.; Fall, M.J. Effect of diazoalkane structure on the stereoselectivity of rhodium(II) (S)-N-(arylsulfonyl)Prolinate catalyzed cyclopropanations. Tetrahedron Lett. 1996, 37, 4133–4136. [Google Scholar] [CrossRef]
- Deng, L.; Giessert, A.J.; Gerlitz, O.O.; Dai, X.; Diver, S.T.; Davies, H.M. Metal carbene-promoted sequential transformations for the enantioselective synthesis of highly functionalized cycloheptadienes. J. Am. Chem. Soc. 2005, 127, 1342–1343. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Davies, H.M. Rhodium-catalyzed [3 + 2] annulation of indoles. J. Am. Chem. Soc. 2009, 132, 440–441. [Google Scholar] [CrossRef] [PubMed]
- Briones, J.F.; Hansen, J.; Hardcastle, K.I.; Autschbach, J.; Davies, H.M.L. Highly enantioselective Rh2(S-DOSP)4-catalyzed cyclopropenation of alkynes with styryldiazoacetates. J. Am. Chem. Soc. 2010, 132, 17211–17215. [Google Scholar] [CrossRef] [PubMed]
- Parr, B.T.; Green, S.A.; Davies, H.M. Rhodium-catalyzed conversion of furans to highly functionalized pyrroles. J. Am. Chem. Soc. 2013, 135, 4716–4718. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Hansen, T.; Churchill, M.R. Catalytic asymmetric C-H activation of alkanes and tetrahydrofuran. J. Am. Chem. Soc. 2000, 122, 3063–3070. [Google Scholar] [CrossRef]
- Davies, H.M.; Ren, P. Catalytic Asymmetric C-H Activation of Silyl Enol Ethers as an Equivalent of an Asymmetric Michael Reaction. J. Am. Chem. Soc. 2001, 123, 2070–2071. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Venkataramani, C. Catalytic Enantioselective Synthesis of β2-Amino Acids. Angew. Chem. Int. Ed. 2002, 41, 2197–2199. [Google Scholar] [CrossRef]
- Davies, H.M.; Walji, A.M. Direct Synthesis of (+)-Erogorgiaene through a Kinetic Enantiodifferentiating Step. Angew. Chem. Int. Ed. 2005, 44, 1733–1735. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Manning, J.R. C-H Activation as a Strategic Reaction: Enantioselective Synthesis of 4-Substituted Indoles. J. Am. Chem. Soc. 2006, 128, 1060–1061. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Dai, X.; Long, M.S. Combined C-H Activation/Cope Rearrangement as a Strategic Reaction in Organic Synthesis: Total Synthesis of (−)-Colombiasin A and (−)-Elisapterosin B. J. Am. Chem. Soc. 2006, 128, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Lee, G.H. Dirhodium(II) Tetra(N-(dodecylbenzenesulfonyl)prolinate) Catalyzed Enantioselective Cyclopropenation of Alkynes. Org. Lett. 2004, 6, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Davies, H.M. Combined C-H functionalization/Cope rearrangement with vinyl ethers as a surrogate for the vinylogous Mukaiyama aldol reaction. J. Am. Chem. Soc. 2011, 133, 11940–11943. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Townsend, R.J. Catalytic asymmetric cyclopropanation of hetereoaryldiazoacetates. J. Org. Chem. 2001, 66, 6595–6603. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Hutcheson, D.K. Enantioselective synthesis of vinylcyclopropanes by rhodium(II)-catalyzed decomposition of vinyldiazomethanes in the presence of alkenes. Tetrahedron Lett. 1993, 34, 7243–7246. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Nagashima, T.; Klino, J.L. Stereoselectivity of Methyl Aryldiazoacetate cyclopropanations of 1,1-diarylethylene. Asymmetric synthesis of cyclopropyl analouge of Tamoxifen. Org. Lett. 2000, 2, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Panaro, S.A. Novel dirhodium tetraprolinate catalysts containing bridging prolinate ligands for asymmetric carbenoid reactions. Tetrahedron Lett. 1999, 40, 5287–5290. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Kong, N. Synthesis and evaluation of a novel dirhodium tetraprolinate catalyst containing bridging prolinate ligands. Tetrahedron Lett. 1997, 38, 4203–4206. [Google Scholar] [CrossRef]
- Qin, C.; Boyarskikh, V.; Hansen, J.H.; Hardcastle, K.I.; Musaev, D.G.; Davies, H.M.L. D2-Symmetric Dirhodium Catalyst Derived from a 1,2,2-triarylcyclopropanecarboxylate ligand: Design, Synthesis and Application. J. Am. Chem. Soc. 2011, 133, 19198–19204. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Davies, H.M.L. Enantioselective synthesis of 2-arylbicyclo[1.1.0]butane carboxylates. Org. Lett. 2013, 15, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Davies, H.M.L. Rh2(R-TPCP)4-catalyzed Enantioselective [3 + 2]-cycloaddition between nitrones and vinyldiazoacetates. J. Am. Chem. Soc. 2013, 135, 14516–14519. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Watanabe, N.; Sato, T.; Shiro, M.; Ikegami, S. Enhancement of Enantioselectivity in Intramolecular C-H Insertion Reactions of α-Diazo-β-Keto Esters Catalyzed by Chiral Dirhodium(II) Carboxylates. Tetrahedron Lett. 1993, 34, 5109–5112. [Google Scholar] [CrossRef]
- Tsutsui, H.; Abe, T.; Nakamura, S.; Anada, M.; Hashimoto, S. Practical Synthesis of Dirhodium(II) Tetrakis[N-phthaloyl-(S)-tert-leucinate]. Chem. Pharm. Bull. 2005, 53, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Chiral Crown Conformation of Rh2(PTTL)4: Enantioselective Cyclopropanation with α-Alkyl-α-diazoesters. J. Am. Chem. Soc. 2009, 131, 7230–7231. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Saito, H.; Tsutsui, H.; Nambu, H.; Anada, M.; Hashimoto, S. Highly Enantio- and Diastereoselective Construction of 1,2-Disubstituted Cyclopentane Compounds by Dirhodium(II) Tetrakis[N-phthaloyl-(S)-tert-leucinate]-Catalyzed C-H Insertion Reactions of α-Diazo Esters. Adv. Synth. Catal. 2005, 347, 1483–1487. [Google Scholar] [CrossRef]
- Awata, A.; Arai, T. Catalytic Asymmetric Cyclopropanation with Diazooxindole. Synlett 2013, 24, 29–32. [Google Scholar]
- Lindsay, V.N.G.; Nicolas, C.; Charette, A.B. Asymmetric Rh(II)-Catalyzed Cyclopropanation of Alkenes with Diacceptor Diazo Compounds: p-Methoxyphenyl Ketone as a General Stereoselectivity Controlling Group. J. Am. Chem. Soc. 2011, 133, 8972–8981. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Takada, K.; Anada, M.; Ando, K.; Hashimoto, S. Enantio- and diastereoselective cyclopropanation with tert-butyl α-diazopropionate catalyzed by dirhodium(II) tetrakis[N-tetrabromophthaloyl-(S)-tert-Leucinate]. Tetrahedron Lett. 2011, 52, 4200–4203. [Google Scholar] [CrossRef]
- Goto, T.; Takada, K.; Shimada, N.; Nambu, H.; Anada, M.; Shiro, M.; Ando, K.; Hashimoto, S. Highly enantioselective cyclopropenation reaction reaction of 1-Alkynes with α-alkyl-α-diazoesters catalyzed by rhodium(II) carboxylates. Angew. Chem. Int. Ed. 2011, 50, 6803–6808. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Deng, Y.; Yim, D.N.; Zavalij, P.Y.; Doyle, M.P. Enantioselective cis-β-lactam synthesis by intramolecular C-H functionalization from enoldiazoacetamides and derivative donor–acceptor cyclopropenes. Chem. Sci. 2015, 6, 2196–2201. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Matsuura, M.; Makino, K.; Nakamura, S.; Nakajima, M.; Kitagaki, S.; Hashimoto, S. Enantioselective Tandem Formation and [2,3]-Sigmatropic Rearrangement of Cyclic Propargylic Oxonium Ylides Catalyzed by Dirhodium(II) Tetrakis[Naphthaloyl-(S)-tert-leucinate]. Isr. J. Chem. 2001, 41, 283–295. [Google Scholar] [CrossRef]
- Watanabe, N.; Ohtake, Y.; Hashimoto, S.; Shiro, M.; Ikegami, S. Asymmetric creation of quaternary carbon centers by enantiotopically selective aromatic C-H insertion catalyzed by chiral dirhodium(II) carboxylates. Tetrahedron Lett. 1995, 36, 1491–1494. [Google Scholar] [CrossRef]
- DeAngelis, A.; Boruta, D.T.; Lubin, J.-B.; Plampin, J.N.; Yap, G.P.A.; Fox, J.M. The chiral crown conformation in paddlewheel complexes. Chem. Commun. 2010, 46, 4541–4543. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, E.; Ventura, D.L.; Brekan, J.A.; Davies, H.M.L. Controlling factors for C-H functionalization versus cyclopropanation of dihydronaphthalenes. J. Org. Chem. 2010, 75, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Maddalena, J.; Ghanem, A. Rh2(S-1,2-NTTL)4: A Novel Rh2(S-PTTL)4 Analog With Lower Ligand Symmetry for Asymmetric Synthesis of Chiral Cyclopropylphosphonates. Chirality 2014, 26, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S. Enantiocontrol in Tandem Carbonyl Ylide Formation and Intermolecular 1,3-Dipolar Cycloaddition of α-Diazo Ketones Mediated by Chiral Dirhodium(II) Carboxylate Catalyst. J. Am. Chem. Soc. 1999, 121, 1417–1418. [Google Scholar] [CrossRef]
- Denton, J.R.; Sukumaran, D.; Davies, H.M.L. EnantioSelective Synthesis of trifluoromethyl-Substituted Cyclopropanes. Org. Lett. 2007, 9, 2625–2628. [Google Scholar] [CrossRef] [PubMed]
- Denton, J.R.; Cheng, K.; Davies, H.M.L. Stereoselective construction of nitrile-substituted cyclopropanes. Chem. Commun. 2008, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Denton, J.R.; Davies, H.M.L. Enantioselective reactions of donor/acceptor carbenoids derived from α-aryl-α-diazoketones. Org. Lett. 2009, 11, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Chepiga, K.M.; Qin, C.; Alford, J.S.; Chennamadhavuni, S.; Gregg, T.M.; Olson, J.P.; Davies, H.M.L. Guide to enantioselective dirhodium(II)-catalyzed cyclopropanation with aryldiazoacetates. Tetrahedron 2013, 69, 5765–5771. [Google Scholar] [CrossRef] [PubMed]
- Briones, J.F.; Davies, H.M.L. Rh2(S-PTAD)4 catalyzed asymmetric cyclopropanation of aryl alkynes. Tetrahedron 2011, 67, 4313–4317. [Google Scholar] [CrossRef]
- Wang, H.; Guptill, D.M.; Varela-Alvarez, A.; Musaev, D.G.; Davies, H.M.L. Rhodium-catalyzed enantioselective cyclopropanation of electron deficient alkenes. Chem. Sci. 2013, 4, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
- Werlé, C.; Goddard, R.; Philipps, P.; Farès, C.; Fürstner, A. Stabilization of a Chiral Dirhodium Carbene by Encapsulation and a Discussion of the Stereochemical Implications. Angew. Chem. Int. Ed. 2016, 55, 10760–10765. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Gardiner, M.G.; Ghanem, A. Design and Synthesis of Novel Chiral Dirhodium(II) Carboxylate Complexes for Asymmetric Cyclopropanation Reactions. Chem.-Eur. J. 2016, 22, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Mattiza, J.T.; Fohrer, J.G.G.; Duddeck, H.; Gardiner, M.G.; Ghanem, A. Optimizing dirhodium(II) tetrakiscarboxylates as chiral NMR auxiliaries. Org. Biomol. Chem. 2011, 9, 6542–6550. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M.G.; Ghanem, A. X-ray crystal structures of bis(EtOAc) and (MeOH)(H2O) adducts of Rh2(S-PTPA)4 complex. Unpublished work. 2010. [Google Scholar]
- Müller, P.; Ghanem, A. Rh(II)-Catalyzed Enantioselective Cyclopropanation of Olefins with Dimethyl Malonate via in Situ Generated Phenyliodonium Ylide. Org. Lett. 2004, 6, 4347–4350. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Gardiner, M.G.; Williamson, R.M.; Müller, P. First X-ray Structure of a N-Naphthaloyl-Tethered Chiral Dirhodium(II) Complex: Structural Basis for Tether Substitution Improving Asymmetric Control in Olefin Cyclopropanation. Chem.-Eur. J. 2010, 16, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Ghanem, A. Polymer monolith-supported dirhodium(II)-catalyzed continuous flow cyclopropanation in capillary format. Tetrahedron Lett. 2016, 57, 852–857. [Google Scholar] [CrossRef]
- Lindsay, V.N.G.; Charette, A.B. Design and Synthesis of Chiral Heteroleptic Rhodium(II) Carboxylate Catalysts: Experimental Investigations of Halogen Bond Rigidification Effects in Asymmetric Cyclopropanation. ACS Catal. 2012, 2, 1221–1225. [Google Scholar] [CrossRef]
- Boruta, D.T.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Rh2(S-PTTL)3TPA—A mixed-ligand dirhodium(II) catalyst for enantioselective reactions of α-alkyl-α-diazoesters. Chem. Sci. 2012, 3, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
















© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adly, F.G. On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights. Catalysts 2017, 7, 347. https://doi.org/10.3390/catal7110347
Adly FG. On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights. Catalysts. 2017; 7(11):347. https://doi.org/10.3390/catal7110347
Chicago/Turabian StyleAdly, Frady G. 2017. "On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights" Catalysts 7, no. 11: 347. https://doi.org/10.3390/catal7110347
APA StyleAdly, F. G. (2017). On the Structure of Chiral Dirhodium(II) Carboxylate Catalysts: Stereoselectivity Relevance and Insights. Catalysts, 7(11), 347. https://doi.org/10.3390/catal7110347