1. Introduction
Organometallic rare earths (REs) chemistry was first introduced at the beginning of the 20th century [
1] but for some time it attracted little interest due to the lack of characterization—and, thus, understanding—of these highly-reactive compounds. However, since the 1970s, with the advent of modern techniques of analysis and synthesis, it became possible to better apprehend the structure and reactivity of organometallic RE complexes, which promoted the growth of research in this field of chemistry and its application to catalysis. Thenceforth, chemists realized that the thorough study of the reactivity of allyl derivatives of the rare earths, besides alkyl and hydride derivatives, could greatly benefit the comprehension of mechanisms involved in rare earth-catalyzed polymerization of olefins and conjugated dienes [
2,
3,
4,
5,
6].
Rare earth-based allyl complexes are a relatively new area of organometallic chemistry of the RE elements and it was not until 1975 that the first series of rare earth complexes bearing an allyl ligand, Cp
2RE(C
3H
5) (RE = Sm, Er, Ho and Cp = C
5H
5), was successfully synthesized by Tsutsui and Ely [
7]. Thereafter, allylic-substituted rare earths, ranging from mono- to tetra-substituted allyl complexes, have been explored, and the group of Taube was probably the most successful in this area during the 1980s and 1990s [
8]. In terms of reactivity, the allyl moiety is of specific interest because it makes it possible to carry out a certain number of elementary organometallic reactions, such as those involved in catalytic processes (insertion reactions [
9], hydrogenolysis [
10,
11], hydrosilylation [
12], alkyl exchange [
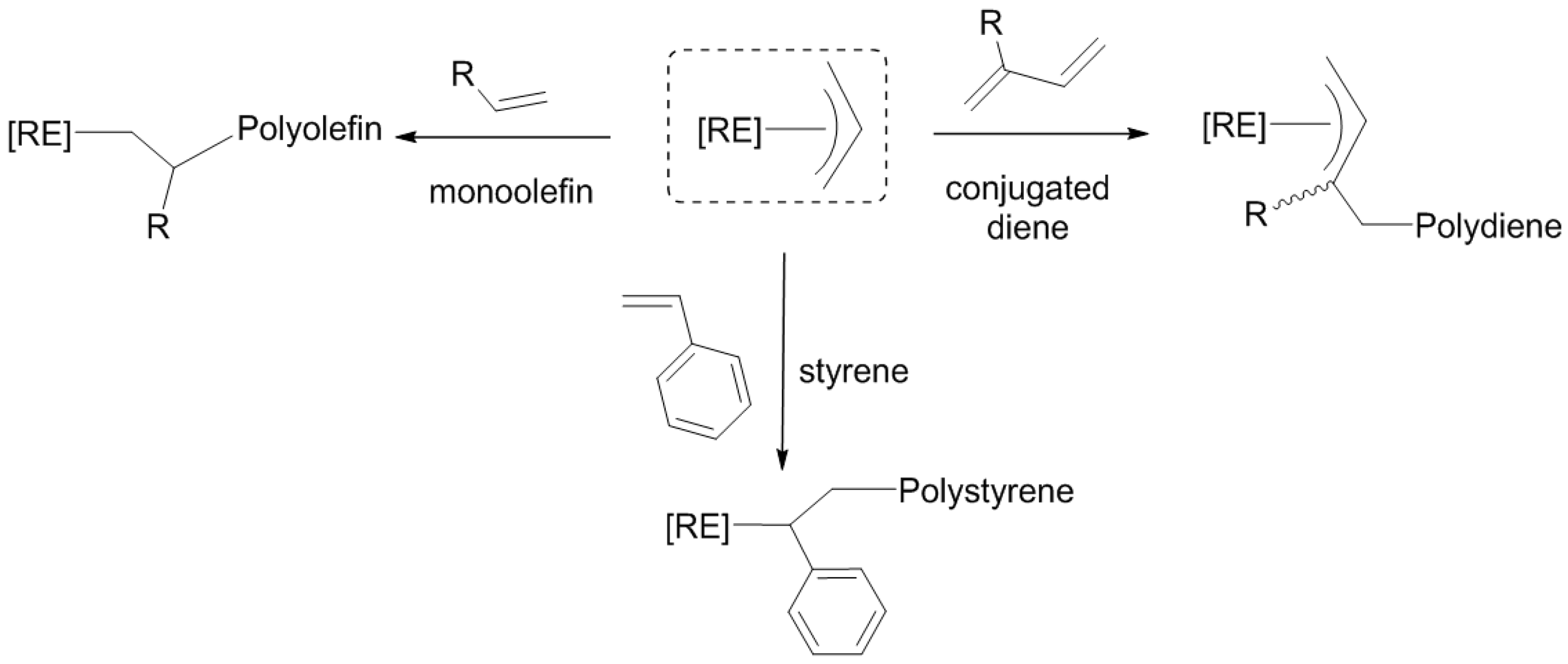
13], etc.). Consequentially, the [RE-(allyl)] species has demonstrated its ability to catalyze polymerization reactions, with a particular behavior towards non-polar monomers (
Scheme 1), some of which are highly stereo-selective [
14,
15,
16]. The [RE-(allyl)] moiety has also been studied as a model for the chain initiation in olefin polymerization [
17], and the coordination of the allyl ligand within the complexes, as well as the specificity of the rare earth metals used have shown to vary the outcome of the polymerizations.
In 2010, Carpentier et al. [
18] reviewed allyl rare earth complexes that had been studied over the past decades, along with their reactivity. Since then, a number of reports have been published that enlarge the knowledge on this particular class of compounds. In this review, we focus on the very recent development dealing with RE allyl compounds, highlighting their ability to catalyze the polymerization of non-polar monomers, such as butadiene, isoprene, styrene, and related co-polymerizations.
2. Allyl Complexes for the Polymerization of Butadiene
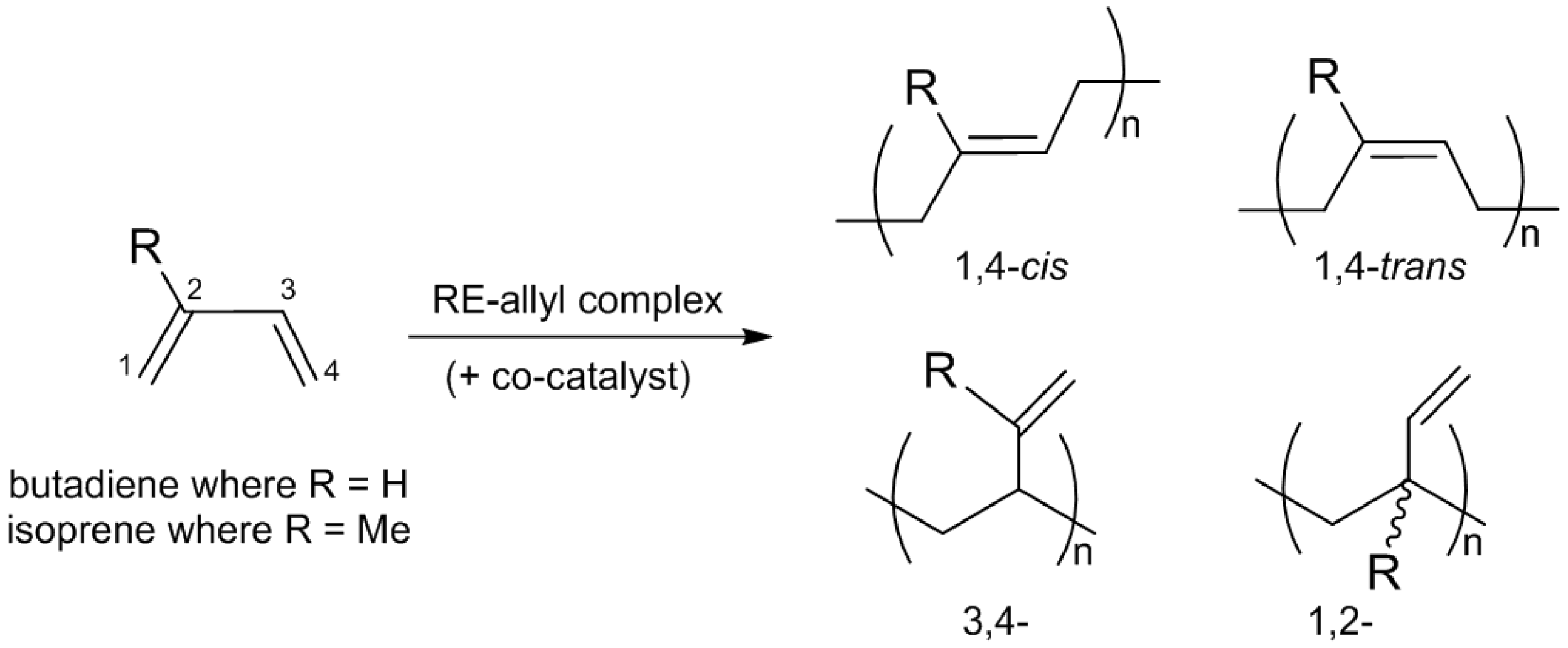
Polybutadiene (PB), arising from the polymerization of butadiene, a petro-sourced monomer, was first synthesized in the 20th century by using a sodium-based catalyst. The most notable advancement regarding the polymerization of dienes was the discovery of the Ziegler-Natta catalysts, which are still being used for industrial scale process for more than half a century, producing highly stereoregular
cis- or
trans-PB (
Scheme 2) [
19]. Rare earth complexes, and among them allyl-based catalysts, took their part in this context showing their ability to produce highly stereoregular PB with high activities, especially with the studies done by Taube’s group [
8,
14,
20].
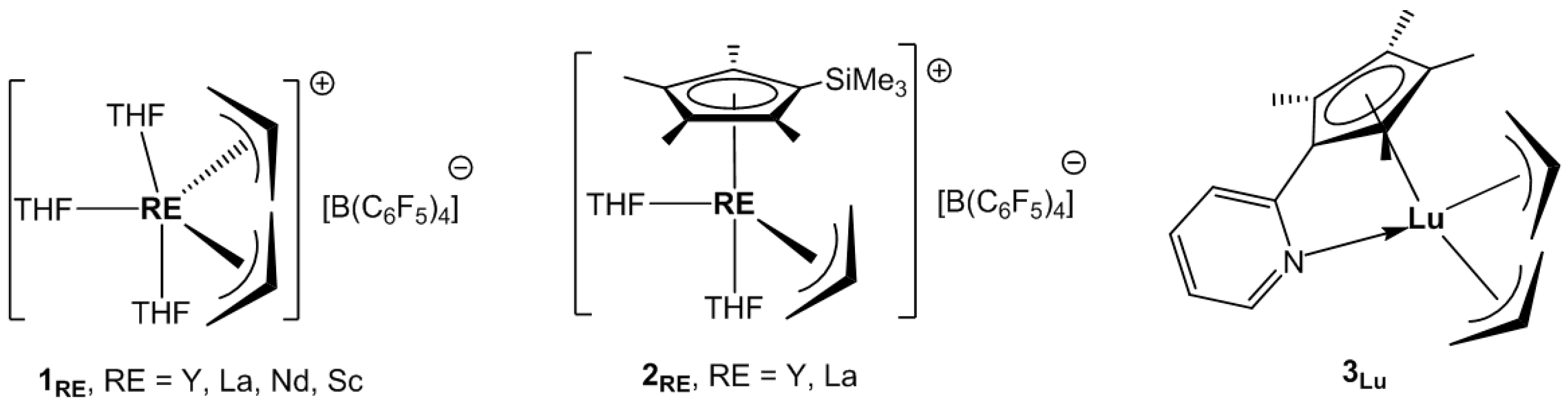
At the end of the 2000s, Okuda and co-workers reported the synthesis of a series of mono-cationic bis-allyl complexes [RE(
η3-C
3H
5)
2(THF)
3]
+[B(C
6F
5)
4]
− (
1RE, RE = Y, La, Nd, THF = tetrahydrofuran,
Scheme 3) by reacting the tris-allyl complexes RE(
η3-C
3H
5)
3(dioxane) described by Taube with one equivalent of {[HNMe
2Ph][B(C
6F
5)
4]} in THF [
21,
22]. The monocationic bis-allyl analogues bearing a non-perfluorinated counteranion [RE(
η3-C
3H
5)
2(THF)
3]
+[BPh
4]
− (
1’RE, RE = Y, La, Nd) were prepared similarly, but with {[HNEt
3Ph][BPh
4]}. In these complexes, the cationic allylic counterpart displays the same molecular structure as in complexes
1RE. X-ray crystal study of the monocationic yttrium complex
1’Y showed that all three allyl ligands are
η3 coordinated with similar bond lengths between each allyl ligand and yttrium metal. Crystals of
1’La and
1’Nd were found to contain a fourth THF molecule, whereas elemental analysis was consistent with three-THF adducts after drying under vacuum. Two independent sets of ionic pairs were observed in
1’La and
1’Nd, with allyl groups in a paddle-wheel fashion, or arranged as pincer-like towards each other. Shorter metal-allyl bonds were noticed by comparison with those of the parent compounds for both
1’La and
1’Nd, revealing a higher Lewis acidity of the rare earth metal from neutral to cationic species. The NMR (Nuclear Magnetic Resonance) analysis of complexes
1Y and
1’Y displayed two signals for the allyl group corresponding to fast
syn/
anti exchange on the NMR timescale. In contrast, three distinct signals were seen for
1La (and
1’La) and
1Nd (and
1’Nd), typical of slow
syn/
anti exchange.
The monocationic allyl complexes
1RE showed no activity towards the polymerization of 1,3-butadiene at room temperature [
21]. In contrast, these complexes were active when combined with Al(
iBu)
3 as co-catalyst. The catalyst made from the yttrium complex
1Y was found to be the most active (TOF 10,000 h
−1) leading to the formation of PB with the highest 1,4-
cis stereoregularity of 90% in comparison to those made from
1La and
1Nd complexes (low yields and 33% and 75% of 1,4-
cis units, respectively). This is a rare example that contradicts the well-known “neodymium effect” [
6]. The in situ addition of one extra equivalent of {[NPhMe
2H][B(C
6F
5)
4]} to the former mono-cationic system in the polymerization mixture led to an increase in both the activity (up to TOF (Turn-Over Frequency) 12,000 h
−1) and the selectivity with 92.5% of 1,4-
cis-PB in the case of
1Y. However, the polymerizations carried out with
1Y as precatalyst gave much broader dispersity, i.e., a less controlled process. Interestingly, the lanthanum-based precatalyst
1La showed reverse stereo-selectivity when used with Al(
iBu)
3 only and with the dual Al(
iBu)
3/{[NPhMe
2H][B(C
6F
5)
4]} combination, switching from 63.3% 1,4-
trans to 80.5% 1,4-
cis-selectivity, respectively. The in situ formation of monoallyl dicationic species, as proposed by the authors, was likely to be responsible for the better reactivity. However, isolated [RE(
η3-C
3H
5)(THF)
6]
2+{[B(C
6F
5)
4]
−}
2 (RE = La, Nd) from bulk scale syntheses were found to be rather unreactive.
The scandium congener [Sc(
η3-C
3H
5)
2(THF)
3]
+[B(C
6F
5)
4]
− (
1Sc) was prepared similarly as
1RE (RE = Y, La, Nd) from the newly synthesized tris-allyl scandium, which was lacking in the family of analogous complexes of rare earths until Okuda and coworkers succeeded to isolate it [
23]. Complex
1Sc could also be synthesized by reacting K[Sc(C
3H
5)
4] with 2 equiv. {[HNEt
3][BPh
4]}.
1H NMR analysis of
1Sc revealed dynamic behavior of the allyl group while the crystallographic distances were typical of the
η3 coordination mode. This scandium allyl complex was assessed towards polymerization of 1,3-butadiene. When it was combined with 1 equiv. of Al(
iBu)
3 as co-catalyst under similar conditions as for
1RE (RE = Y, La, Nd), it gave rise to a little amount of PB (7% yield, TOF 280 h
−1) with low stereoregularity (<60% 1,4-
cis). The activity was improved (TOF 1150 h
−1) with the addition of one equivalent of [HNMe
2Ph][B(C
6F
5)
4] in the polymerization mixture, but no change in the selectivity was observed.
The same research group synthesized the half-sandwich mono-allyl complexes [RE(
η5-C
5Me
4SiMe
3)(
η3-C
3H
5)(THF)
2]
+[B(C
6F
5)
4]
− (
2RE, RE = Y, La,
Scheme 3) by protonation of the bis-allyl complexes RE(
η5-C
5Me
4SiMe
3)(
η3-C
3H
5)
2(THF) with one equivalent of {[NPhMe
2H][B(C
6F
5)
4]} [
21]. Little rigidity was noticed for the allyl group of
2La by
1H NMR, whereas
2Y displayed higher fluxionality with fast
syn/anti exchange. The activity of these mono-cationic complexes was screened towards the polymerization of 1,3-butadiene in the presence of Al(
iBu)
3 (5 equiv.) as a co-catalyst, since the complexes were found to be inactive on their own. The lanthanum system showed lower activity (TOF 1600 h
−1), in comparison to the yttrium one which gave quantitative conversion for the same reaction time at room temperature in toluene (TOF 12,000 h
−1) and moderate selectivity (86% 1,4-
cis).
Jian et al. prepared the pyridyl-functionalized half-sandwich of lutetium, (C
5Me
4-C
5H
4N)Lu(
η3-C
3H
5)
2 3Lu by the clean protonolysis reaction of Lu(
η3-C
3H
5)
3(dioxane) with C
5Me
4H-C
5H
4N in THF [
24]. The X-ray analysis showed that both allyl groups coordinate to the lutetium in a
η3 mode. Due to the coordination of the pyridyl moiety, the complex was isolated as solvent free.
1H NMR displayed the typical 1 (quintet)/4 (doublet) set of signals for allyl groups in dynamic equilibrium. In combination with trityl borate activator, highly active catalyst towards butadiene polymerization was formed (TOF 60,000 h
−1), which was also
cis-1,4-selective up to 97%. When chlorobenzene was used as the solvent, a drop in catalytic activity was noticed, but with the benefit of the stereo-conversion of
cis-1,4 PB (99%).
3. Allyl Complexes for the Polymerization of Isoprene
The coordination polymerization of isoprene can lead to the formation of polyisoprene (PI) containing four different isomers: 1,4
-cis, 1,4-
trans, 3,4-, and 1,2- (
Scheme 2). Natural rubber extracted from the
Hevea tree is composed predominantly of
cis- units, whereas the one extracted from Gutta Percha is mainly
trans-. Both
cis-and
trans-PI found numerous applications in the fields of adhesives, sports equipment, or the tire industry [
25].
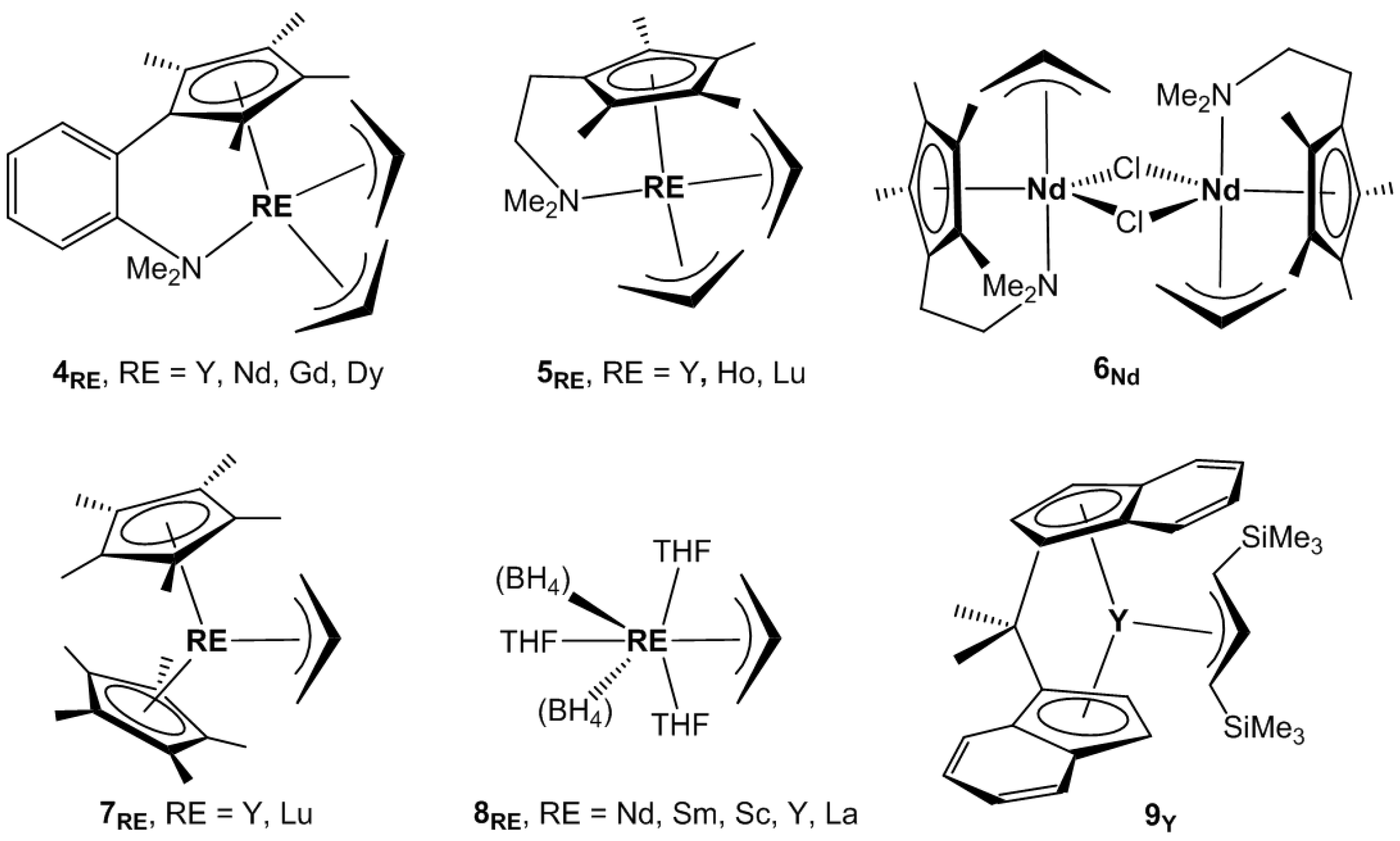
Cui, Hou, and co-workers explored constrained-geometry-conformation (CGC) allyl complexes of the rare earths as catalysts for isoprene polymerization [
26]. They synthesized the aminophenyl-cyclopentadienyl complexes (C
5Me
4–C
6H
4-
o-NMe
2)RE(
η3-C
3H
5)
2 (
4RE, RE = Y, Nd, Gd, Dy,
Scheme 4) by first reacting (C
5Me
4–C
6H
4-
o-NMe
2)Li with RECl
3(THF)
n and then adding the Grignard reagent, C
3H
5MgCl. By
1H NMR, the allyl group in
4Y was found fluxional with the typical 1H/4H resonances. The yttrium, gadolinium, and dysprosium complexes were characterized by X-ray crystallography as solvent-free—even though the reaction was performed in THF solvent—isostructural complexes and it showed that both allyl moieties coordinate in the
η3 mode. The activity of these bis(allyl) complexes was assessed towards the polymerization of isoprene in the presence of AlR
3 (mainly Al(
iBu)
3) and {[PhMe
2NH][B(C
6F
5)
4]} in toluene at 20 °C. The neodymium complex
4Nd had the highest activity (TOF 3000 h
−1), followed by the gadolinium
4Gd (1000 h
−1) and then the dysprosium
4Dy analogue (110 h
−1). The yttrium complex
4Y was almost inert towards this polymerization under these conditions, while low to medium activity (TOF 140–800 h
−1) was noticed with {[Ph
3C][B(C
6F
5)
4]}/AlR
3 (R = Me, Et,
iBu). The gadolinium complex
4Gd afforded the highest
cis-regular PI at 99.2% (at 0 °C), along with the living character of the polymerization. It was observed that when the Al/Gd ratio was increased, a typical catalyzed chain growth (CCG) process was operating, with regular decrease of the molecular weight of the PI while the molecular weight distribution remained unchanged. Al(
iBu)
3 behaved partially as a chain transfer agent, with ca. eight growing PI chains per RE metal, while no drop of stereo-selectivity was evident (ca. 98%
cis-units).
Jende et al. synthesized the allyl half-sandwich complexes of small-size rare earths, Cp
NMe2RE(
η3-C
3H
5)
2 (Cp
NMe2 = C
5Me
4CH
2CH
2NMe
2;
5RE, RE = Y, Ho, Lu,
Scheme 4) having a more flexible (
N,N-dimethylamino)ethyl-functionalized cyclopentadienyl ligand than in the
4RE complexes [
27]. The reaction was conducted in two steps by first reacting RECl
3(THF)
x with Cp
NMe2Li at room temperature, and then adding two equivalents of the Grignard reagent C
3H
5MgCl. X-ray analysis showed that the
5RE complexes were under a bis(allyl) half-sandwich monomeric form, and were all isostructural. One of the allyl groups showed similar bond lengths between terminal and central carbon atoms, while the second allyl group showed a significantly longer bond length between the rare earth metal and the terminal carbon. The overall moiety arrangement was likened to that of
4Y [26]. The
1H NMR of both diamagnetic complexes
5Y and
5Lu showed a similar quintet (1H)/doublet (4H) set of signals characteristic of dynamic exchange of the allylic protons, along with splitting of the quintet in the case of
5Y being attributable to the coupling with
89Y. When the same synthetic procedure as for
5RE was done in the case of the larger size neodymium element, it gave a monoallyl chloro derivative [Cp
NMe2Nd(
η3-C
3H
5)(
μ-Cl)]
2 (
6Nd,
Scheme 4) instead of the expected bis allyl half-sandwich. Complex
6Nd was found to be dimeric through (
μ-Cl) bridges with one substantially longer Nd-Cl bond than the other one, anticipating a possible reactivity. [Allyl]/[Cl] exchange was observed when complexes
5Y and
5Ho were reacted with AlEt
2Cl, affording multi(
μ-chlorido) hexametallic [RE
6Cl
12] clusters. This was in agreement with the observation that the combination of any
5RE with AlEt
2Cl was found inert towards isoprene polymerization.
When activated with either {[Ph3C][B(C6F5)4]} or {[PhNMe2H][B(C6F5)4]} borates, the half-sandwiches 5Y and 5Ho were found to be poorly active (TOF 100 h−1) towards the polymerization of isoprene, while 5Lu displayed higher activity (TOF 500 h−1). The yttrium (5Y) and holmium (5Ho) complexes afforded predominantly 3,4-PI with both co-catalysts. The lutetium complex 5Lu afforded non stereo-regular PI. The dispersities were very narrow (1.04–1.17) for all the precatalysts used, accounting for unique active species. In addition, in the presence of 10 equiv. AlMe3, the 5Y/borate and 5Ho/borate combinations afforded much more active catalysts (TOF up to 2000 h−1) with a switch in selectivity towards trans-1,4 selectivity (71% Y, 72% Ho). In contrast, when 10 equiv. AliBu3 were added to the 5Y/borate and 5Ho/borate systems, it afforded a major selectivity towards cis-1,4 PI (74% Y, 74% Ho) along with improved activity (TOF 1000 h−1). For the lutetium complex 5Lu, when AlMe3 or Al(iBu)3 was added to the system the main effect was a similar gain of activity (TOF up to 2000 h−1), but with no improvement of the stereocontrol. A decrease in the polymer molecular weights with narrow distributions was also noticed, which indicated a chain transfer to aluminum. The activity of the chloroallyl neodymium complex 6Nd was also assessed towards the polymerization of isoprene with the use of either {[Ph3C][B(C6F5)4]} or {[PhNMe2H][B(C6F5)4]} as a borate activator. There was no activity when the [Nd]:[borate] ratio was 1:1. However, when 1 equiv. of activator was added to the dinuclear complex ([Nd]:[borate] is 2:1), PI, with mainly 3,4-motives of up to 66%, was isolated with good activity (TOF 500 h−1), along with narrow dispersities (1.10–1.11). When 10 equiv. of AlMe3 was added to the system along with {[PhNMe2H][B(C6F5)4]}, there was a switch in stereo-selectivity and trans-1,4-PI (85%) was obtained, while the addition of Al(iBu)3 (10 equiv.) gave rise to 3,4-PI (85%). The activity was improved by a factor of four (TOF 2000 h−1) and of two (TOF 1000 h−1) by the addition of AlMe3 and AliBu3, respectively. In all cases with complexes bearing this CpNMe2 ligand (5RE and 6Nd), the use of AliBu3 vs. AlMe3 provided reversible transfer between the RE metal and the aluminum during the polymer chain growing process. Allyl(RE)-alkyl(Al) exchange was evidenced by 1H NMR experiments to support the formation of the polymerization active species. None of these allyl complexes 5RE and 6Nd were found active on their own without activator/co-catalyst.
The photopolymerization of isoprene mediated by (C
5Me
5)
2RE(
η3-C
3H
5) (
7RE, RE = Y, Lu,
Scheme 4) was assessed [
28]. These complexes had been previously synthesized and characterized [
13]. In particular, the
1H NMR analysis established non-fluxional allyl group with 1H/2H/2H allyl signals. In neat monomer, the reaction resulted in obtaining low molecular weight PI with 3,4-units being slightly predominant, which was consistent with radical polymerization, according to the authors. In the absence of irradiation, no polymerization was observed with the yttrium complex
7Y.
Bonnet, Visseaux, and co-workers synthesized the first RE complexes bearing both allyl and borohydride ligands, RE(BH
4)
2(
η3-C
3H
5)(THF)
3 (
8RE, RE = Nd, Sm,
Scheme 4) by reacting RE(BH
4)
3(THF)
3 with half an equivalent of Mg(C
3H
5)
2(L)
n (L = THF, dioxane) in THF at room temperature [
29]. From
1H NMR analysis, the allyl moiety appeared as a 1H/2H/2H set of resonances for both
8RE complexes, revealing no dynamic behavior at the
1H NMR timescale. X-ray analyses showed that both complexes were monomeric and isostructural, with the two borohydride ligands being tridentate. The activity of these mixed borohydrido-allyl rare earth complexes RE(BH
4)
2(
η3-C
3H
5)(THF)
3 was assessed towards the polymerization of isoprene. Whereas the samarium complex showed no reactivity, its neodymium analogue was found to be active, either on its own, due to the presence of the Nd-allyl bond, or combined with various alkylating reagents. When the latter was tested alone, highly
trans-regular PI with 92.2%
trans-selectivity along with Ð = 1.54 was obtained with moderate activity (TOF 177 h
−1). In the presence of one equivalent of Mg(
nBu)(Et) with respect to
8Nd, the activity was substantially increased (TOF 425 h
−1), but also the
trans-selectivity of the reaction was improved (95.5%
trans). With aluminum-based co-catalysts, such as Al(
iBu)
3 or MAO (MethylAlumOxane), the activities were greatly improved with TOF of 1000 h
−1, however, the
trans-selectivity was affected (78.7% and 68.2%, respectively). This family of complexes was recently extended to scandium (bis-THF adduct), yttrium, and lanthanum [
30].
The
ansa-lanthanidocene allyl
rac-{Me
2C(Ind)
2}Y[
η3-1,3-(SiMe
3)
2C
3H
3] (
9Y) (Ind = 2-indenyl) was assessed for the polymerization of isoprene [
32]. This compound was synthesized previously and initially evaluated for styrene polymerization as single-component catalyst [
31]. Towards isoprene, and again in the absence of co-reagent, complex
9Y afforded 1,4-
trans PI (87–91%) with moderate activity (TOF 70 h
−1). It is noteworthy that, up to now, this is the unique example of a single-component yttrium catalyst for the
trans-stereo-selective polymerization of isoprene. With di(ethyl)zinc in excess, it was established that reversible Y/Zn chain transfer was operating, with a comparable activity (TOF 76 h
−1), while maintaining the 1,4-
trans selectivity (ca. 90%). In turn, the polymerization of isoprene mediated by
9Y/Mg(
nBu)
2 occurred with a good level of transfer, but at the expense of the 1,4-
trans selectivity (up to 47% 3,4 units).
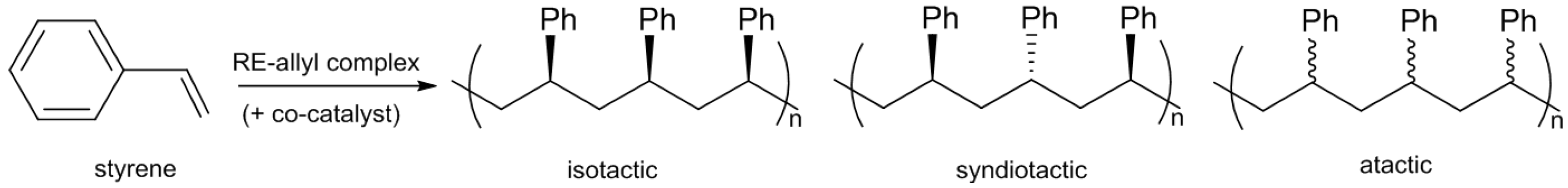
4. Allyl Complexes for the Polymerization of Styrene
Polystyrene (PS) is a thermoplastic polymer mostly known for its applications in long-lasting packaging. Styrene, when polymerized by coordination-insertion polymerization, can give rise to PS under three different forms: isotactic, syndiotactic, or atactic (
Scheme 5).
The scandium bis-allyl mono-cationic complex [Sc(
η3-C
3H
5)
2(THF)
3]
+[B(C
6F
5)
4]
− (
1Sc), which was proved to be efficient towards butadiene polymerization when combined with Al(
iBu)
3 and {[HNMe
2Ph][B(C
6F
5)
4]} (see above) was also studied for styrene polymerization. It was found to be inactive in this case as a single component, while displaying low activity in the presence of Al(
iBu)
3 (TOF up to 95 h
−1) to afford atactic PS. The addition of {[HNMe
2Ph][B(C
6F
5)
4]} as activator did not really improve the catalysis (atactic PS, TOF 114 h
−1) [
23].
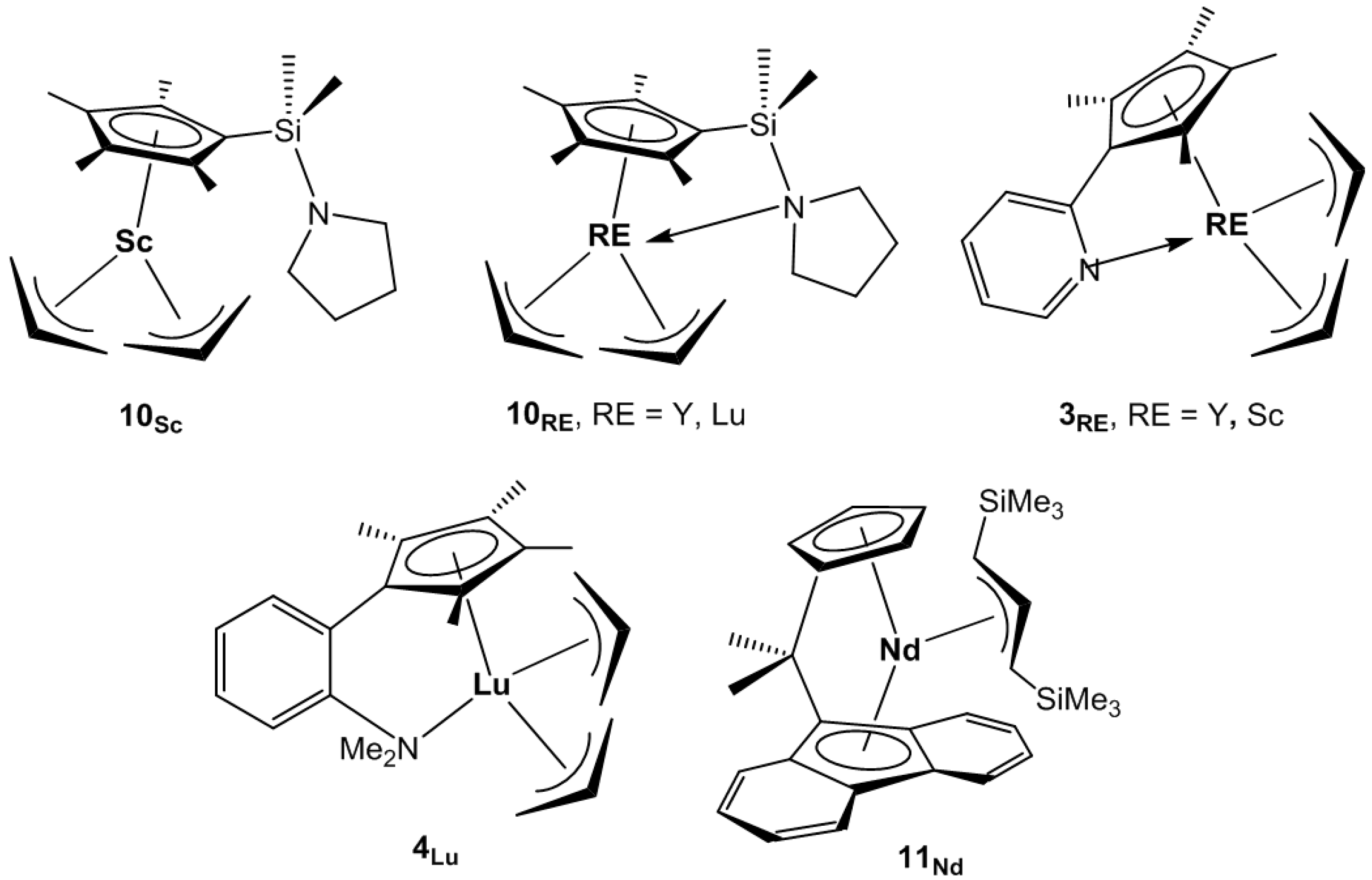
The pyrrolidinyl-functionalized half-sandwich complexes (C
5Me
4SiMe
2NC
4H
8)RE(
η3-C
3H
5)
2 (
10RE, RE = Sc, Y, Lu,
Scheme 6) were synthesized by reacting RECl
3 with one equivalent of C
5Me
4SiMe
2-NC
4H
8Li followed by the addition of two equivalents of C
3H
5MgCl in THF at room temperature [
33]. The
1H NMR spectra of the three complexes indicated the fluxional allyl ligand in solution, with one sharp doublet signal for the terminal allylic protons and one multiplet for the central allylic protons. X-ray analysis showed that for the scandium complex
10Sc, the pendant pyrrolidinyl ligand does not coordinate to the metal center through the nitrogen, whereas this coordination was present in the yttrium
10Y and lutetium
10Lu complexes due to higher size of the RE
3+ cation [
36]. In
10Sc, the two allyl moieties were coordinated to the central metal in
η3 mode with one allyl group prone and the other supine. The half sandwich complex
10Sc was found to be highly active towards styrene polymerization when activated with one equivalent of {[Ph
3C][B(C
6F
5)
4]} in toluene at room temperature, producing pure syndiotactic PS (TOF 250 h
−1). Yttrium complex
10Y was much less active (70 h
−1) while
10Lu only produced traces of the polymer under the same conditions. When an excess of Al(
iBu)
3 was added to
10Sc/{[Ph
3C][B(C
6F
5)
4]}, the activity increased drastically (TOF 1500 h
−1).
The pyridyl-functionalized half-sandwich complexes (C
5Me
4-C
5H
4N)RE(
η3-C
3H
5)
2 (
3RE, RE = Y, Sc,
Scheme 6), analogs of complex
3Lu, were prepared. The syntheses were conducted by metathetic reaction of (C
5Me
4-C
5H
4N)Li with 1 equiv. RECl
3, followed by addition of 2 equiv. allyl MgCl in THF at room temperature [
35]. Alternatively, the target compounds could be also obtained by the acid base reaction between RE(C
3H
5)
3(1,4-dioxane) and the pyridyl-cyclopentadiene C
5Me
4H-C
5H
4N, as same as previously done for
3Lu. The X-ray structure displayed the expected CGC-geometry with
η5(Cp)/
κ1(Py) coordination to the RE center. In contrast to what was observed in complexes
2Y [
21] and
4Y [
26], the
1H NMR of
3Y displayed a 1H/2H/2H pattern resonances for the allyl ligands, thus suggesting poor fluxionality in solution, while in the case of
3Sc only two allyl signals (1H/4H) were observed. In combination with {[Ph
3C][B(C
6F
5)
4]} in toluene, complex
3Y showed moderate activity (TOF 120 h
−1) but syndiotactic enriched PS (
rrrr = 88%). This catalytic system made of yttrium was much more active in chlorobenzene (TOF 2000 h
−1) but less stereoselective. In turn, the scandium analogue
3sc was found to display under the same conditions exceptionally high performances affording PS perfectly syndiotactic (
rrrr > 99%) with TOF value of 60,000 h
−1 (complete conversion of 1000 equivalents of monomer in 1 min at 20 °C in toluene) [
37] and narrow dispersity (Ð = 1.40–1.50). However, lower activity/selectivity was noticed in chlorobenzene for
3Sc. Although the process was less controlled (Ð = 1.94), the lutetium complex
3Lu gave the same notable results as
3Sc in terms of catalytic capability, and remarkably, the former complex exhibited a rare dual catalysis ability [
24,
38] in both syndiotactic styrene polymerization and
cis-selective butadiene polymerization. Contrary to what advanced for
4Lu, the smaller Cp
cent-RE-N bite angle in pyridyl-Cp complexes
3RE, along with a more electron withdrawing effect of the ligand, was proposed to explain the catalytic efficiency of the latter complexes.
Cui, Hou, and co-workers extended the family of constrained geometry catalysts
4RE [
26] to the lutetium derivative (C
5Me
4–C
6H
4-
o-NMe
2)Lu(
η3-C
3H
5)
2 (
4Lu,
Scheme 6), by a synthetic procedure similar to that used for the latter complexes [
35]. Upon activation of this lutetium complex with {[Ph
3C][B(C
6F
5)
4]}, or with the {[Ph
3C][B(C
6F
5)
4]}/Al(
iBu)
3 combination, the product was inert for the polymerization of styrene. The same was observed for
4Y under the same conditions. According to the authors, this could be due to the large Cp
cent-Lu-N bite angle in
4RE, in comparison with the value determined in complexes
3RE, which hinders the coordination and insertion of the styrene monomer.
Carpentier and colleagues found that the combination of bulky allyl
ansa-lanthanidocenes
rac-{Me
2C(Ind)
2}Y[
η3-1,3-(SiMe
3)
2C
3H
3] (
9Y) and {Me
2C(Cp)(Flu)}Nd[
η3-1,3-(SiMe
3)
2C
3H
3] (
11Nd) (Flu = 9-fluorenyl,
Scheme 6) with di(n-butyl)magnesium in excess behaved efficiently as binary catalytic systems for the stereo-controlled coordinative polymerization of styrene under reversible chain transfer regime (CCTP, coordinative chain transfer polymerization). Isotactic PS was produced with
9Y/Mg(
nBu)
2 while 11
Nd/Mg(
nBu)
2 yielded syndiotactic PS, both with high activities (TOF up to 2100 h
−1 and 2500 h
−1, respectively [
34]. By adjusting the amount of Mg(
nBu)
2, up to 200 polymer chains can be generated per RE center. Complex
9Y was previously shown to be active as a single-component catalyst [
32], but the dispersity was improved in the presence of excess Mg(
nBu)
2. Mechanistic investigations, also confirmed by the support of theoretical studies, demonstrated that the initiation of the polymerization resulted from the insertion of styrene into the RE-allyl (single component) or RE-alkyl (chain transfer) moiety, and that an enantiomorphic site control mechanism (ECM) was operative to account for the isoselectivity observed [
39]. As for the
ansa derivative
11Nd, it was synthesized by ionic metathesis between K[1,3-(SiMe3)2C3H3] and [{Me
2C(Cp)(Flu)}Nd(μ-Cl)]
2 and was found unsolvated. It was shown that
11Nd acts as a single-component catalyst for the polymerization of styrene, and produces
sPS albeit at a much lower rate (TOF = 20–60 h
−1) than the regular allyl compounds {Me
2C(Cp)(Flu)}RE(
η3-C
3H
5)(THF) (RE = Y, La, Nd, 1000–17,000 h
−1) [
40]. Using DFT (Density Functional Theory) studies, the origin of the syndiospecificity control, due to a chain-end control mechanism (CEM), was proposed to result from the conjunction of the minimization of two repulsion effects: the classical phenyl (incoming monomer)-phenyl (last unit inserted) one during the growing of the polymer chain, and also of the repulsion between the fluorenyl ligand and the incoming styrene unit [
41].
6. Concluding Remarks
In the last eight years, a number of new allyl rare earth complexes have been synthesized and assessed towards the (co-)polymerization of non-polar monomers (
Table 1). It must be noted that when browsing through the recent reports of the field gathered in this review, it appears that the utilization of such compounds for polymerization is limited to conjugated dienes and styrene, while none deal with ethylene, although allyl species of the rare earths are known to mediate the polymerization of that latter monomer [
3,
6].
In some cases towards conjugated dienes or styrene, an allyl rare earth complex is active by itself, i.e., no co-catalyst is necessary to initiate the polymerization. However, this is limited to the case of neodymium, or bulky ansa-metallocenes. In general, the performances are improved when an alkylating reagent and/or an activator are associated to the allylic compound. Regarding conjugated dienes, allyl complexes afford catalysts that enable the production of polymers with high stereoselectivity, along with very high activities.
The complexes synthesized by most research teams often focus on the “small” REs, i.e., the
late lanthanides—having small size ionic radius—also including yttrium and scandium, especially when they are of the CGC-type. In turn, when it comes to metallocene-like derivatives, “big” RE, i.e., the
early lanthanides, also including lanthanum, are privileged. In general, many scandium complexes in a given series afford the best catalyst, particularly as far as pseudo-cationic processes are concerned, which corroborates a recent theoretical study by Hou and coworkers [
42].
Most allyl complexes of the RE described in this review are cyclopentadienyl derivatives, at the exception of the mixed allyl-borohydrides series recently reported. Such mixed allyl-borohydrides undoubtedly foreshadow a new platform towards novel families of allyl rare earth post-metallocenes.
Although allyl complexes of the rare earths may sometimes be difficult to isolate, their synthesis is clearly worth the effort. Indeed, the hapticity of the allyl ligand can assist in isolating a compound where the alkyl analog is not stable, and also limits the coordination of an additional solvent molecule, which may be detrimental to the catalytic performances. Moreover, as illustrated in this mini-review, allyl complexes may be obtained under a monomeric form, which can favor the reactivity vs. bridged alkyl complexes. So far, the presence of an allyl ligand does not guarantee the reactivity towards the insertion reaction of a monomer molecule and, hence, the polymerization: this allyl moiety must be effectively reactive. The fluxionality of the allyl ligand, as seen by proton NMR, can be an indication of a possible reactivity: it is quite frequently observed that the most active catalyst in a series often corresponds to the complex whose allyl ligand has a certain degree of fluxionality. However, this does not seem to be generalizable for the larger rare earths.
Nevertheless, in most cases the help of a co-catalyst and/or an activator is mandatory, at least to improve the catalytic performances, or to better control the process, especially when the reactions are conducted under reversible chain transfer conditions.
It can be anticipated that further research will be conducted in this area of chemistry for the years to come.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}