Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems

Abstract
:1. Introduction
2. Useful Notations
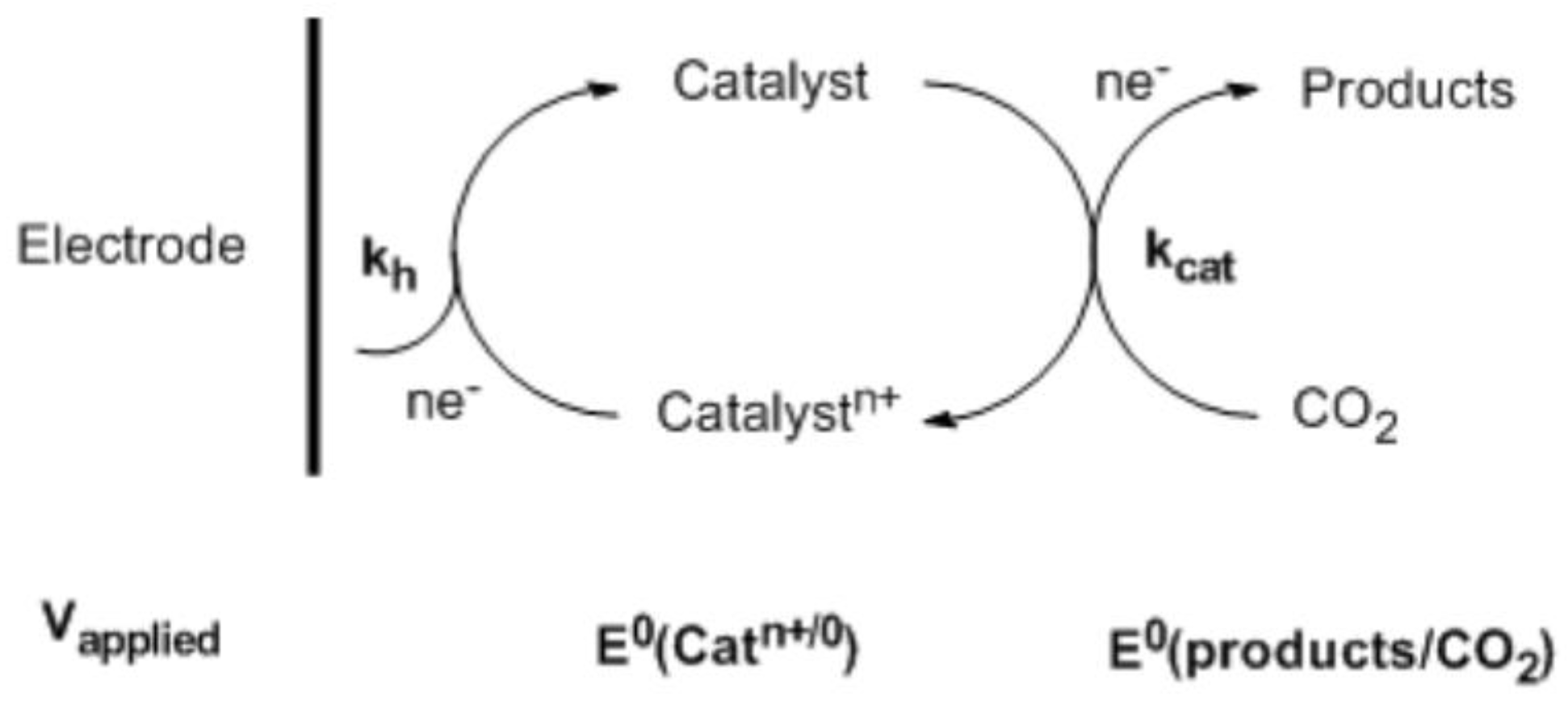
3. Homogeneous CO2 Reduction
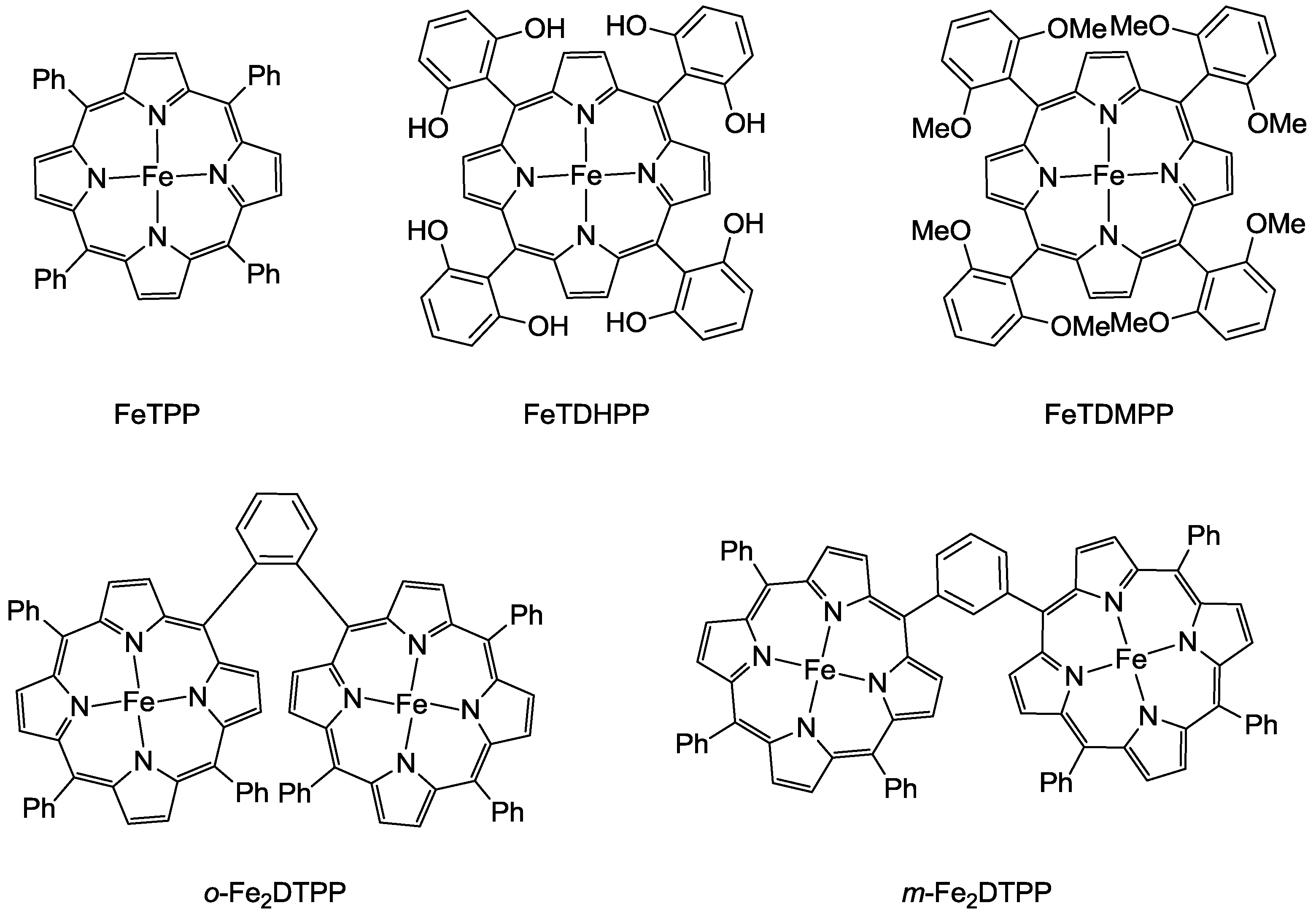
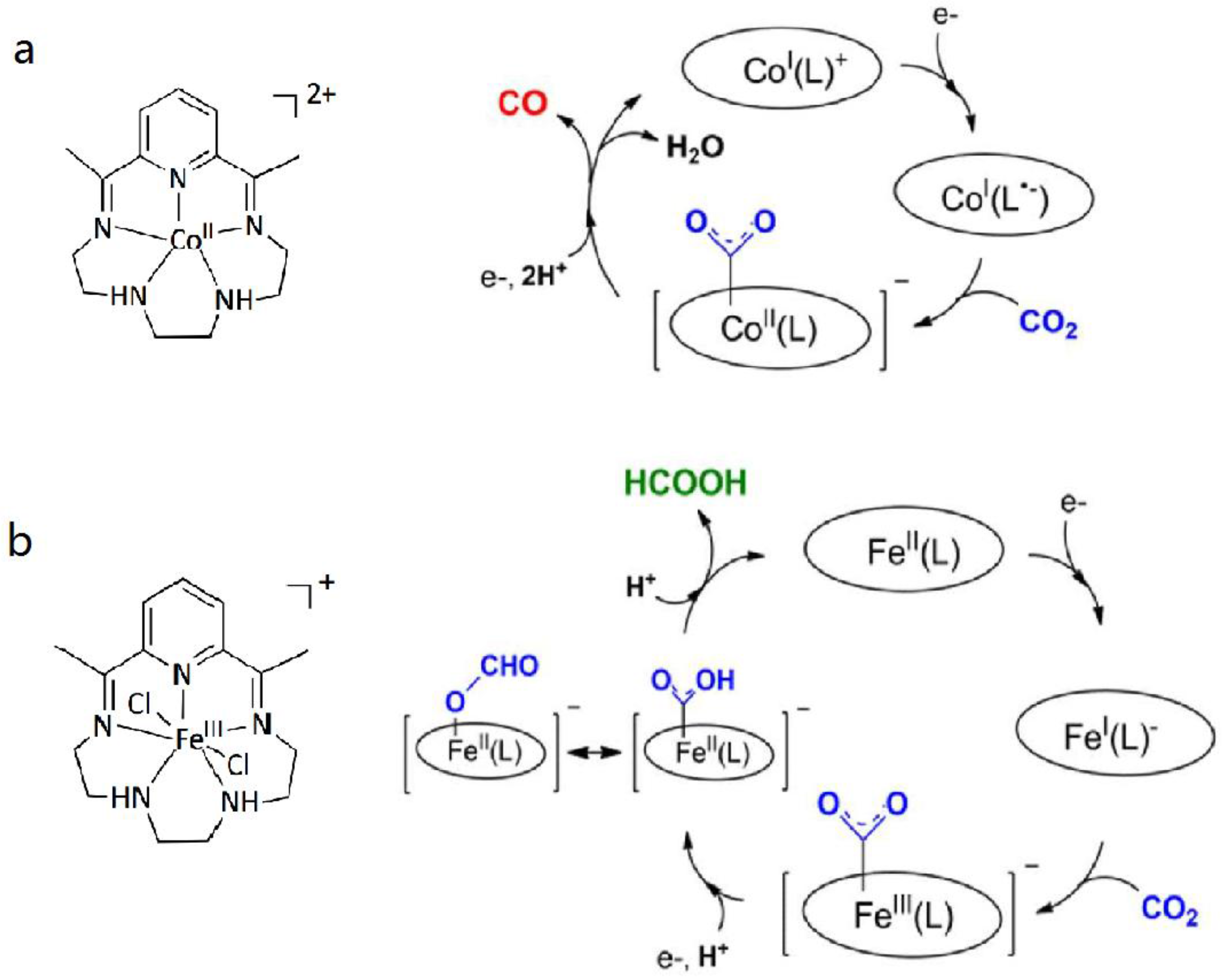
3.1. Metal Complexes with Macrocyclic Ligands
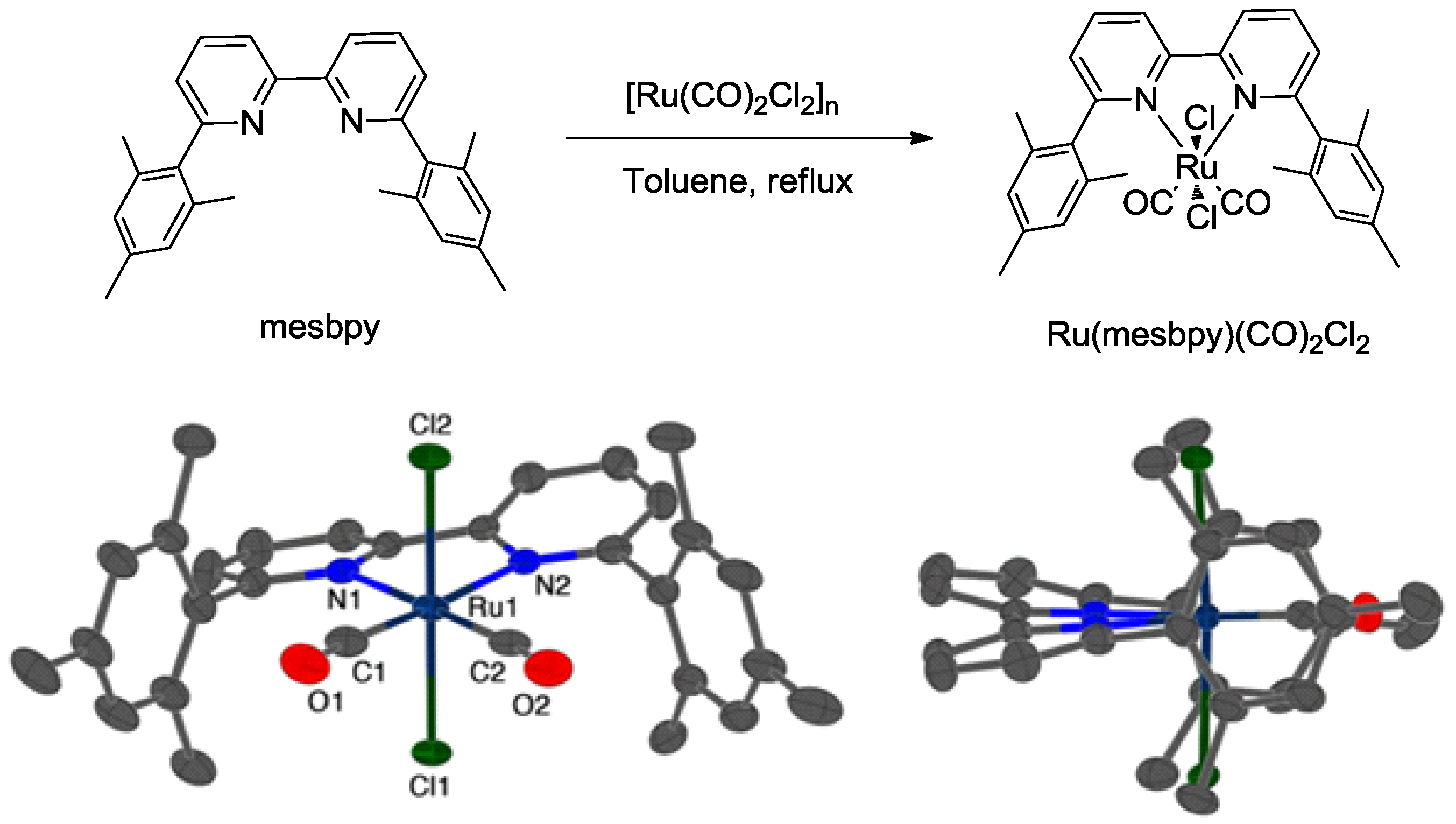
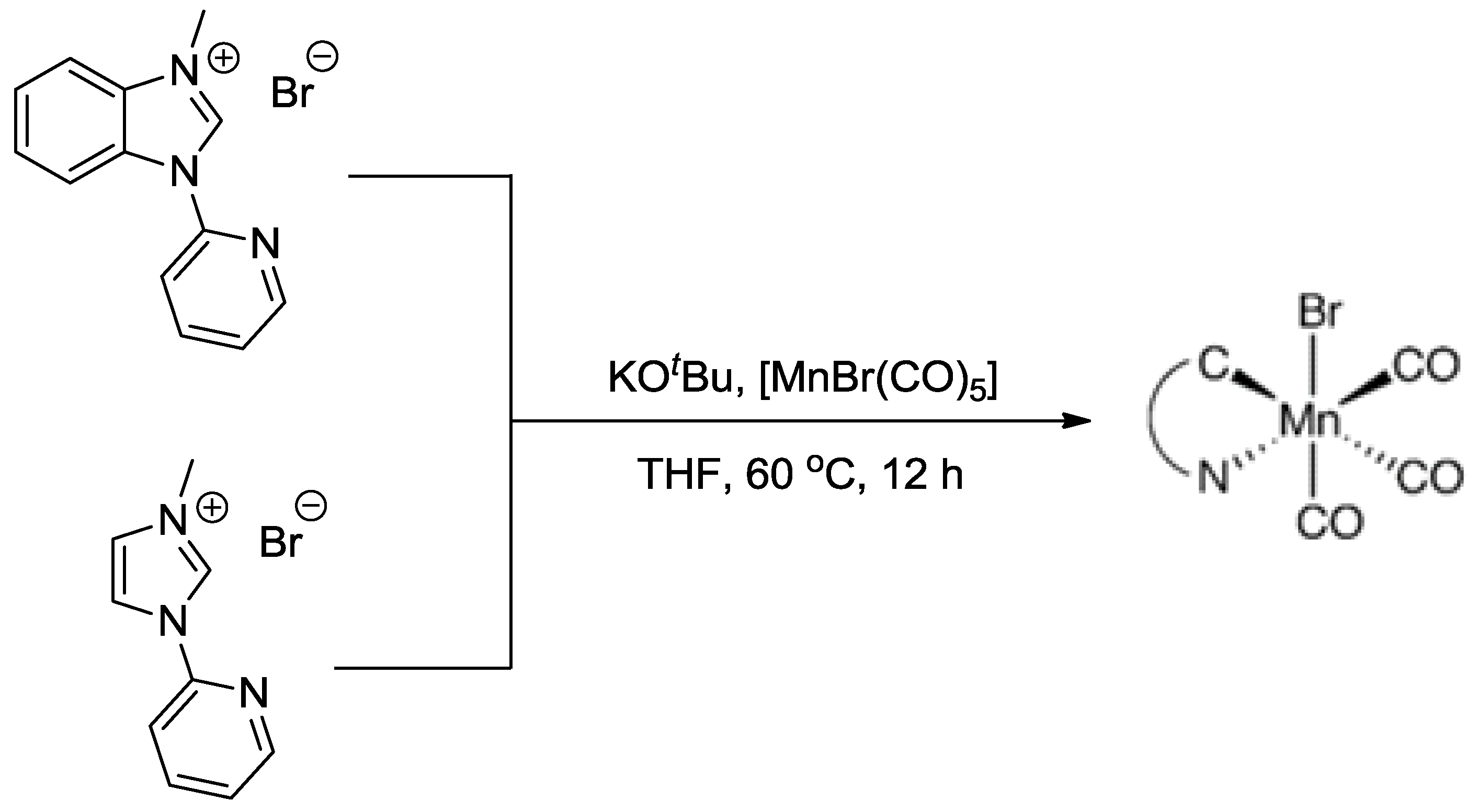
3.2. Metal Complexes with Polydentade Ligands
4. Heterogeneous Electrocatalysis of CO2 Reduction
4.1. Selective Production of Carbon Monoxide
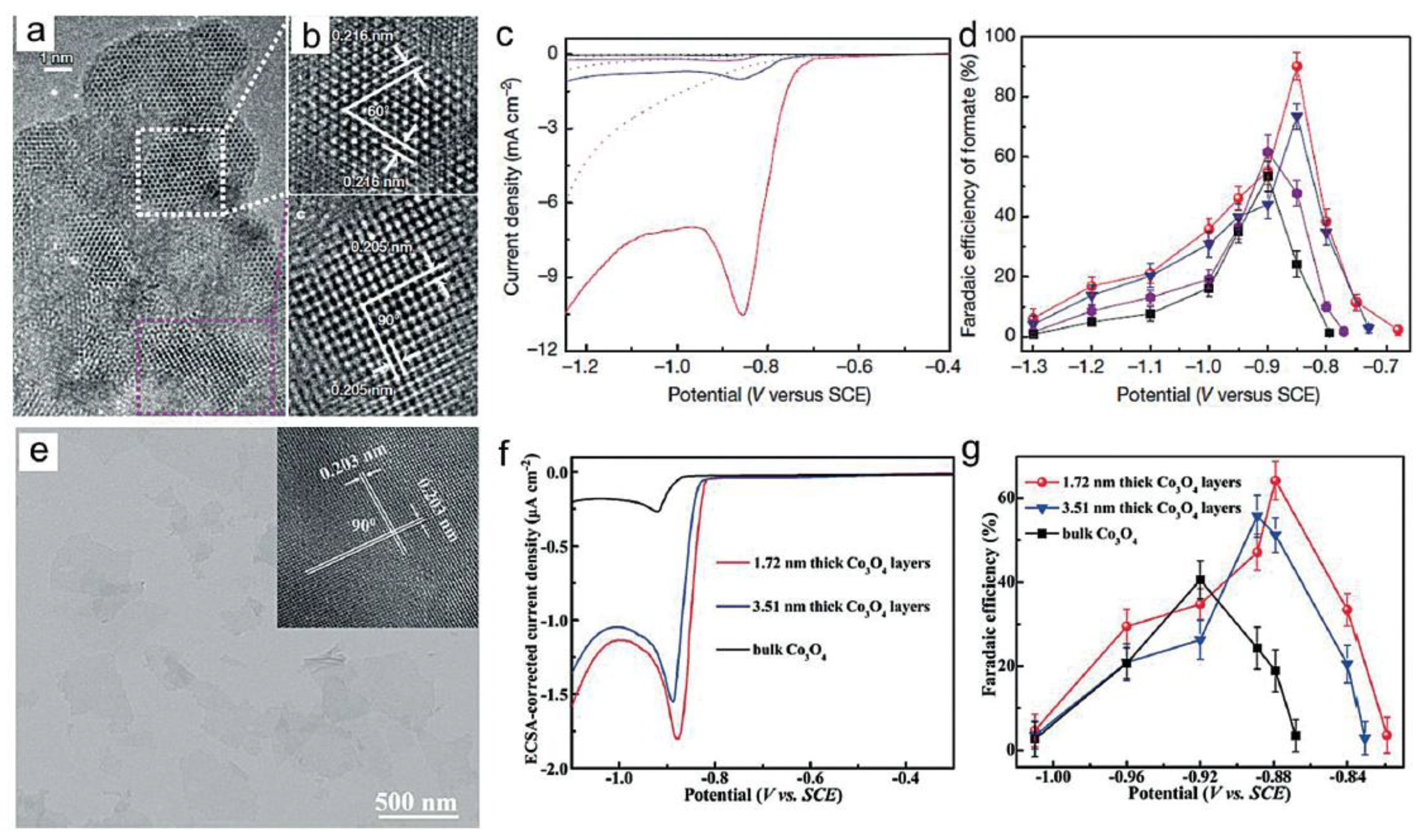
4.2. Selective Production of Formate
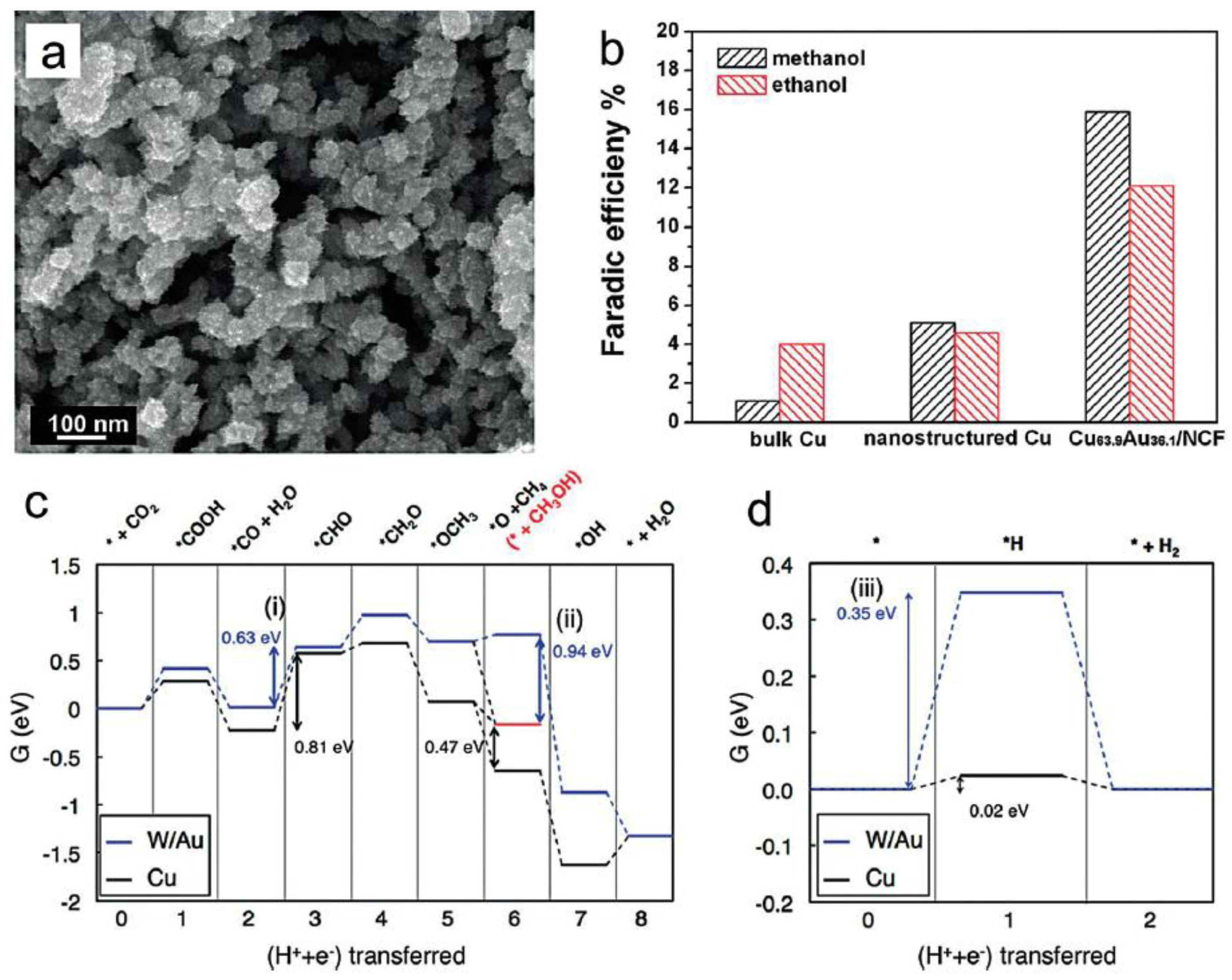
4.3. Selective Production of Methanol
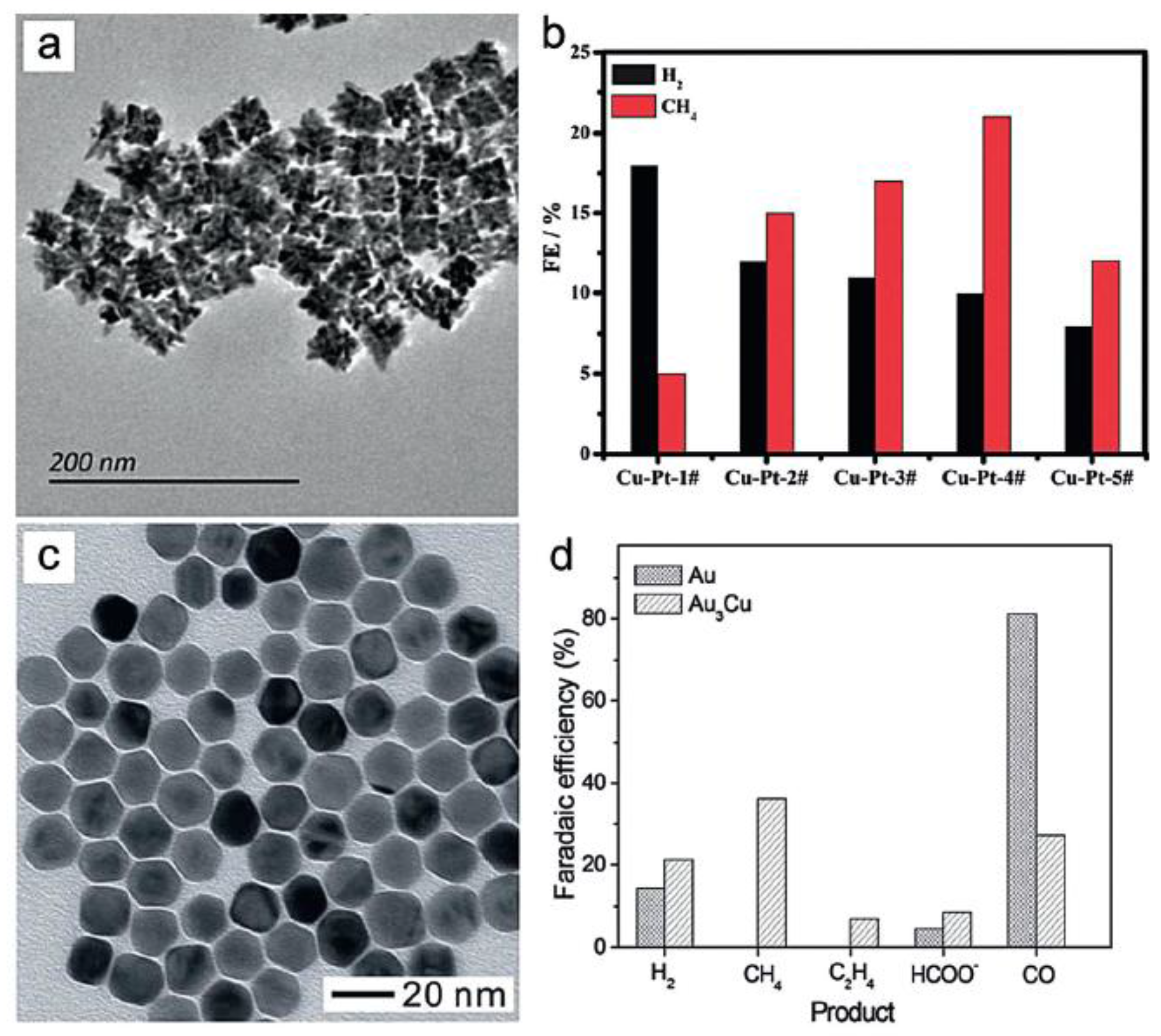
4.4. Selective Production of Methane
4.5. Selective Production of C2 Products
5. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent Progress in the Electrochemical Conversion and Utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Robert, M. Running the Clock: CO2 Catalysis in the Age of Anthropocene. ACS Energy Lett. 2016, 1, 281–282. [Google Scholar] [CrossRef]
- Schrag, D.P. Preparing to Capture Carbon. Science 2007, 315, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Omae, I. Recent Developments in Carbon Dioxide Utilization for the Production of Organic Chemicals. Coord. Chem. Rev. 2012, 256, 1384–1405. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.F.; Muckerman, J.T.; Fujita, E. Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction Catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M. Carbon Dioxide Recovery and Utilization; Springer Netherlands: Berlin, Germany, 2003. [Google Scholar]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Genovese, C.; Ampelli, C.; Marepally, B.C.; Papanikolaou, G.; Perathoner, S.; Centi, G. Electrocatalytic Reduction of CO2 for the Production of Fuels: A Comparison between Liquid and Gas Phase Conditions. Chem. Eng. Trans. 2015, 43, 2281–2286. [Google Scholar]
- Aresta, M.; Nobile, C.F.; Albano, V.G.; Forni, E.; Manassero, M. New Nickel-Carbon Dioxide Complex: Synthesis, Properties, and Crystallographic Characterization of (Carbon dioxide)-bis(tricyclehexyl phosphine)nickel. J. Chem. Soc. Chem. Commun. 1975, 636–637. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Grice, K.A. Carbon Dioxide Reduction with Homogeneous Early Transition Metal Complexes: Opportunities and Challenges for Developing CO2 Catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.-M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular Polypyridine-Based Metal Complexes as Catalysts for the Rreduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Saveant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.; Zahran, Z.N.; Naruta, Y. Efficient Electrocatalytic CO2 Reduction with a Molecular Cofacial Iron Porphyrin Dimer. Chem. Commun. 2015, 51, 16900–16903. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, Z.; Wei, X.G.; Gallenkamp, C.; Bonin, J.; Anxolabenhere-Mallart, E.; Lau, K.C.; Lau, T.C.; Robert, M. Molecular Catalysis of the Electrochemical and Photochemical Reduction of CO2 with Earth-Abundant Metal Complexes. Selective Production of CO vs. HCOOH by Switching of the Metal Center. J. Am. Chem. Soc. 2015, 137, 10918–10921. [Google Scholar] [CrossRef] [PubMed]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef] [PubMed]
- Spall, S.J.P.; Keane, T.; Tory, J.; Cocker, D.C.; Adams, H.; Fowler, H.; Meijer, H.M.; Hartl, F.; Weinstein, J.A. Manganese Tricarbonyl Complexes with Asymmetric 2-Iminopyridine Ligands: Toward Decoupling Steric and Electronic Factors in Electrocatalytic CO2 Reduction. Inorg. Chem. 2016, 55, 12568–12582. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Orio, M.; Molton, F.; Vezin, H.; Duboc, C.; Deronzier, A.; Chardon-Noblat, S. Pulsed-EPR Evidence of a Manganese(II) Hydroxycarbonyl Intermediate in the Electrocatalytic Reduction of Carbon Dioxide by a Manganese Bipyridyl Derivative. Angew. Chem. Int. Ed. 2014, 53, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Shaw, T.W.; Stanton, C.J., 3rd; Majetich, G.F.; Bocarsly, A.B.; Schaefer, H.F., 3rd. NHC-Containing Manganese(I) Electrocatalysts for Two-Electron Reduction of CO2. Angew. Chem. Int. Ed. 2014, 53, 5152–5155. [Google Scholar]
- Sun, C.; Gobetto, R.; Nervi, C. Recent Advances in Catalytic CO2 Reduction by Organometal Complexes Anchored on Modified Electrodes. New J. Chem. 2016, 40, 5656–5661. [Google Scholar] [CrossRef]
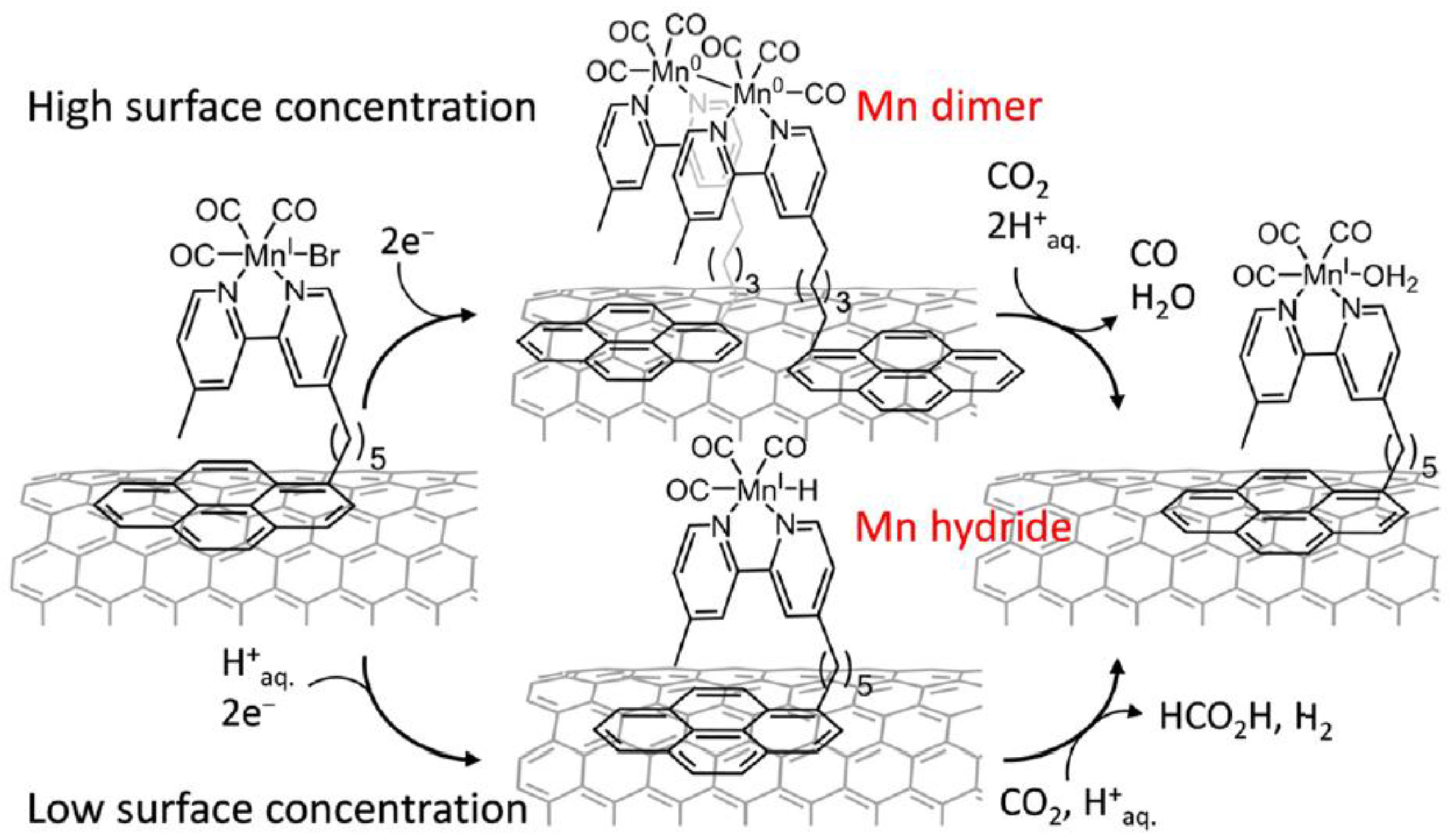
- Reuilard, B.; Ly, K.H.; Rosser, T.E.; Kuehnel, M.F.; Zebger, I.; Reisner, E. Tuning Product Selectivity for Aqueous CO2 Reduction with a Mn(bipyridine)-pyrene Catalyst Immobilized on a Carbon Nanotube Electrode. J. Am. Chem. Soc. 2017, 139, 14425–14435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.D.; Liu, J.L.; Qiao, S.Z. Recent Advances in Inorganic Heterogeneous Electrocatalysts for Reduction of Carbon Dioxide. Adv. Mater. 2017, 28, 3423–3452. [Google Scholar] [CrossRef] [PubMed]
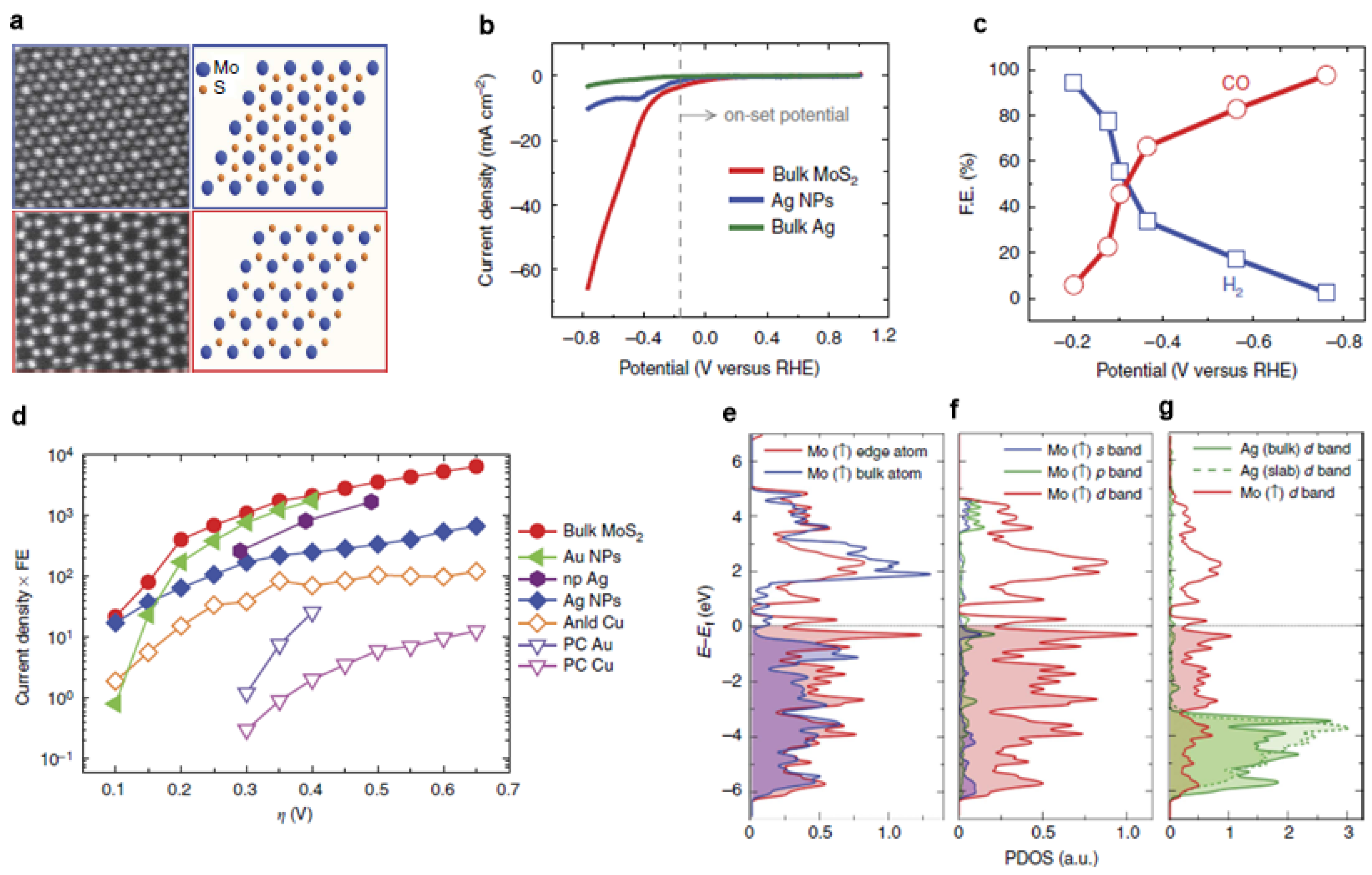
- Asadi, M.; Kim, K.; Liu, C.; Addepalli, A.V.; Abbasi, P.; Yasaei, P.; Phillips, P.; Behranginia, A.; Cerrato, J.M.; Haasch, R.; et al. Nanostructured Transition Metal Dichalcogenide Electrocatalysts for CO2 Reduction in Ionic Liquid. Science 2016, 353, 467–470. [Google Scholar] [CrossRef] [PubMed]
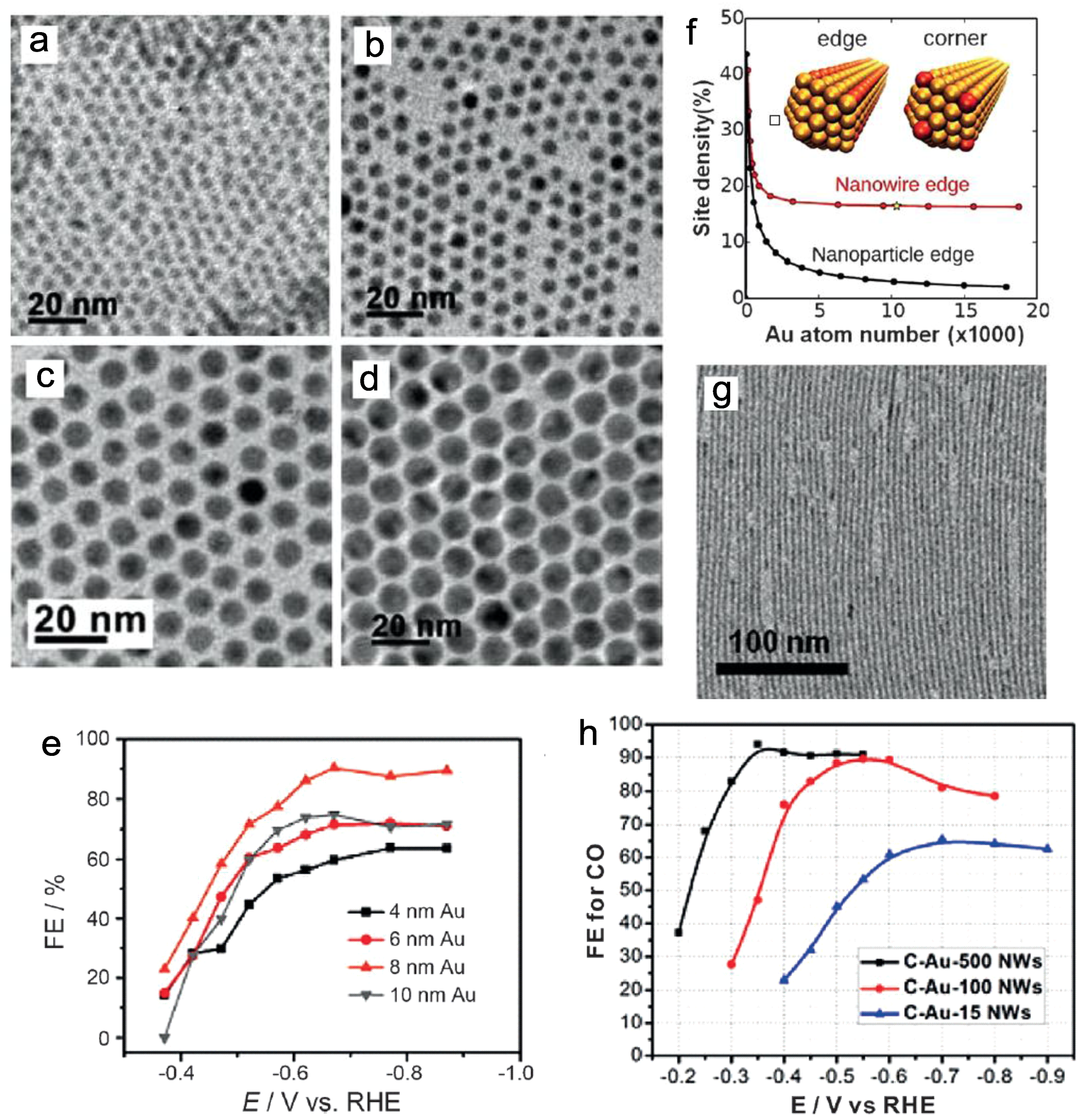
- Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional Size-Dependnet Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-E.; Yang, K.D.; Yoon, S.M.; Ahn, H.-Y.; Lee, Y.Y.; Chang, H.; Jeong, D.H.; Lee, Y.-S.; Kim, M.Y.; Nam, K.T. Concave Rhombic Dodecahedral Au Nanocatalyst with Multiple High-Index Facets for CO2 Reduction. ACS Nano 2015, 9, 8384–8393. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Eletroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef] [PubMed]
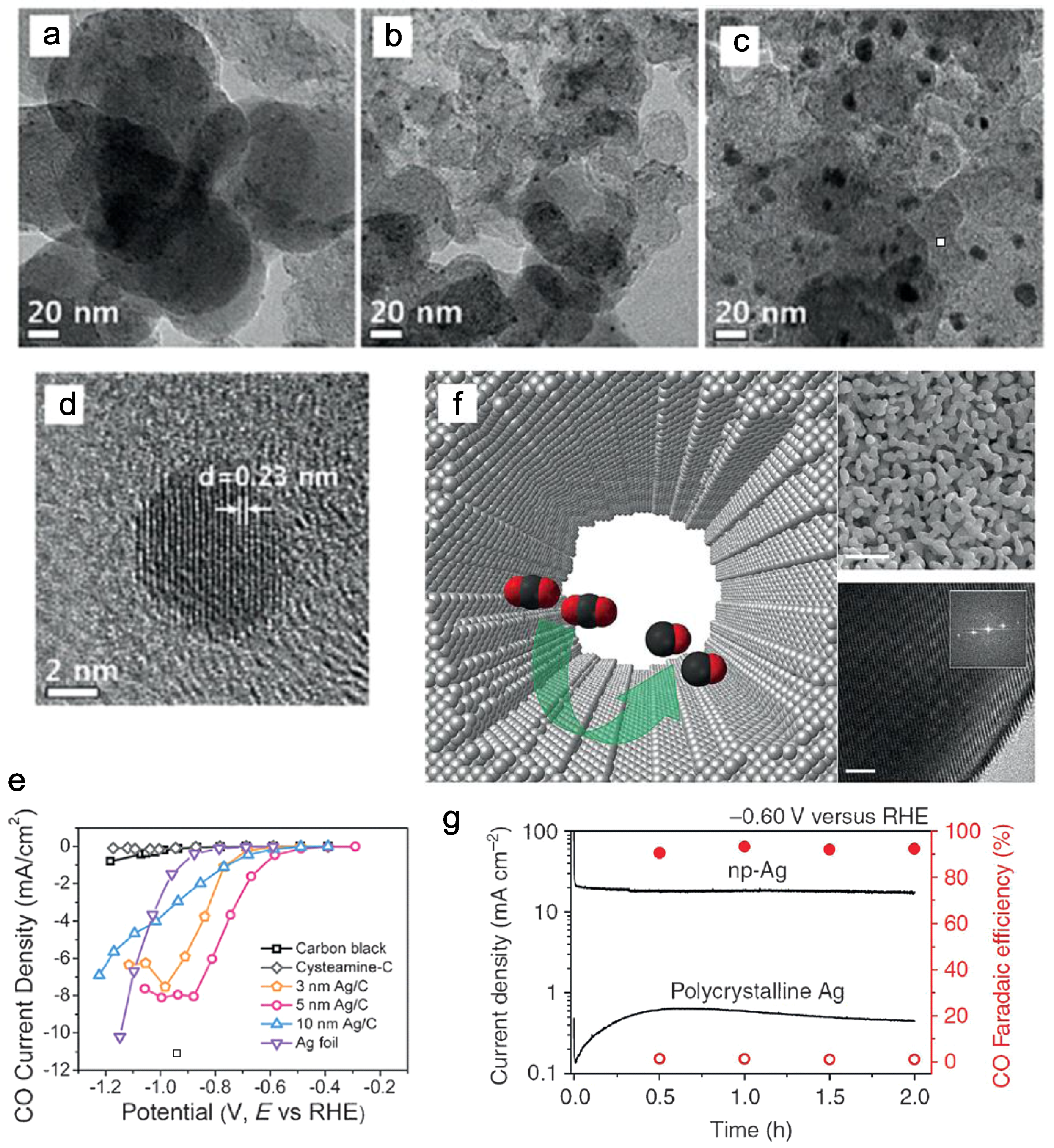
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Khojin, A.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.A.; Masel, R.I. Nanoparticles Silver Catalysts that Show Enhanced Activity for Carbon Dioxide Electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A Selective and Efficient Electrocatalyst for Carbon Dioxide Reduction. Nat. Commun. 2014, 5, 3242–3247. [Google Scholar] [CrossRef] [PubMed]
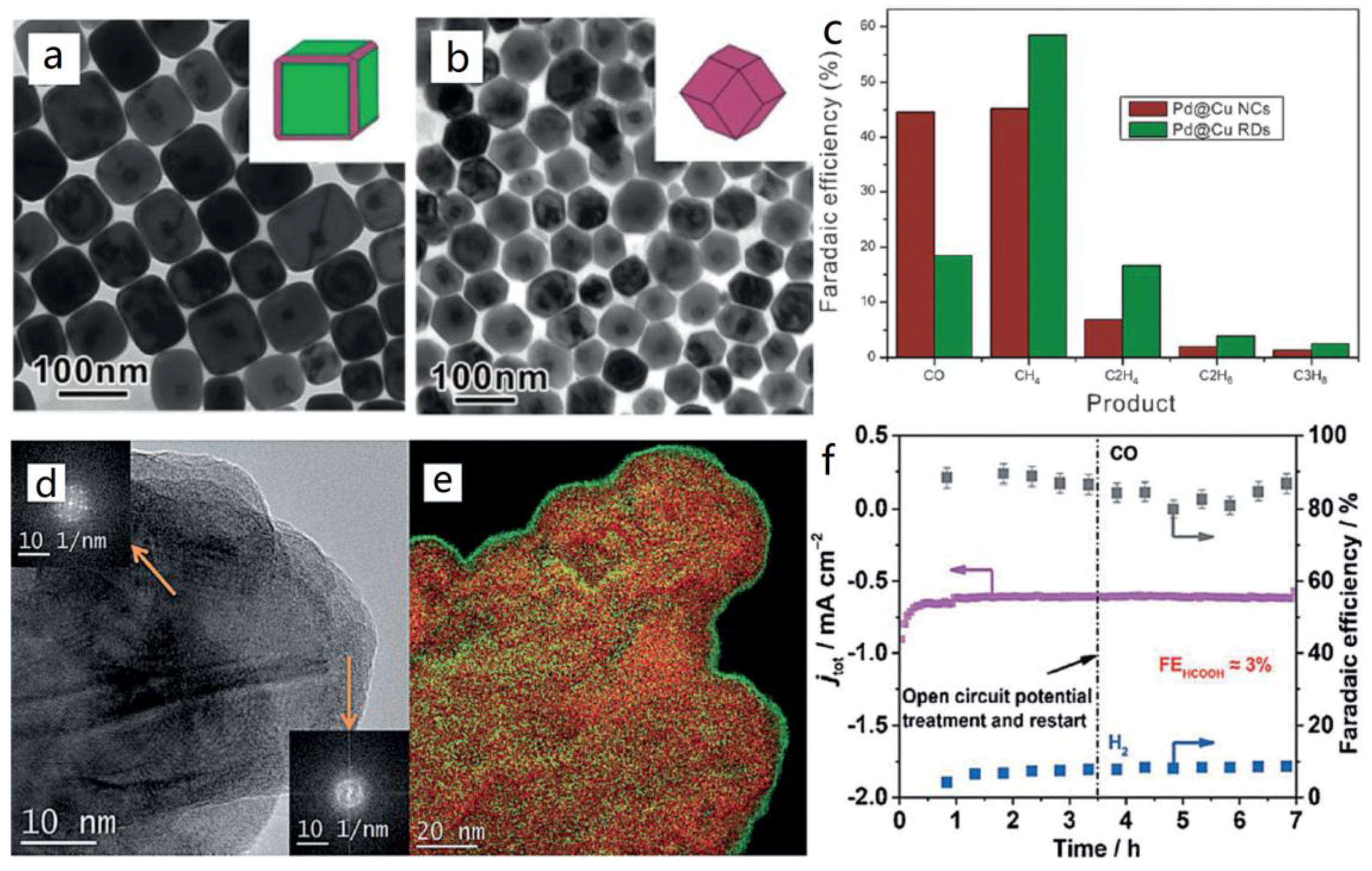
- Gao, D.; Zhou, H.; Wang, J.; Miao, S.; Yang, F.; Wang, G.; Wang, J.; Bao, X. Size-Dependent Electrocatalytic Reduction of CO2 over Pd Nanoparticles. J. Am. Chem. Soc. 2015, 137, 4288–4291. [Google Scholar] [CrossRef] [PubMed]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.R.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, G.; Zhang, Z.; Jin, M.; Yin, Y. Selectivity on Etching: Creation of High-Energy Facets on Copper Nanocrystals for CO2 Electrochemical Reduction. ACS Nano 2016, 10, 4559–4564. [Google Scholar] [CrossRef] [PubMed]
- Rasul, S.; Anjum, D.H.; Jedidi, A.; Minenkov, Y.; Cavallo, L.; Takanabe, K. A Highly Selective Copper–Indium Bimetallic Electrocatalyst for the Electrochemical Reduction of Aqueous CO2 to CO. Angew. Chem. Int. Ed. 2015, 54, 2146–2150. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges. Nat. Commun. 2014, 5, 4470–4477. [Google Scholar] [CrossRef] [PubMed]
- Benck, J.D.; Hellstern, T.R.; Kibsgaard, J.; Chakthranont, P.; Jaramillo, T.F. Catalyzing the Hydrogen Evolution Reaction (HER) with Molybdenum Sulfide Nanomaterials. ACS Catal. 2014, 4, 3957–3971. [Google Scholar] [CrossRef]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially Oxidized Atomic Cobalt Layers for Carbon Dioxide Electroreduction to Liquid Fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jiao, X.; Sun, Z.; Zhang, W.; Sun, Y.; Wang, C.; Hu, Q.; Zu, X.; Yang, F.; Yang, S.; et al. Ultrathin Co3O4 Layers Realizing Optimized CO2 Eletroreduction to Formate. Angew. Chem. Int. Ed. 2016, 55, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Liu, D.; Palmore, G.T.R. Electrochemical Reduction of CO2 at Copper Nanofoams. ACS Catal. 2014, 4, 3091–3095. [Google Scholar] [CrossRef]
- Min, X.; Kanan, M.W. Pd-Catalyzed Electrohydrogenation of Carbon Dioxide to Formate: High Mass Activity at Low Overpotential and Identification of the Deactivation Pathway. J. Am. Chem. Soc. 2015, 137, 4701–4708. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Abram, D.N.; Hansen, H.A.; Hatsukade, T.; Jackson, A.; Johnson, N.C.; Hellstern, T.R.; Kuhl, K.P.; Cave, E.R.; Feastera, J.T.; et al. Synthesis of Thin Film AuPd Alloys and Their Investigation for Electrocatalytic CO2 Reduction. J. Mater. Chem. A 2015, 3, 20185–20194. [Google Scholar] [CrossRef]
- Jia, F.; Yu, X.; Zhang, L. Enhanced Selectivity for the Electrochemical Reduction of CO2 to Alcohols in Aqueous Solution with Nanostructured Cu–Au Alloy as Catalyst. J. Power Sources 2014, 252, 85–89. [Google Scholar] [CrossRef]
- Back, S.; Kim, H.; Jung, Y. On the Selective Heterogeneous CO2 Electroreduction to Methanol. ACS Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Hansen, H.A.; Montoya, J.H.; Zhang, Y.-J.; Shi, C.; Peterson, A.A.; Nørskov, J.K. Electroreduction of Methanediol on Copper. Catal. Lett. 2013, 143, 631–635. [Google Scholar] [CrossRef]
- Manthiram, K.; Beberwyck, B.J.; Alivisatos, A.P. Enhanced Electrochemical Methanation of Carbon Dioxide with a Dispersible Nanoscale Copper Catalyst. J. Am. Chem. Soc. 2014, 136, 13319–13325. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Deng, C.; Li, X.; Xue, Y.; Yan, Y.-M.; Sun, K. Composition Dependent Activity of Cu–Pt Nanocrystals for Electrochemical Reduction of CO2. Chem. Commun. 2015, 51, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yang, L.; Yin, Y.; Jin, M. Thermodynamic Controlled Synthesis of Intermetallic Au3Cu Alloy Nanocrystals from Cu Microparticles. J. Mater. Chem. A 2014, 2, 902–906. [Google Scholar] [CrossRef]
- Kas, R.; Kortlever, R.; Yilmaz, H.; Koper, M.T.M.; Mul, G. Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions. ChemElectroChem 2015, 2, 354–358. [Google Scholar] [CrossRef]
- Tang, W.; Peterson, A.A.; Varela, A.S.; Jovanov, Z.P.; Bech, L.; Durand, W.J.; Dahl, S.; Norskov, J.K.; Chorkendorff, I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Eletroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 Reduction on Cu2O-Derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-F.; Huang, Y.-X.; Li, W.-W.; Song, X.-N.; Xiong, L.; Yu, H.-Q. Efficient Electrochemical CO2 Reduction on a Unique Chrysanthemum-Like Cu Nanoflower Electrode and Direct Observation of Carbon Deposite. Electrochim. Acta 2014, 139, 137–144. [Google Scholar] [CrossRef]
- Torelli, D.A.; Francis, S.A.; Crompton, J.C.; Javie, A.; Thompson, J.R.; Brunschwig, B.S.; Soriaga, M.P.; Lewis, N.S. Nickel-Gallium-Catalyzed Electrochemical Reduction of CO2 to Highly Reduced Products at Low Overpotentials. ACS Catal. 2016, 6, 2100–2104. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation Number | Half-Electrochemical Thermodynamic Reactions | E° (V) |
|---|---|---|
| Equation (1) | CO2 + 2H+ + 2e− = CO(g) + H2O | −0.53 |
| Equation (2) | CO2 + 2H+ + 2e− = HCO2H | −0.61 |
| Equation (3) | CO2 + 4H+ + 4e− = HCHO + H2O | −0.48 |
| Equation (4) | CO2 + 6H+ + 6e− = CH3OH + H2O | −0.38 |
| Equation (5) | CO2 + 8H+ + 8e− = CH4 + 2H2O | −0.24 |
| Equation (6) | 2CO2 + 12H+ + 12e− = C2H4 + 4H2O | −0.34 |
| Equation (7) | CO2 + e− = CO2•− | −1.90 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, D.-M.; Zhu, Y.-P.; Chen, P.; Ma, T.-Y. Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts 2017, 7, 373. https://doi.org/10.3390/catal7120373
Feng D-M, Zhu Y-P, Chen P, Ma T-Y. Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts. 2017; 7(12):373. https://doi.org/10.3390/catal7120373
Chicago/Turabian StyleFeng, Da-Ming, Yun-Pei Zhu, Ping Chen, and Tian-Yi Ma. 2017. "Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems" Catalysts 7, no. 12: 373. https://doi.org/10.3390/catal7120373
APA StyleFeng, D. -M., Zhu, Y. -P., Chen, P., & Ma, T. -Y. (2017). Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts, 7(12), 373. https://doi.org/10.3390/catal7120373