Direct Carboxylation of C(sp3)-H and C(sp2)-H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions

Abstract
:1. Introduction
- (1)
- (2)
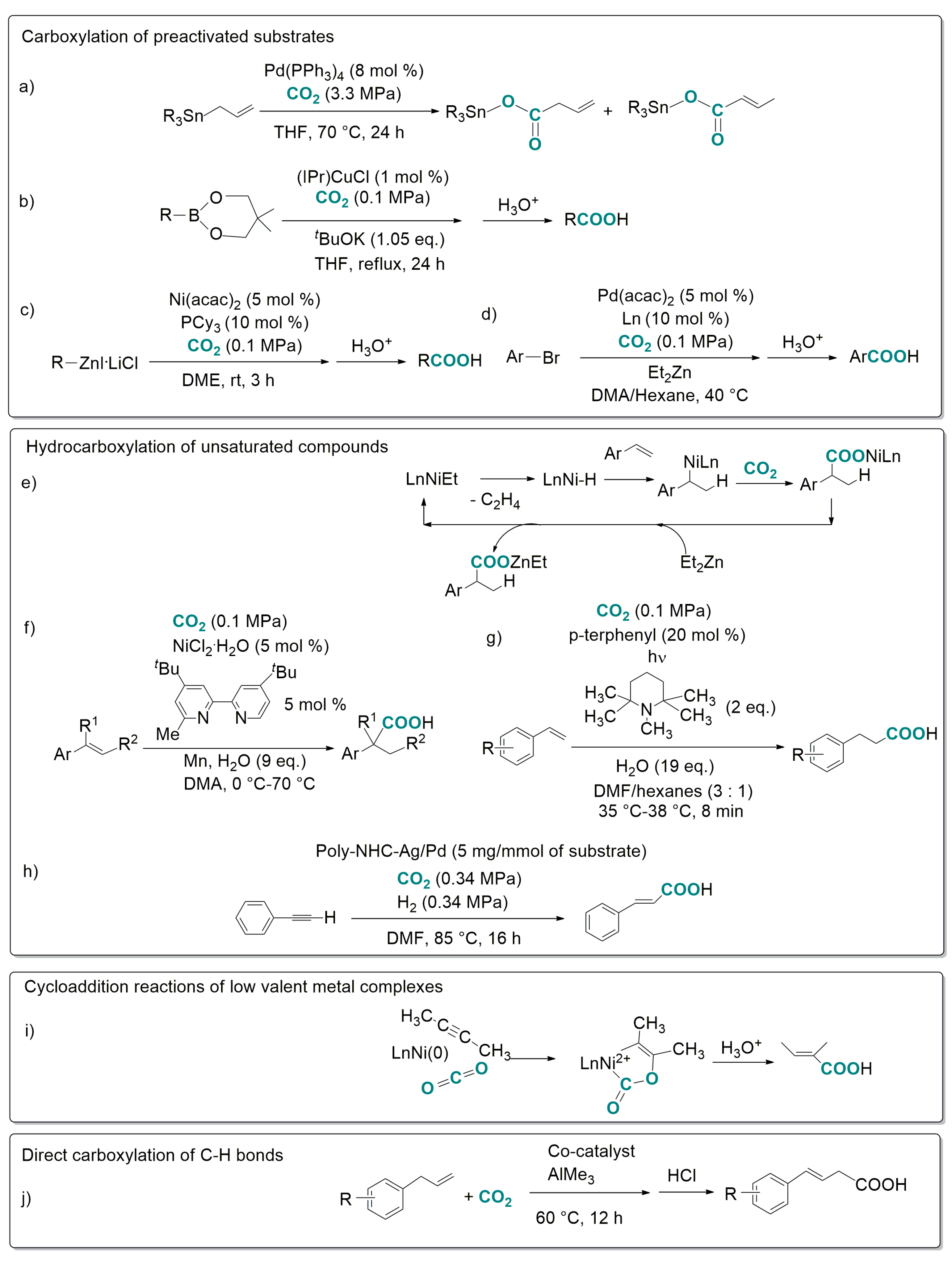
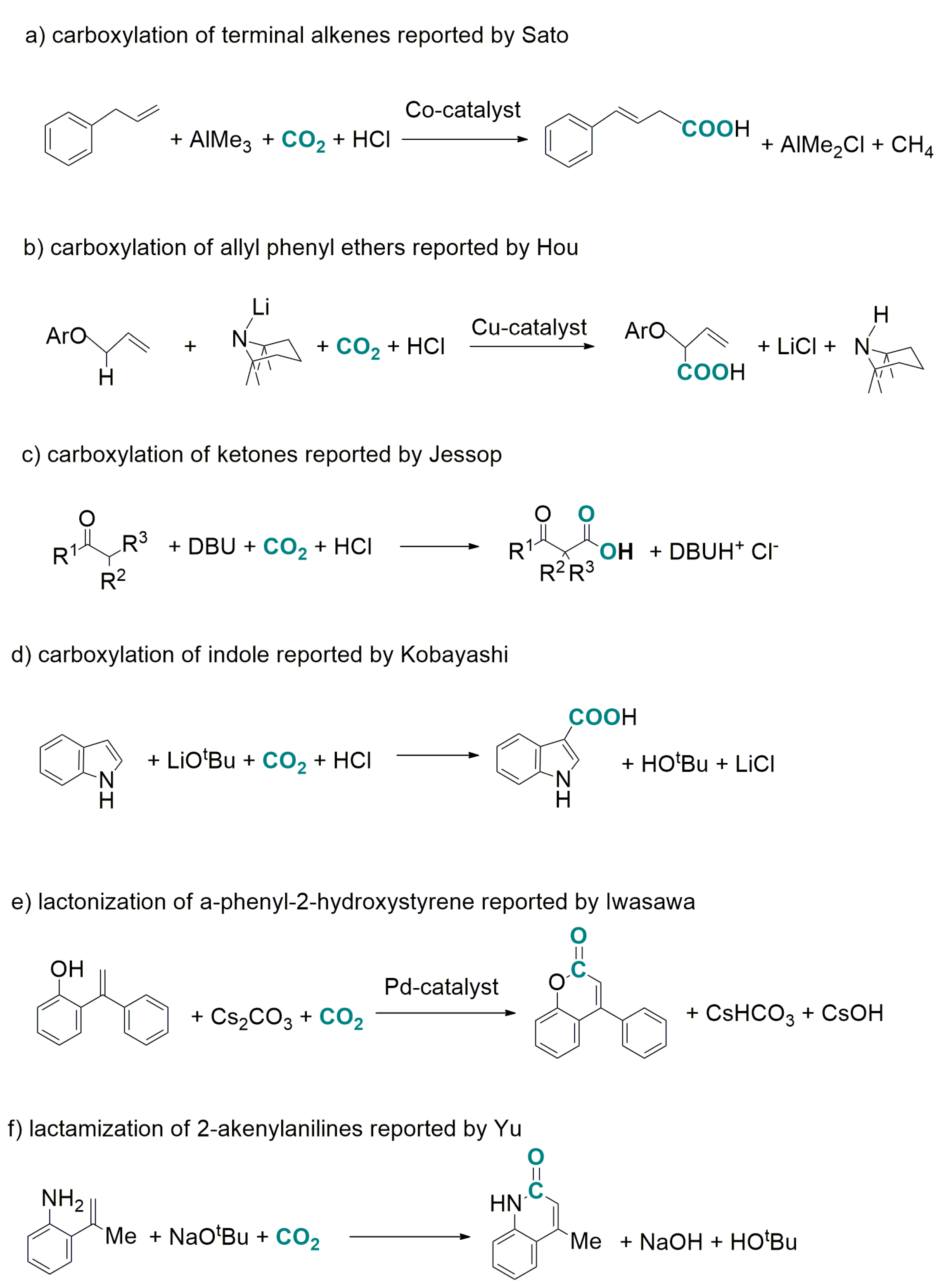
- transition-metal-catalysed hydrocarboxylation of unsaturated compounds (olefins, allenes, alkynes) requiring AlEt3 or ZnEt2 co-reactants to generate a metal-hydride species undergoing insertion of the unsaturated substrate and subsequent carboxylation of the organometallic intermediate [38,39,40,41,42] as exemplified in Scheme 1e. It is worth citing very recent examples of hydrocarboxylation reactions involving substrate reduction by a “formal hydride donor”: (i) in Scheme 1f, [43] hydride is formally generated from H2O and Mn; (ii) in Scheme 1g, [44] the substrate is reduced by photo-induced transfer of 2e− and 2H+ from a sacrificial amine; (iii) in Scheme 1h, [45] the substrate is carboxylated and subsequently reduced with H2 in a Poly-NHC/Ag/Pd mediated process;
- (3)
- (4)
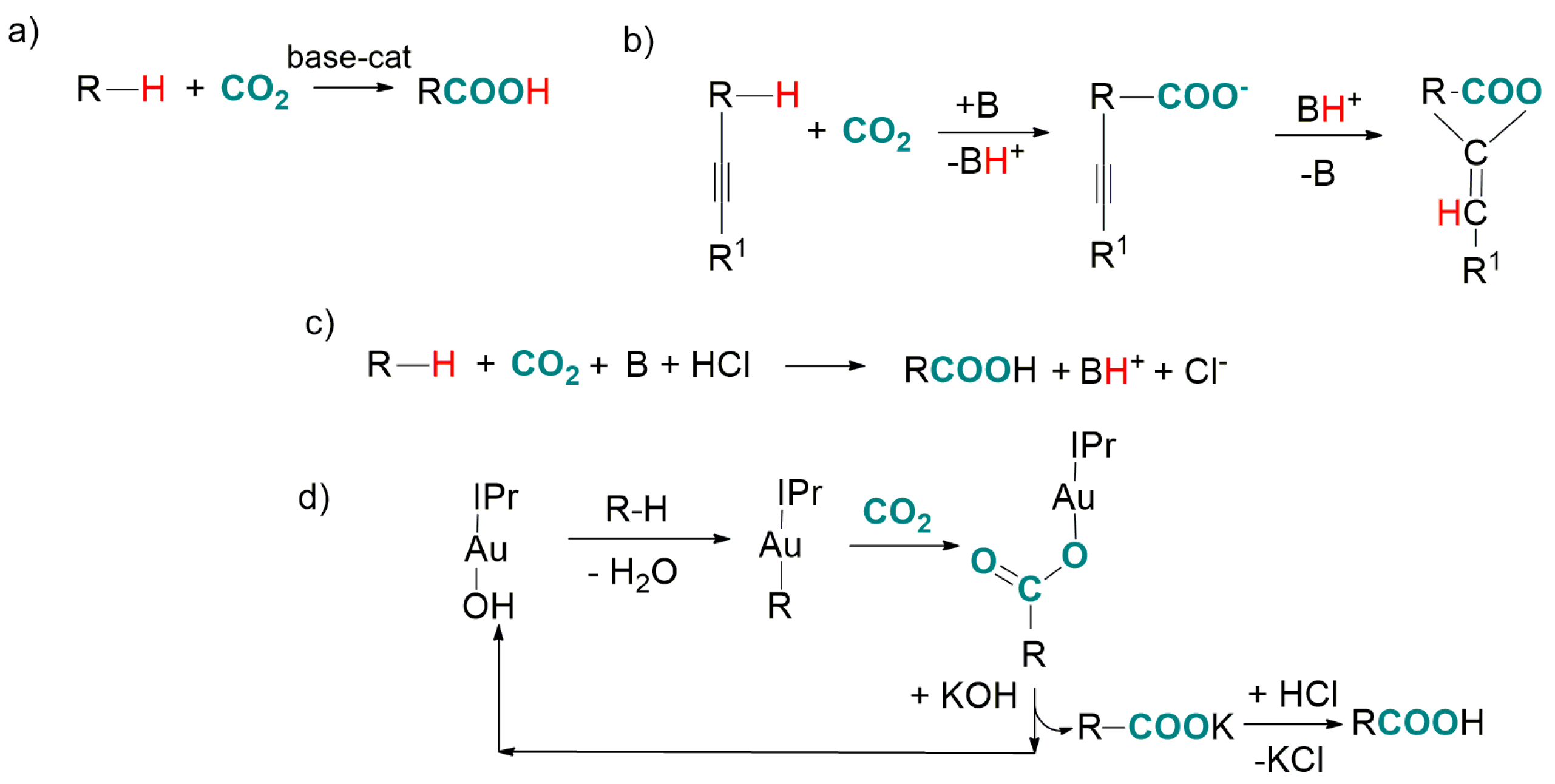
2. Transition-Metal-Catalyzed Carboxylation of C(sp3)-H Bonds with CO2
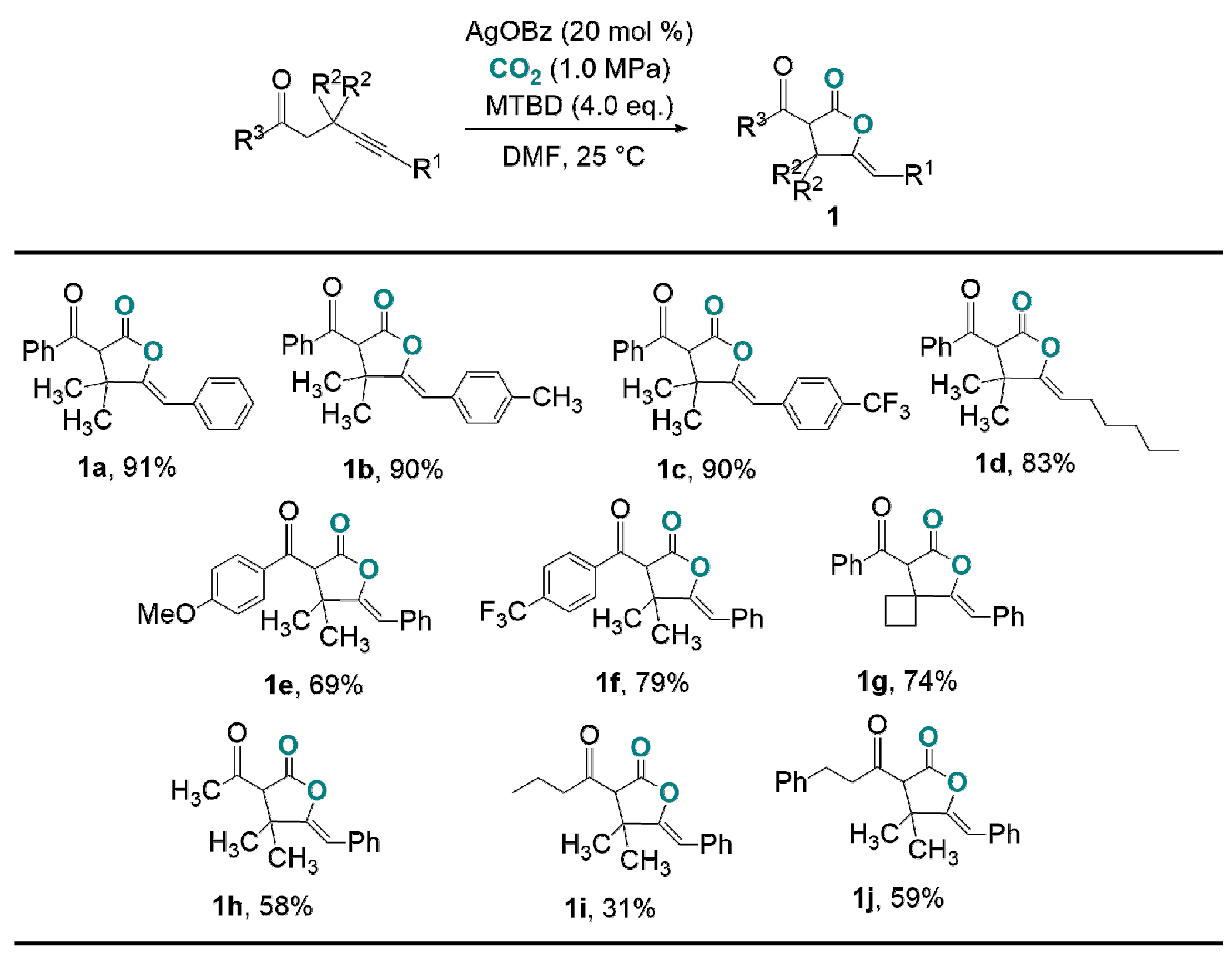
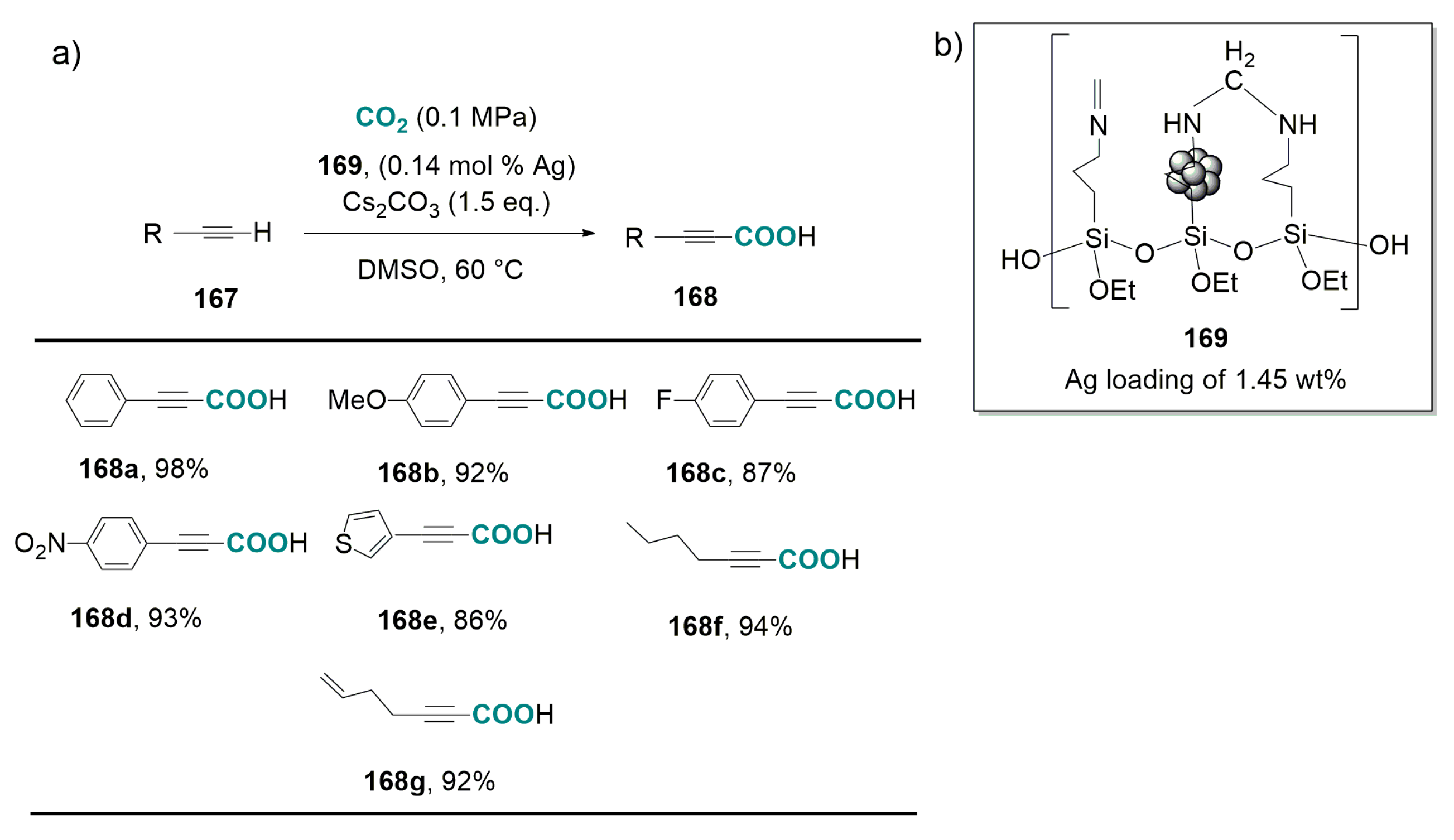
2.1. Carboxylation of Compounds Possessing Activated C(sp3)-H Bonds Catalyzed by Ag-Salts in Conjunction with Strong Bases
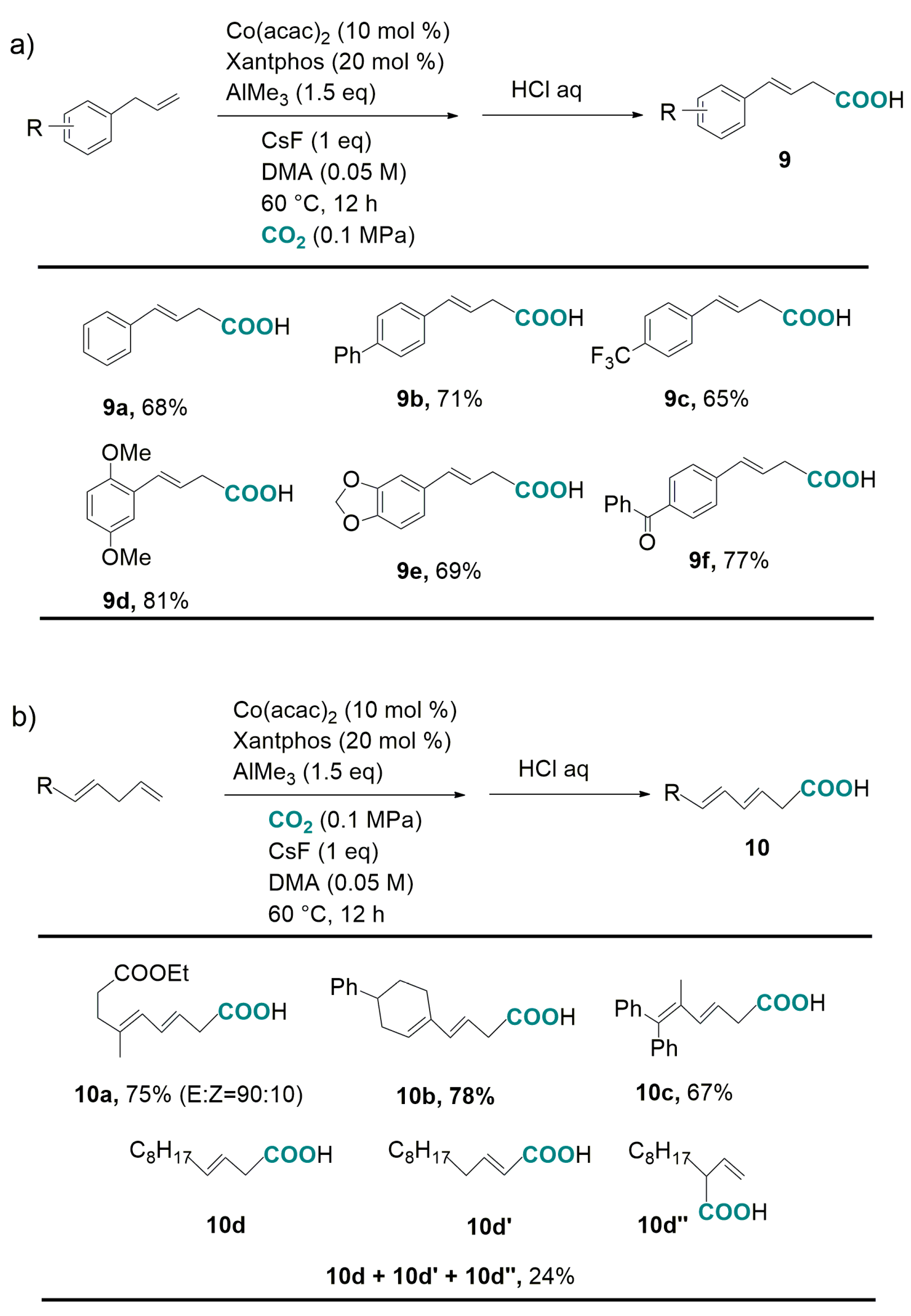
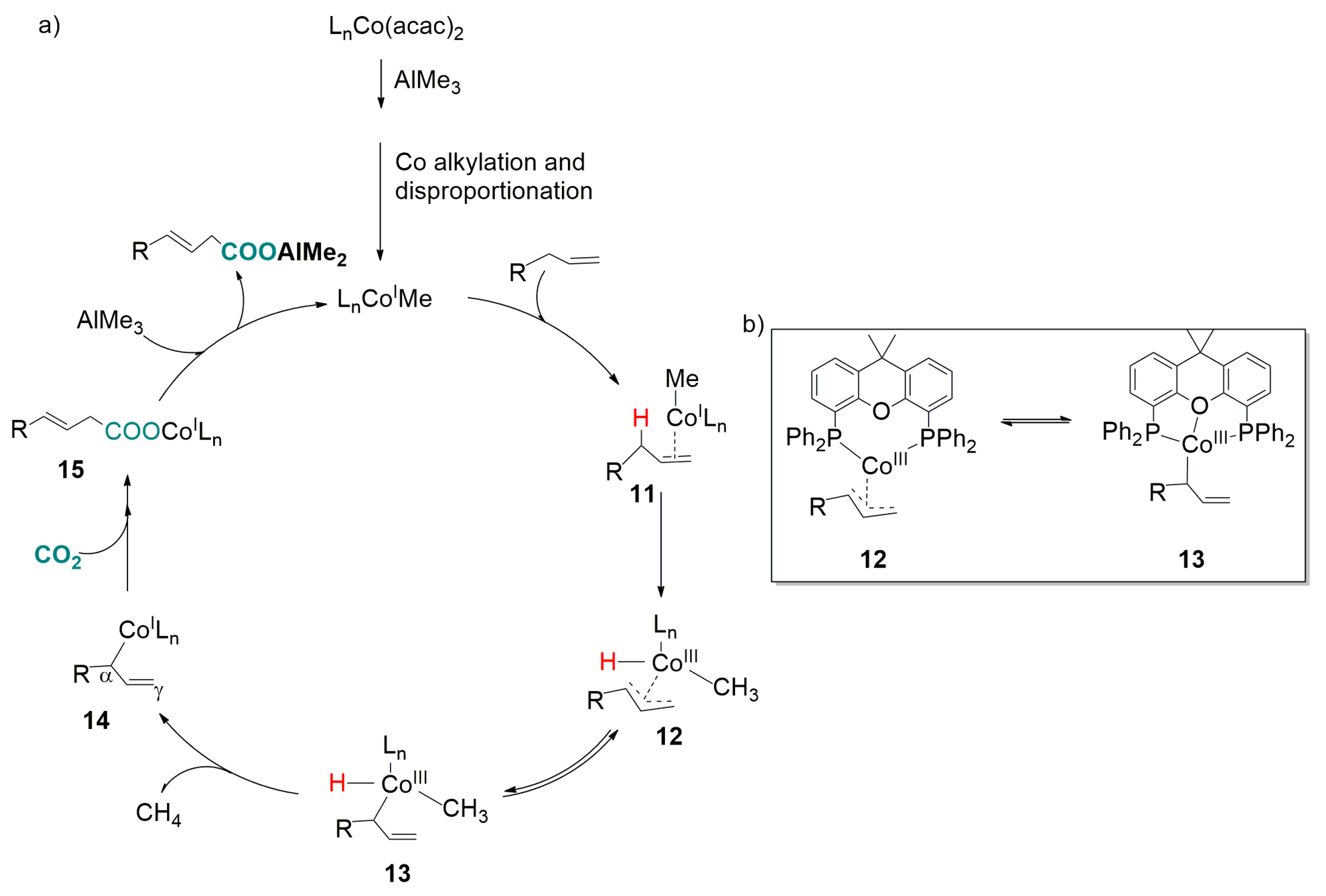
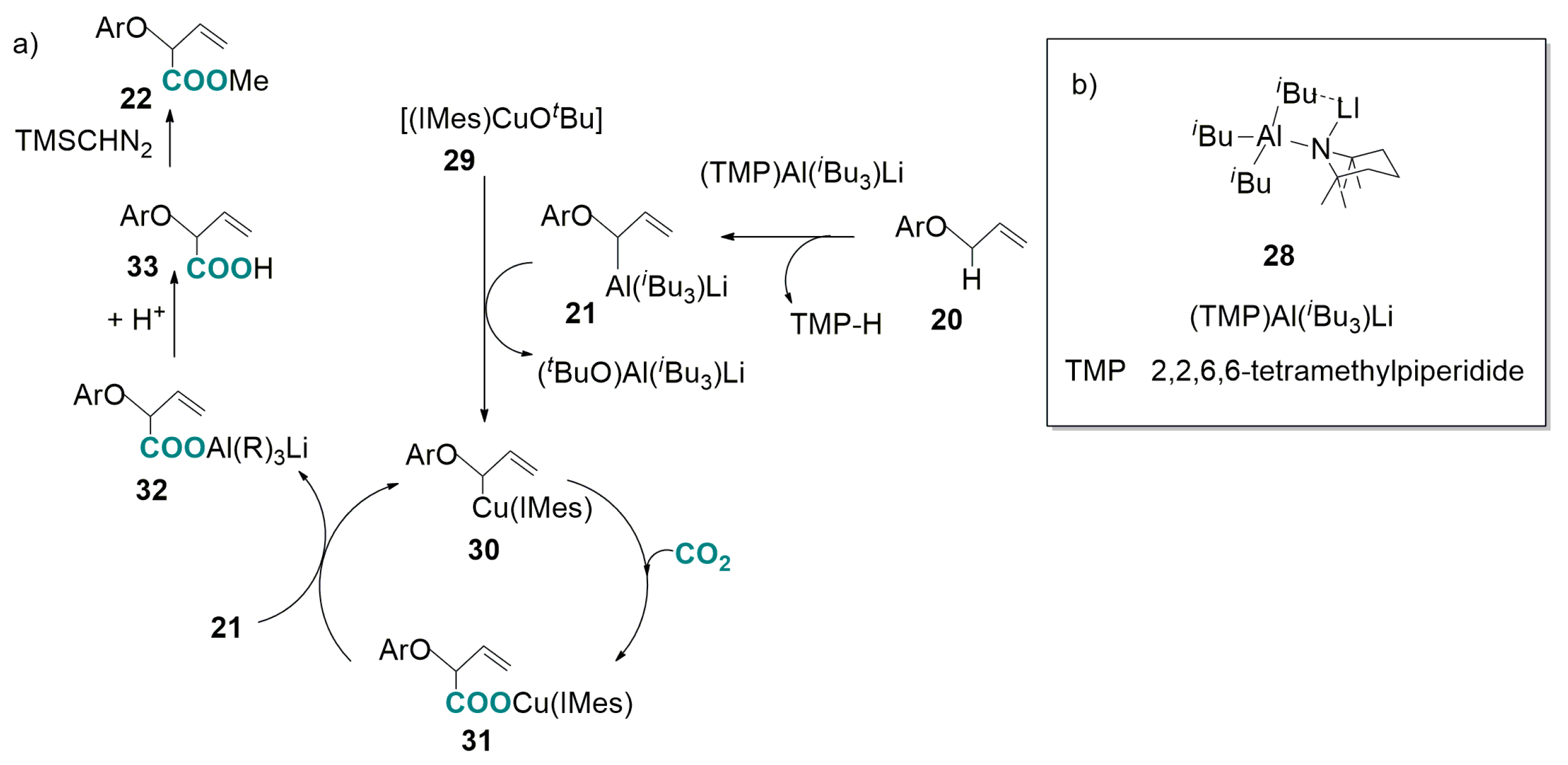
2.2. Carboxylation of Benzylic and Allylic C(sp3)-H Bonds Catalyzed by Transition-Metal-Complexes
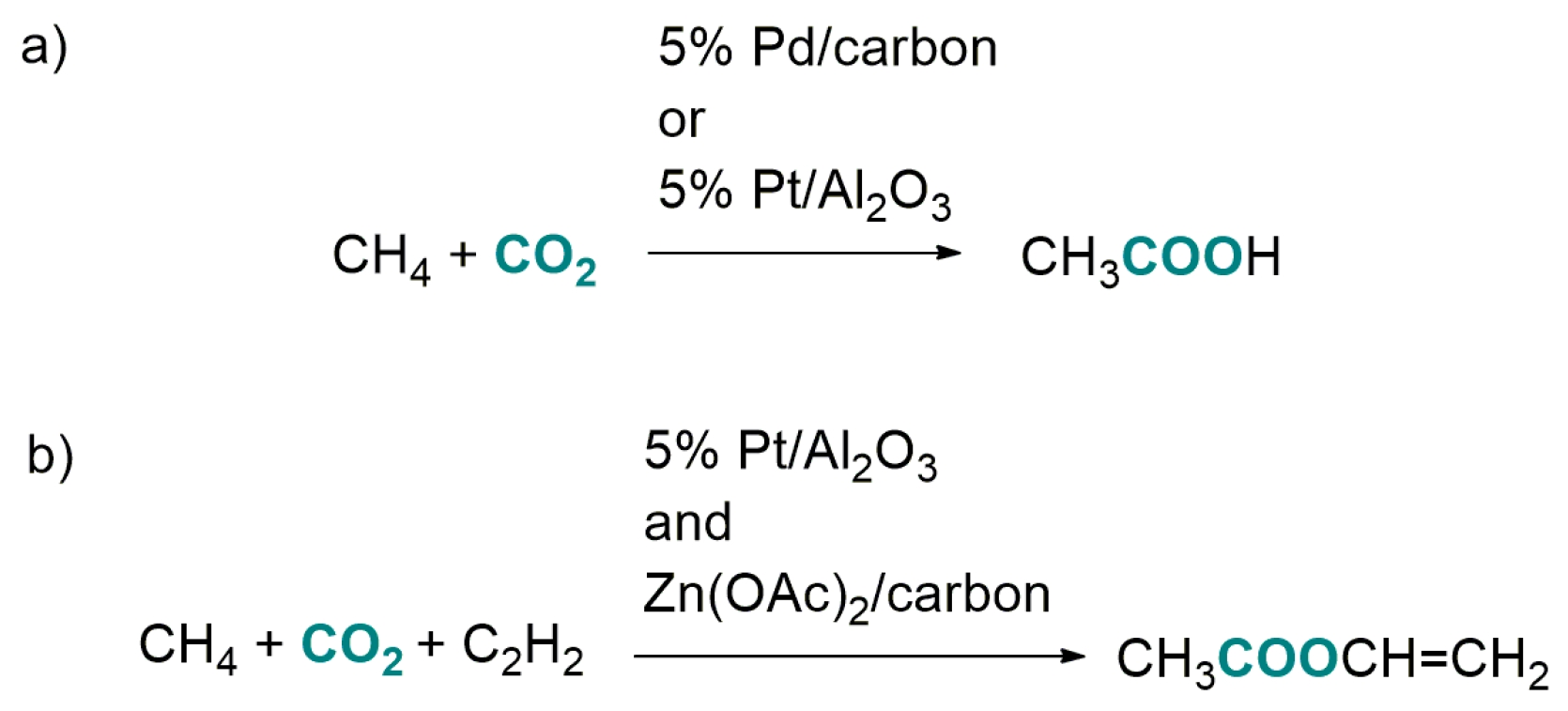
2.3. Carboxylation of CH4 with CO2 by Heterogeneous Catalysis
3. Light-Driven CO2-Based Carboxylation Reactions of C(sp3)-H Bonds
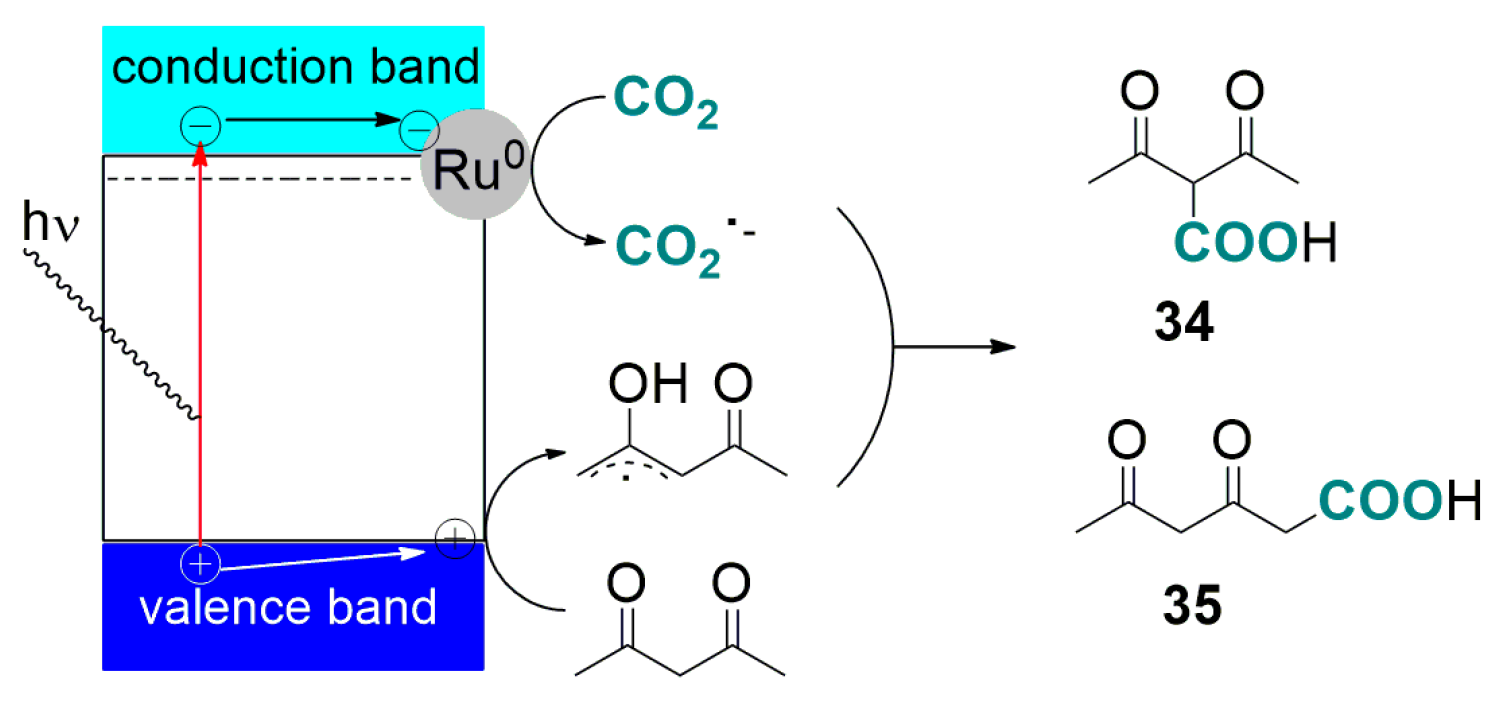
3.1. Photo-Catalysed Carboxylation of Acetylacetone with CO2
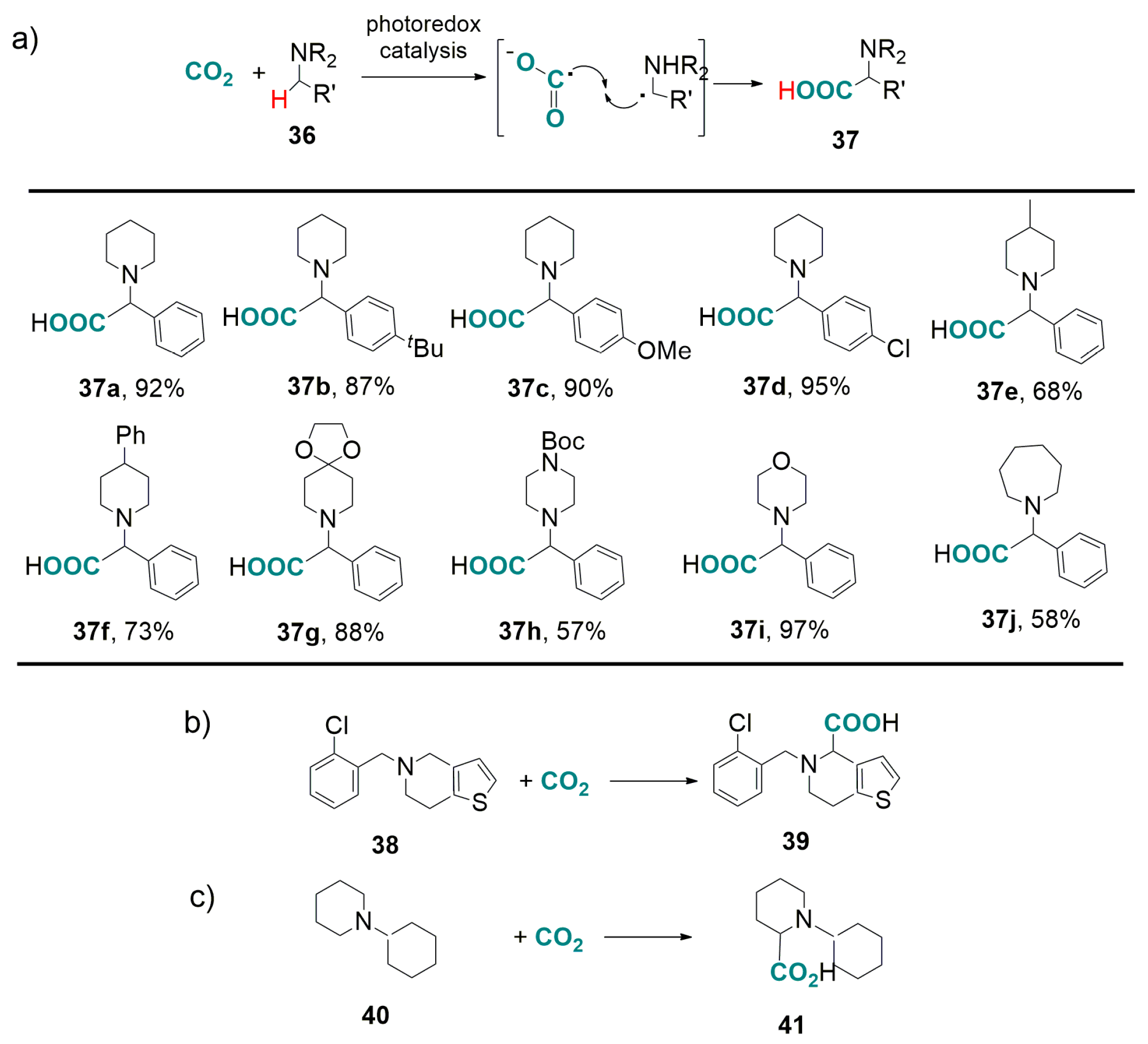
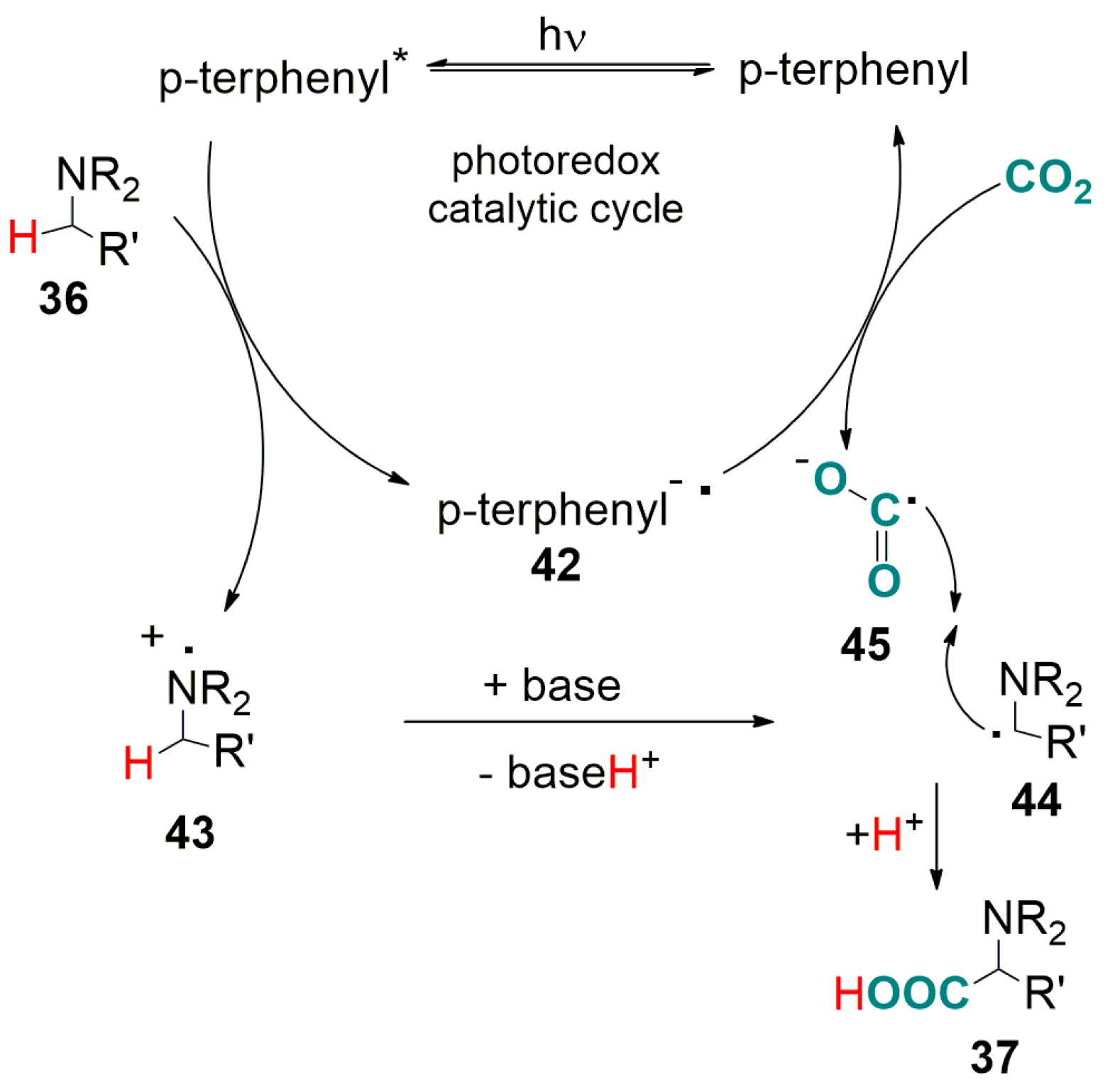
3.2. Light-Driven Carboxylation of Benzylic and Allylic C(sp3)-H Bonds with CO2
4. Brønsted-Base-Mediated Carboxylation of C(sp3)-H Bonds with CO2
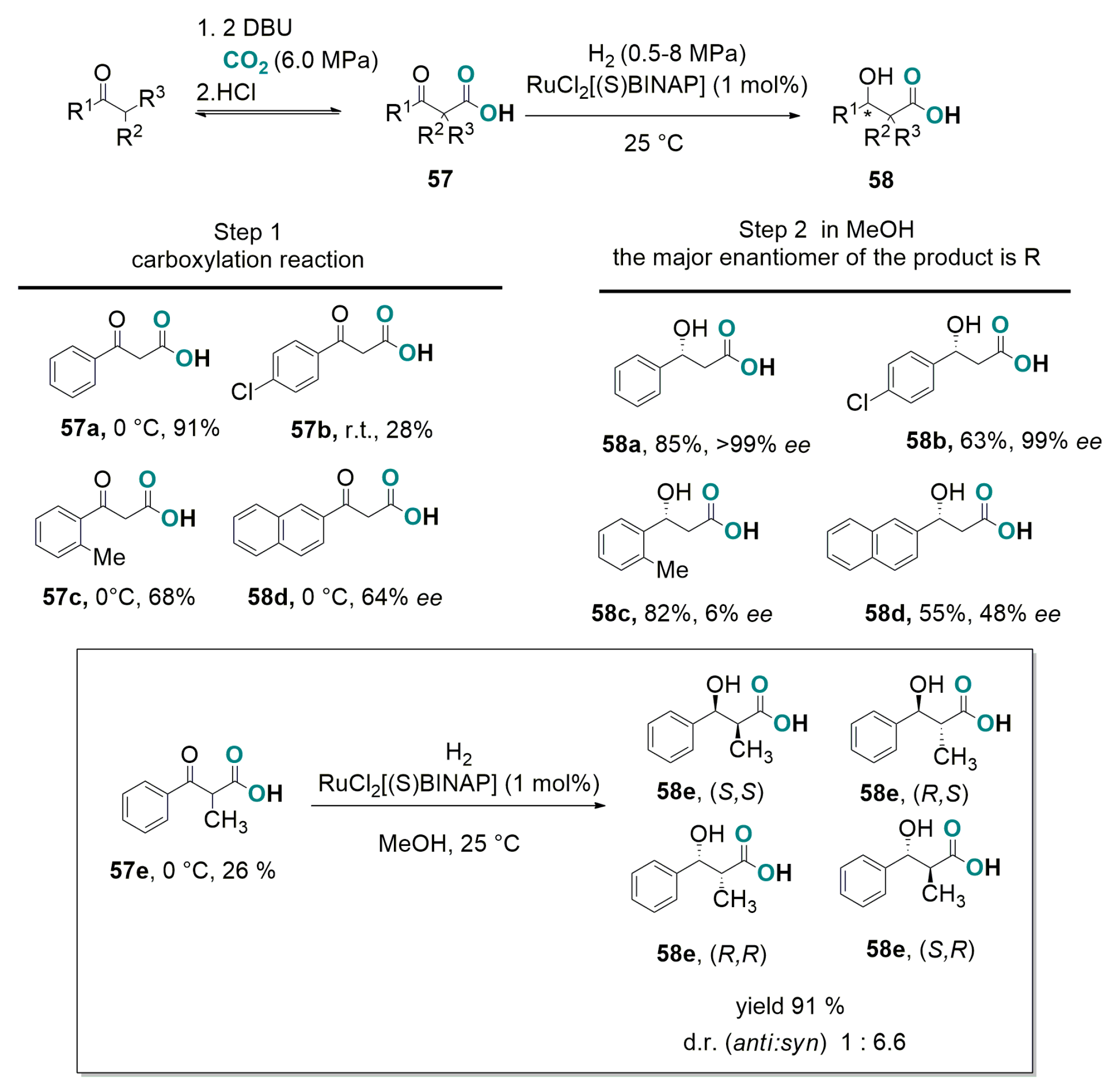
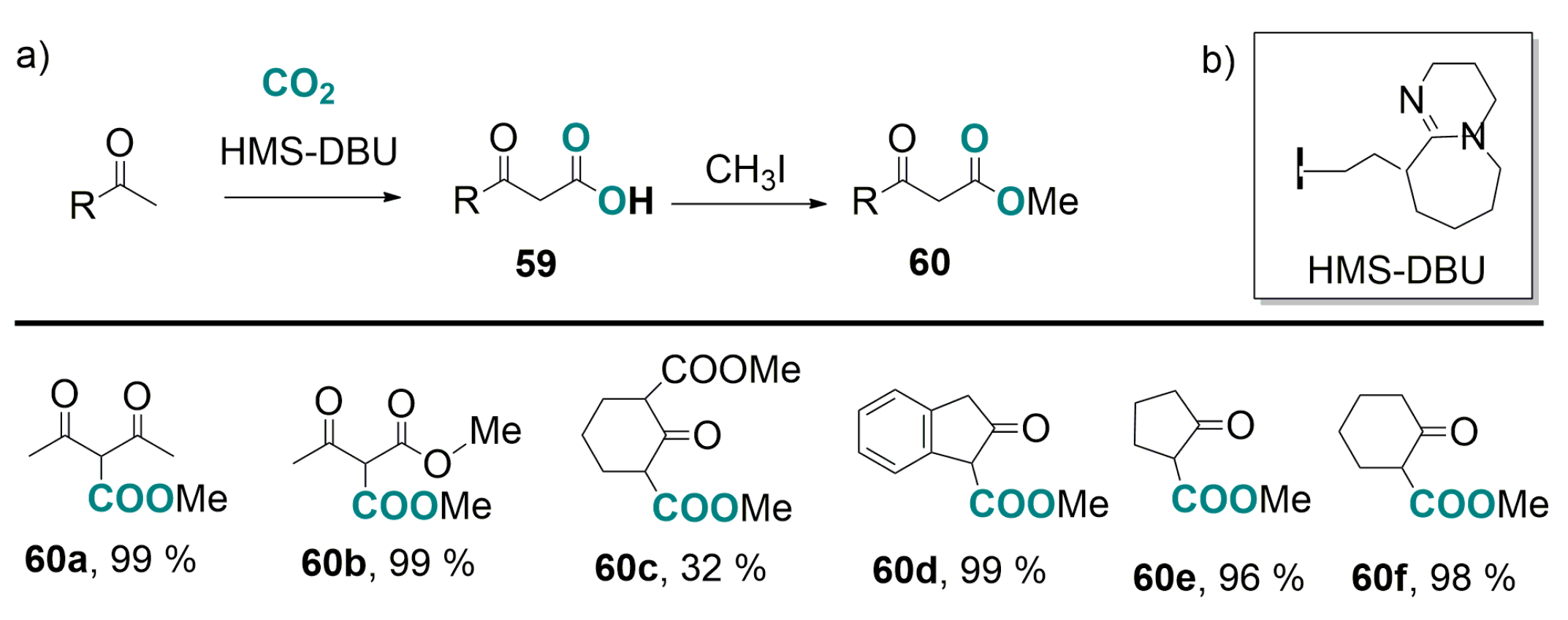
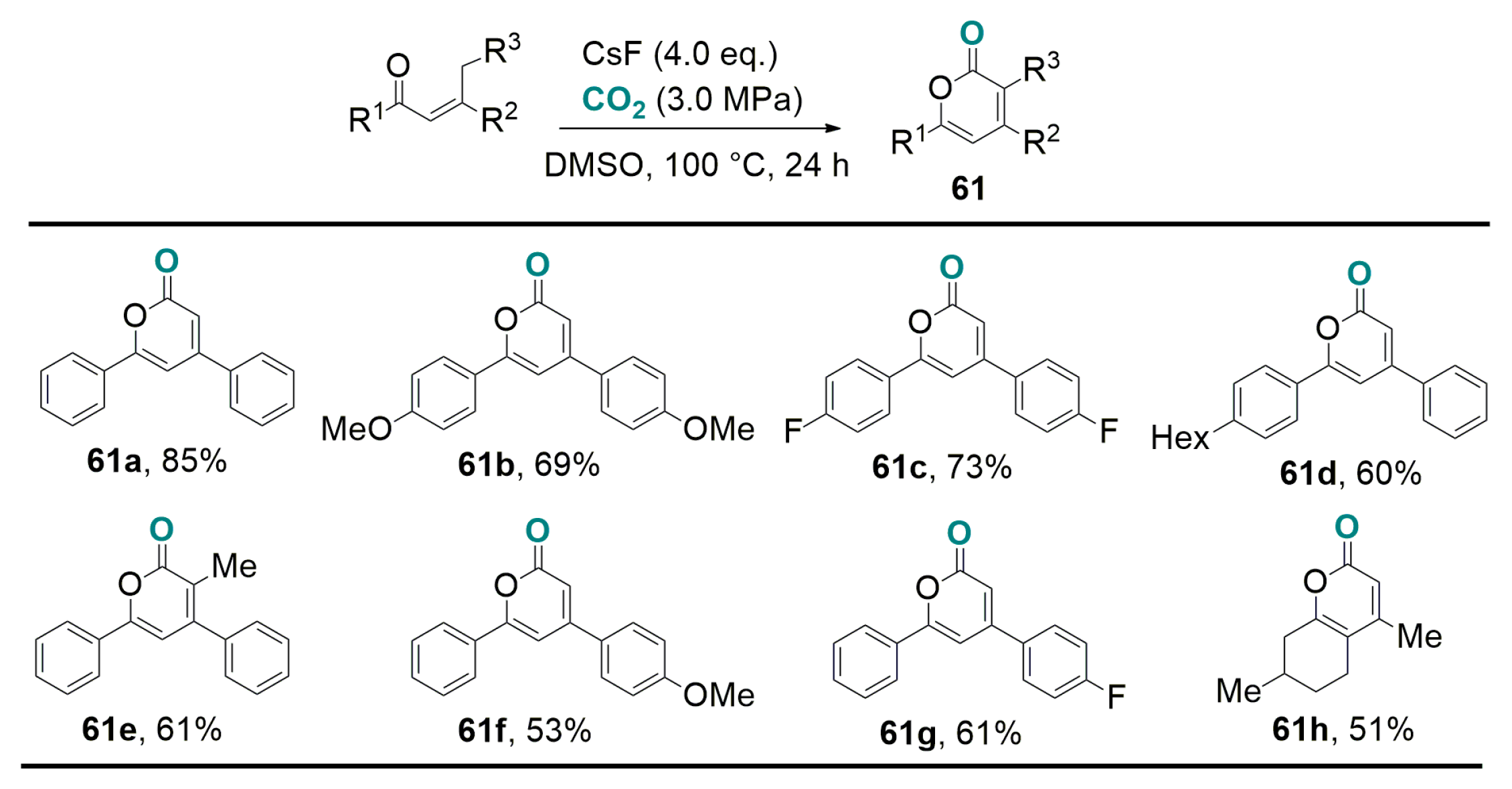
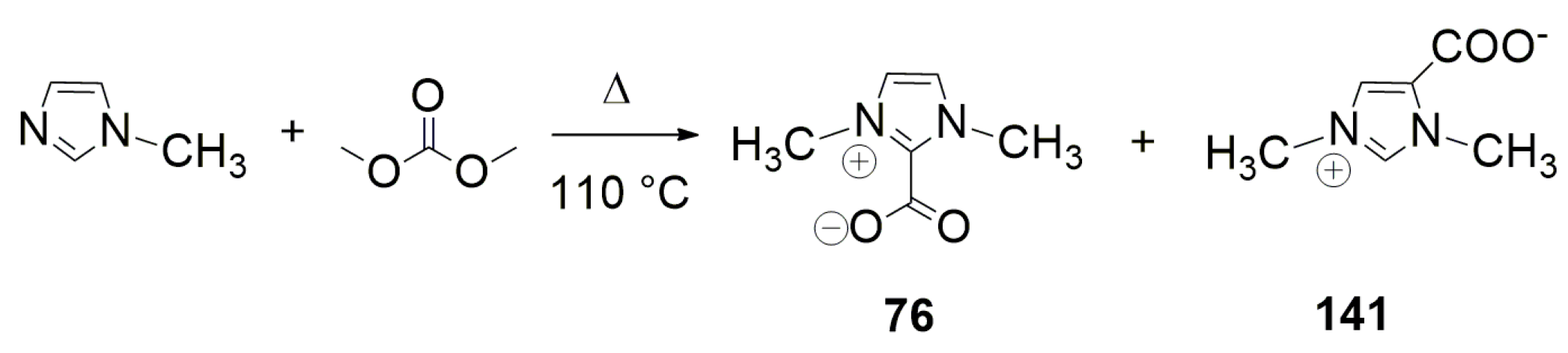
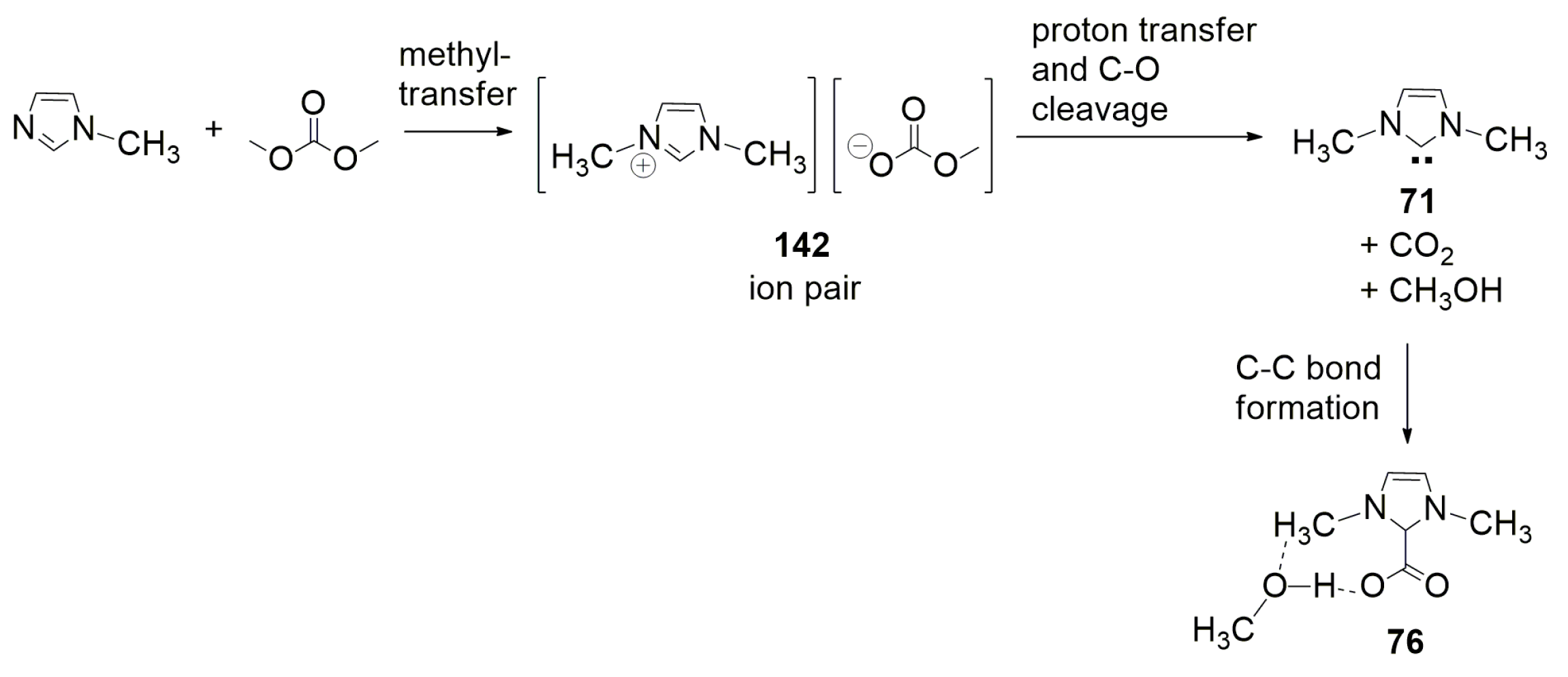
4.1. Recent Advances in Brønsted-Base Mediated Carboxylation of Acidic C(sp3)-H Bond with CO2
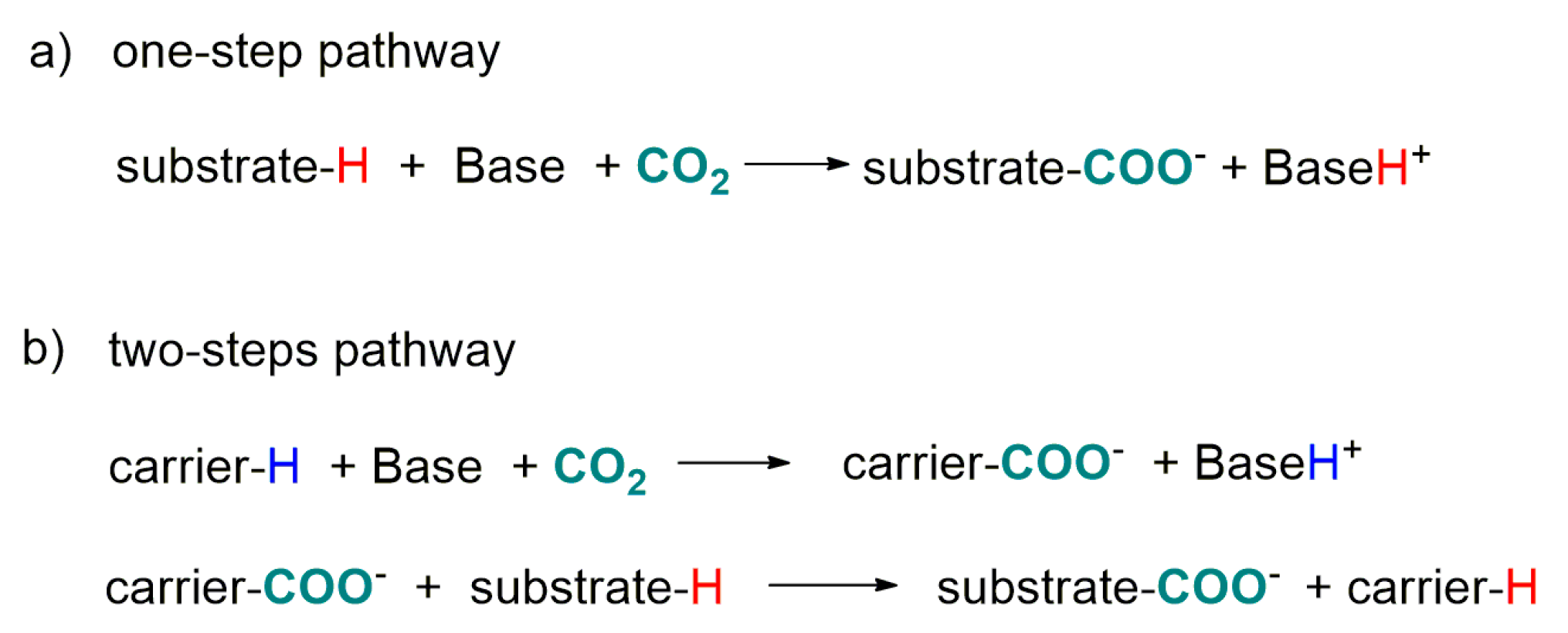
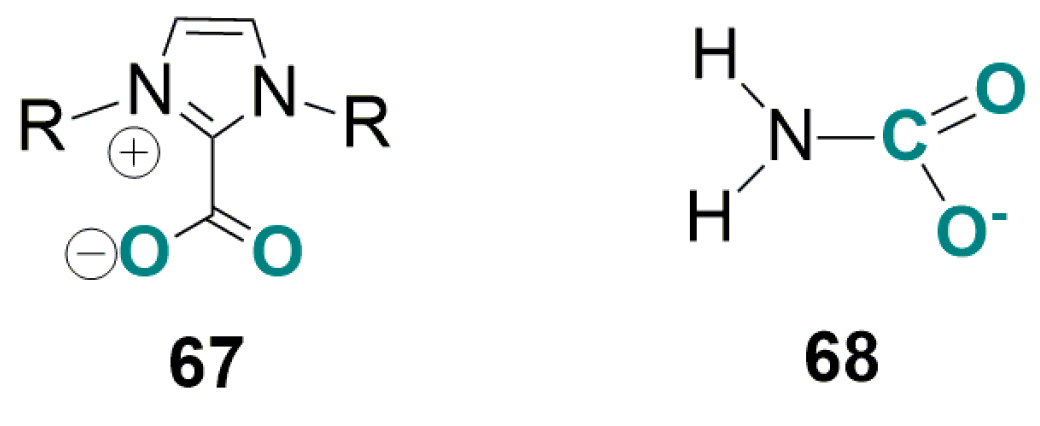
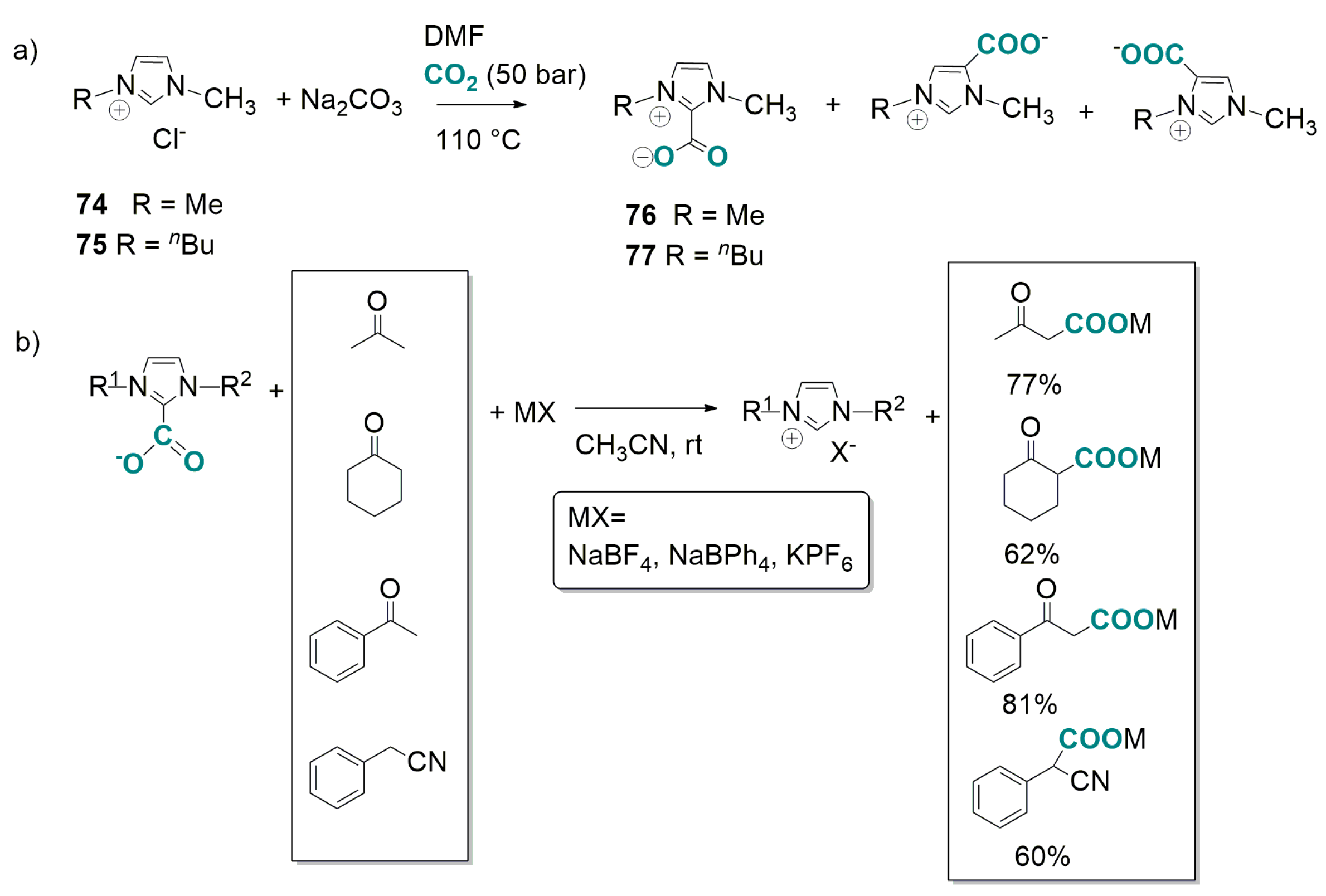
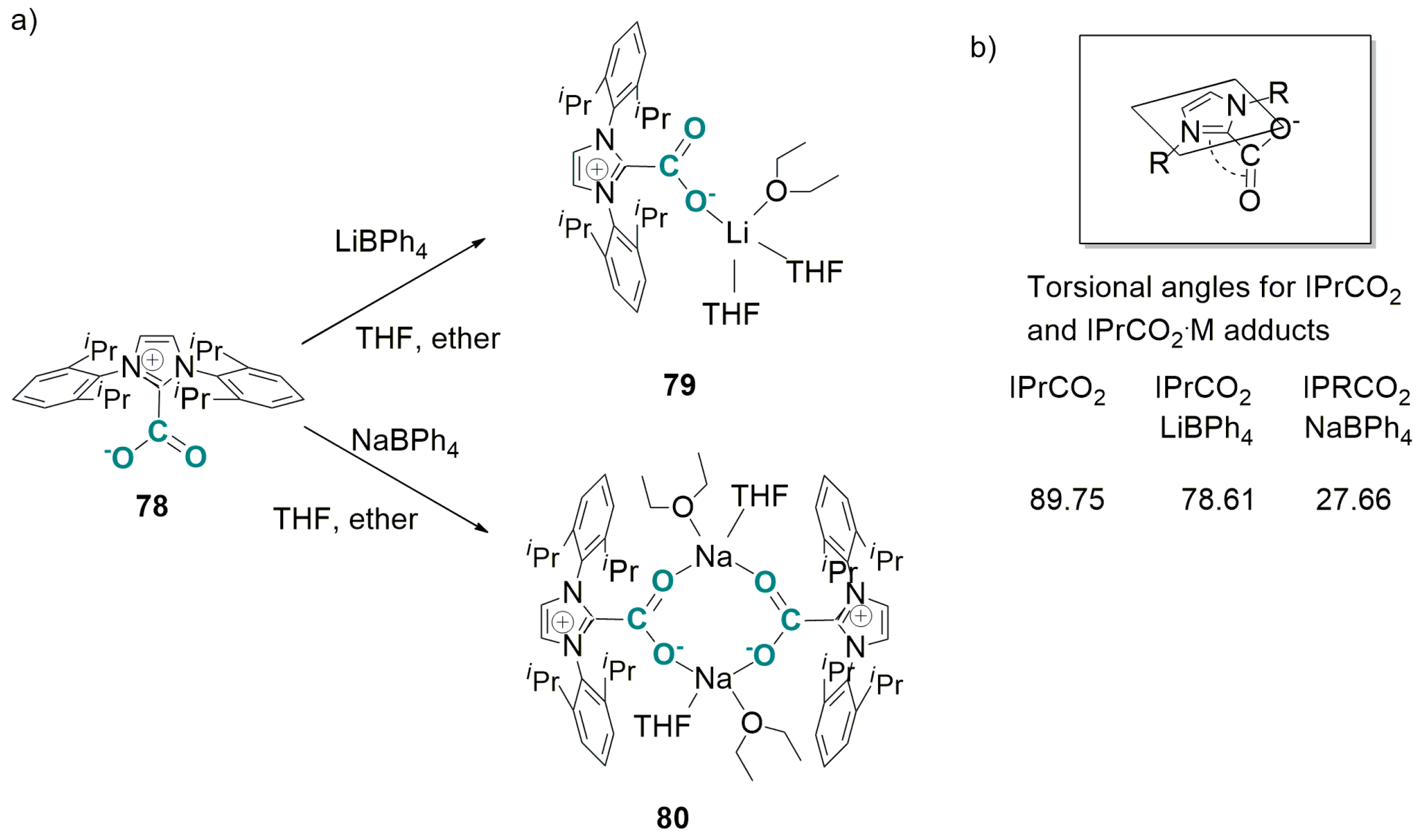
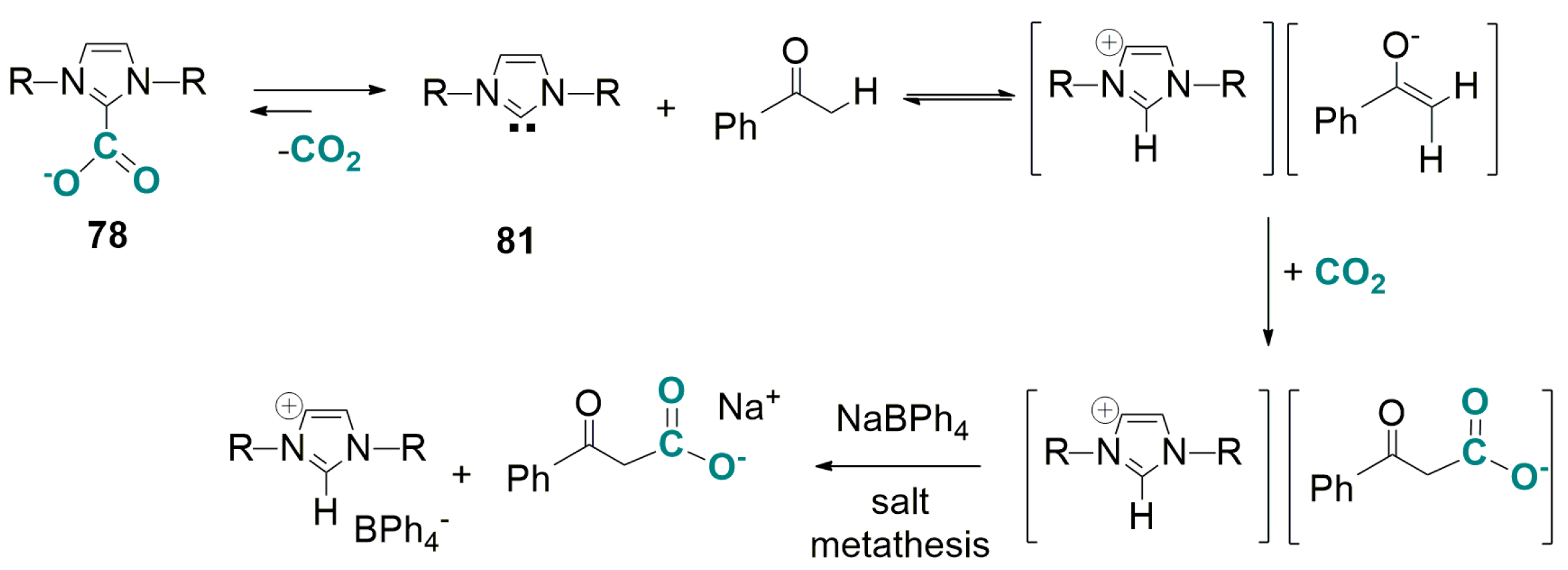
4.2. Carboxylation of C(sp3)-H Active Bonds by Using CO2 Carriers
5. Transition-Metal-Catalyzed Carboxylation of C(sp2)-H Bonds with CO2
5.1 Carboxylation of Alkenyl-C-H Bonds by Pd-Catalysis
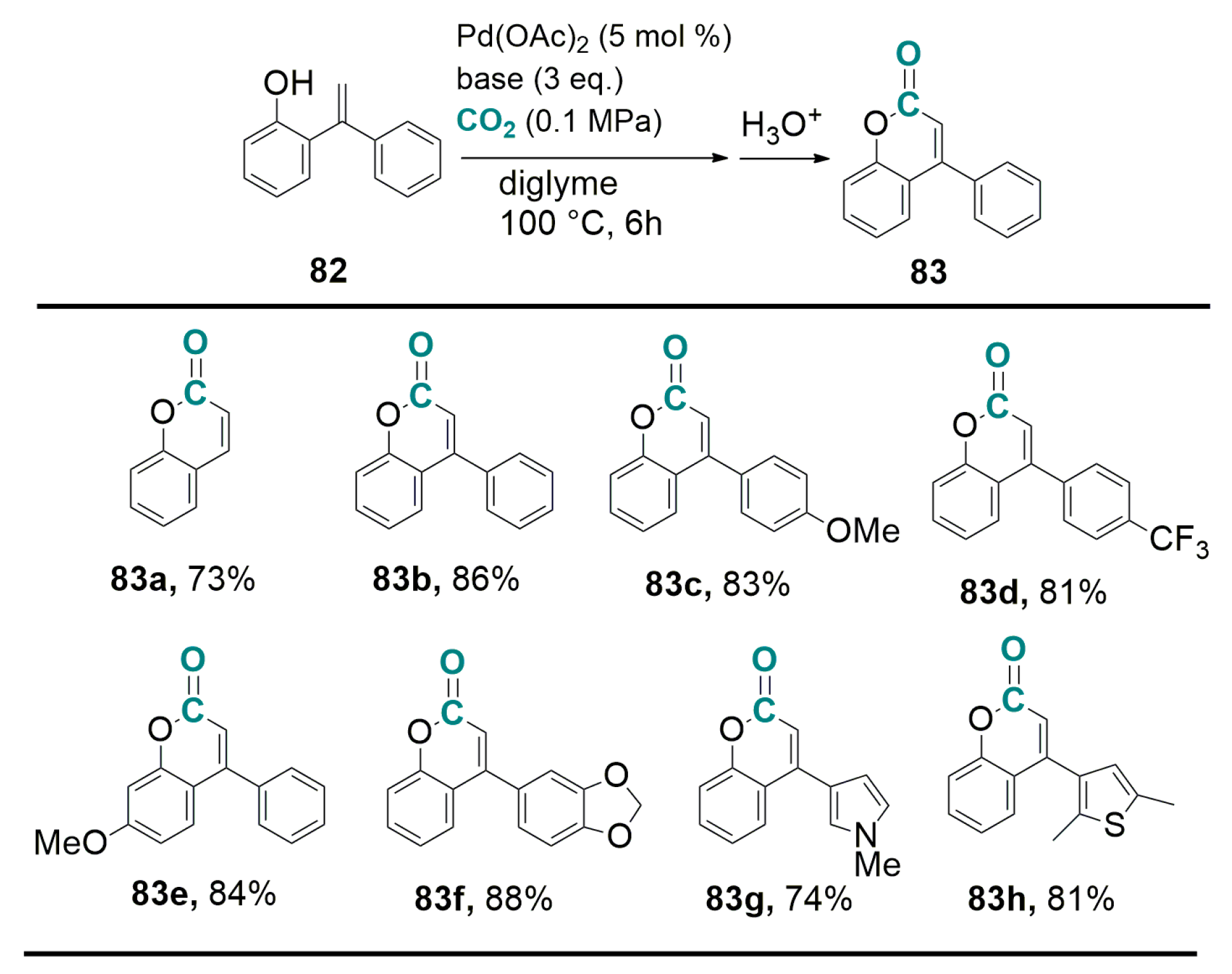
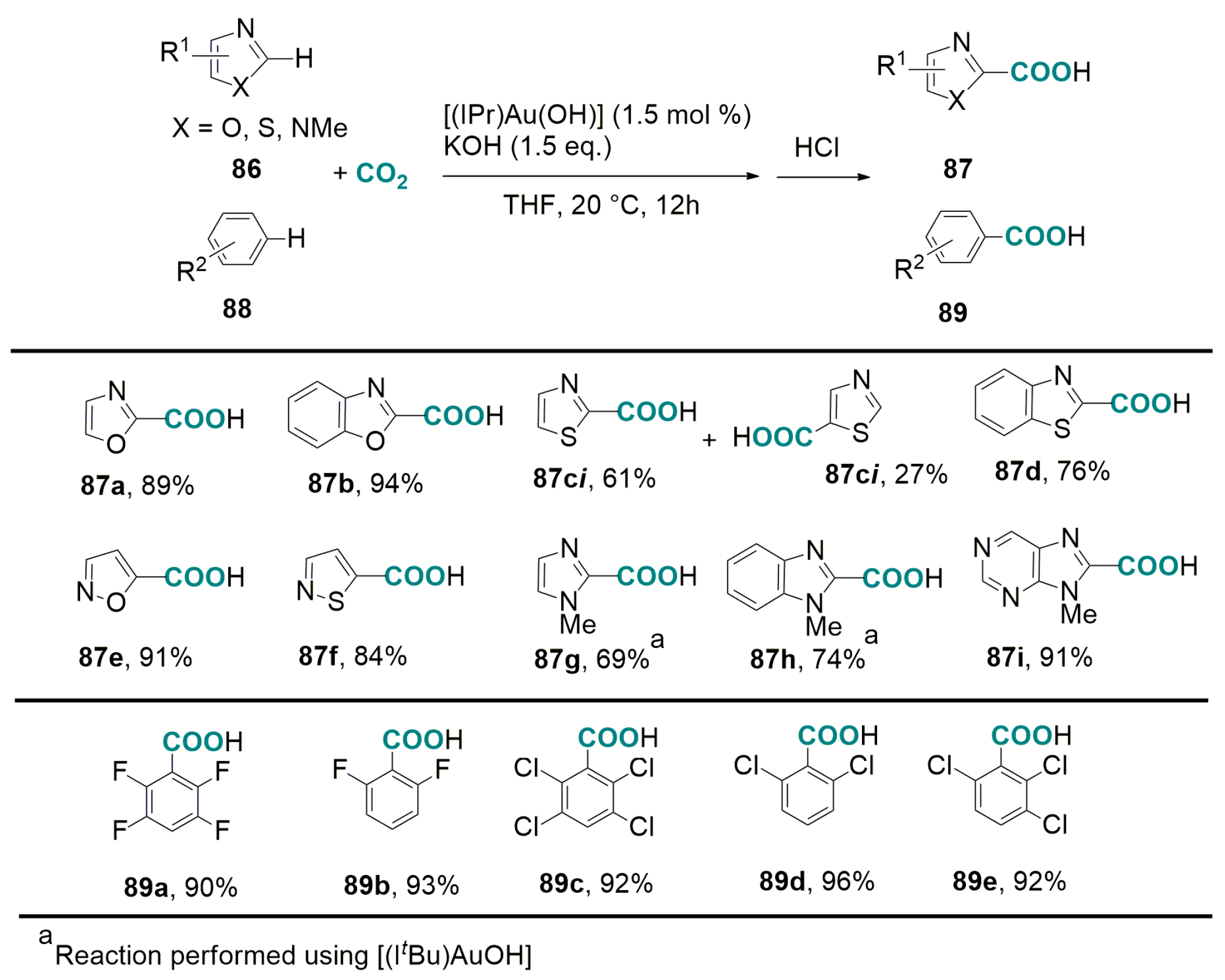
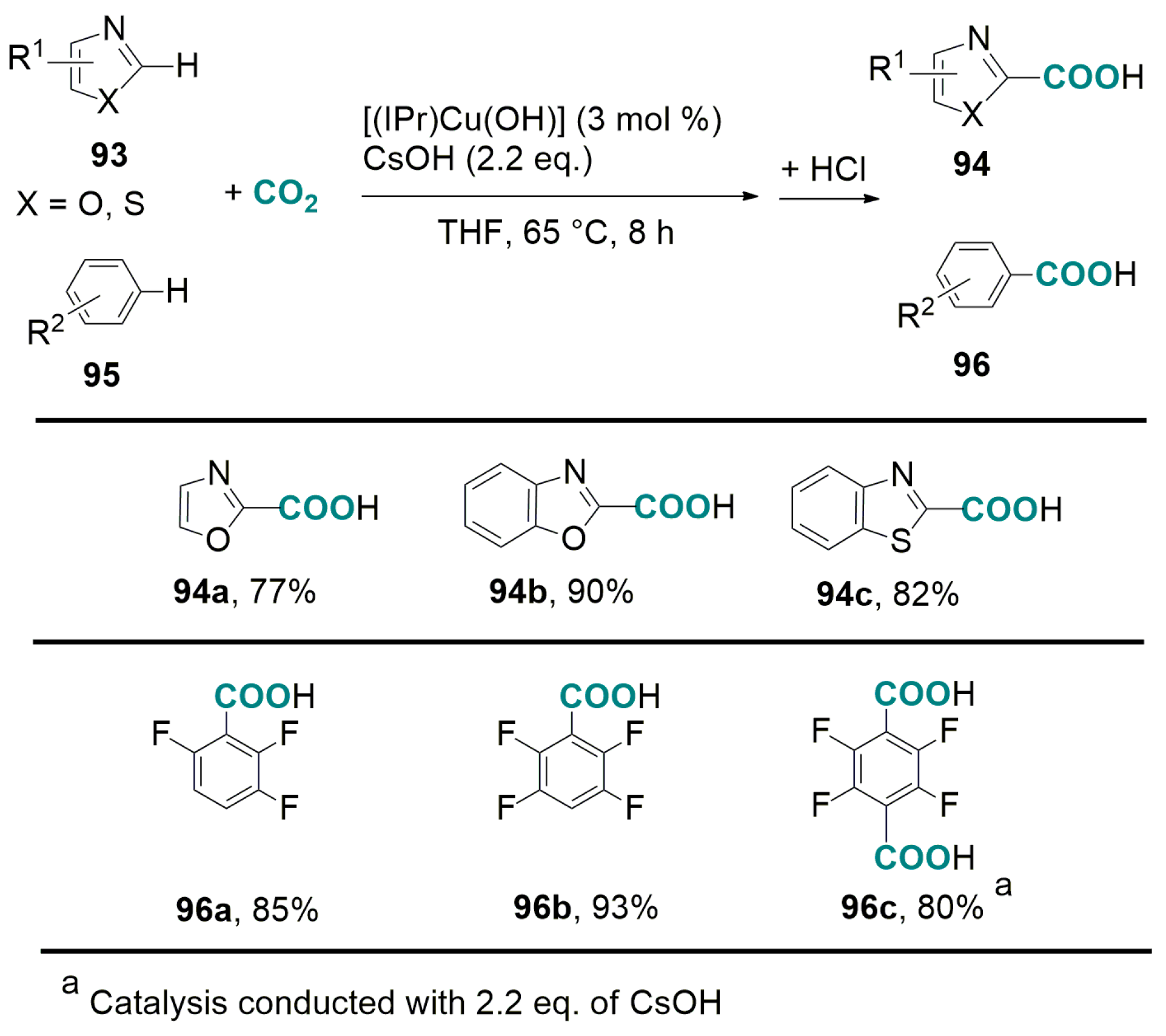
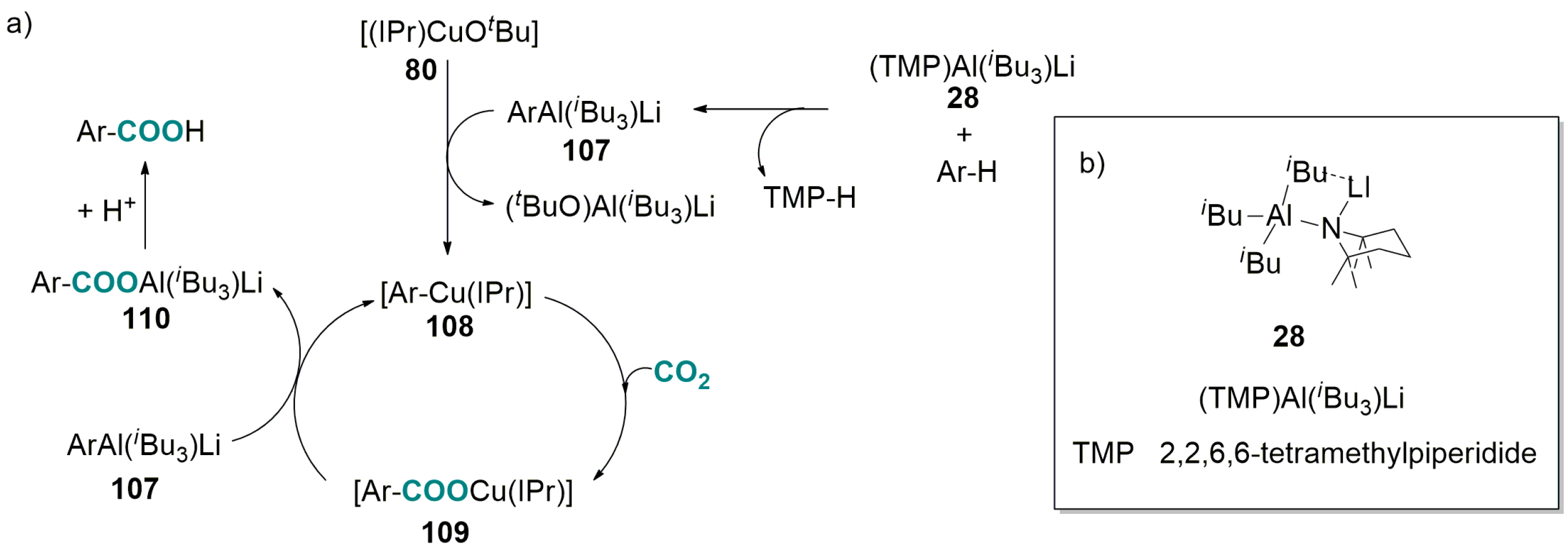
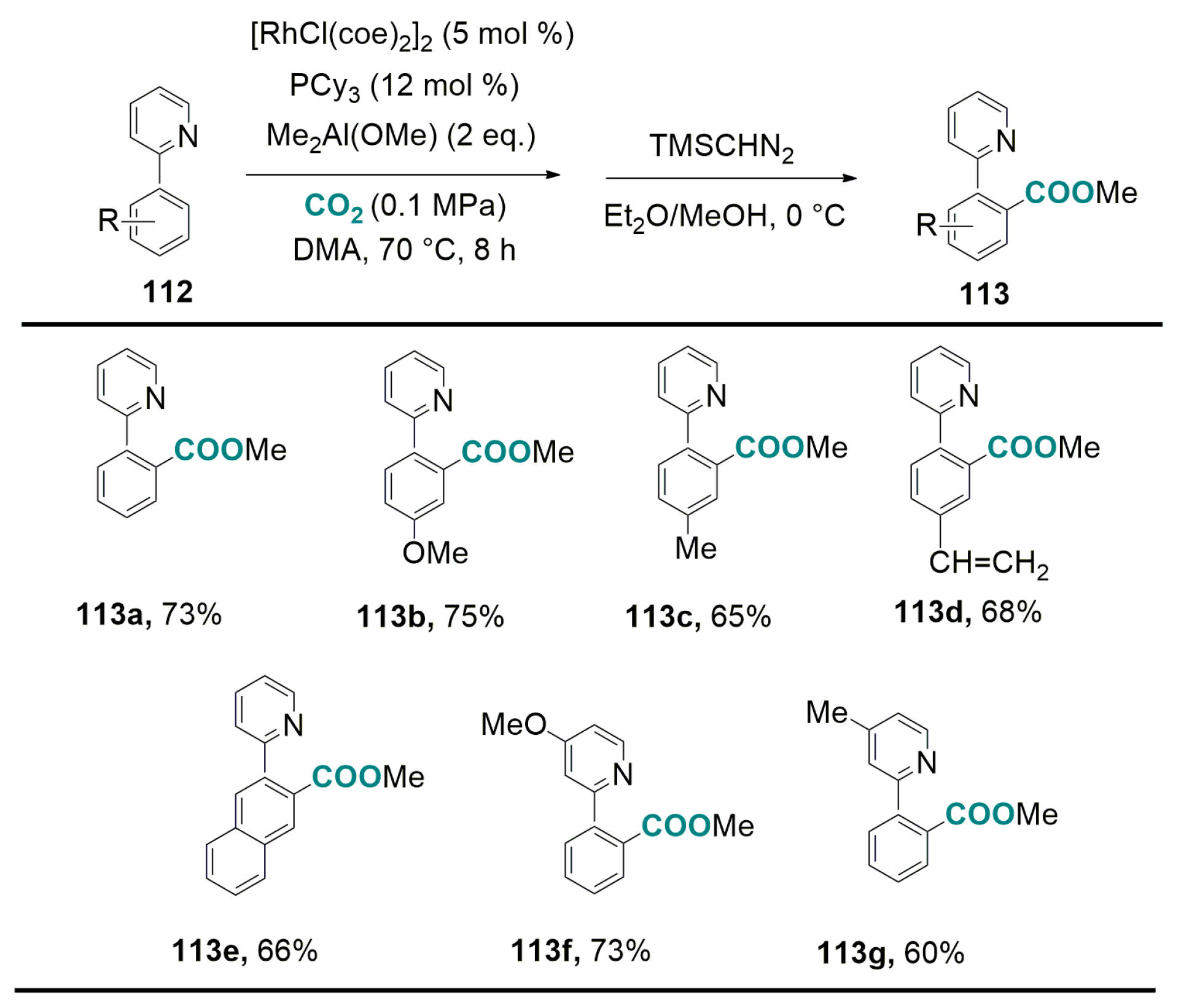
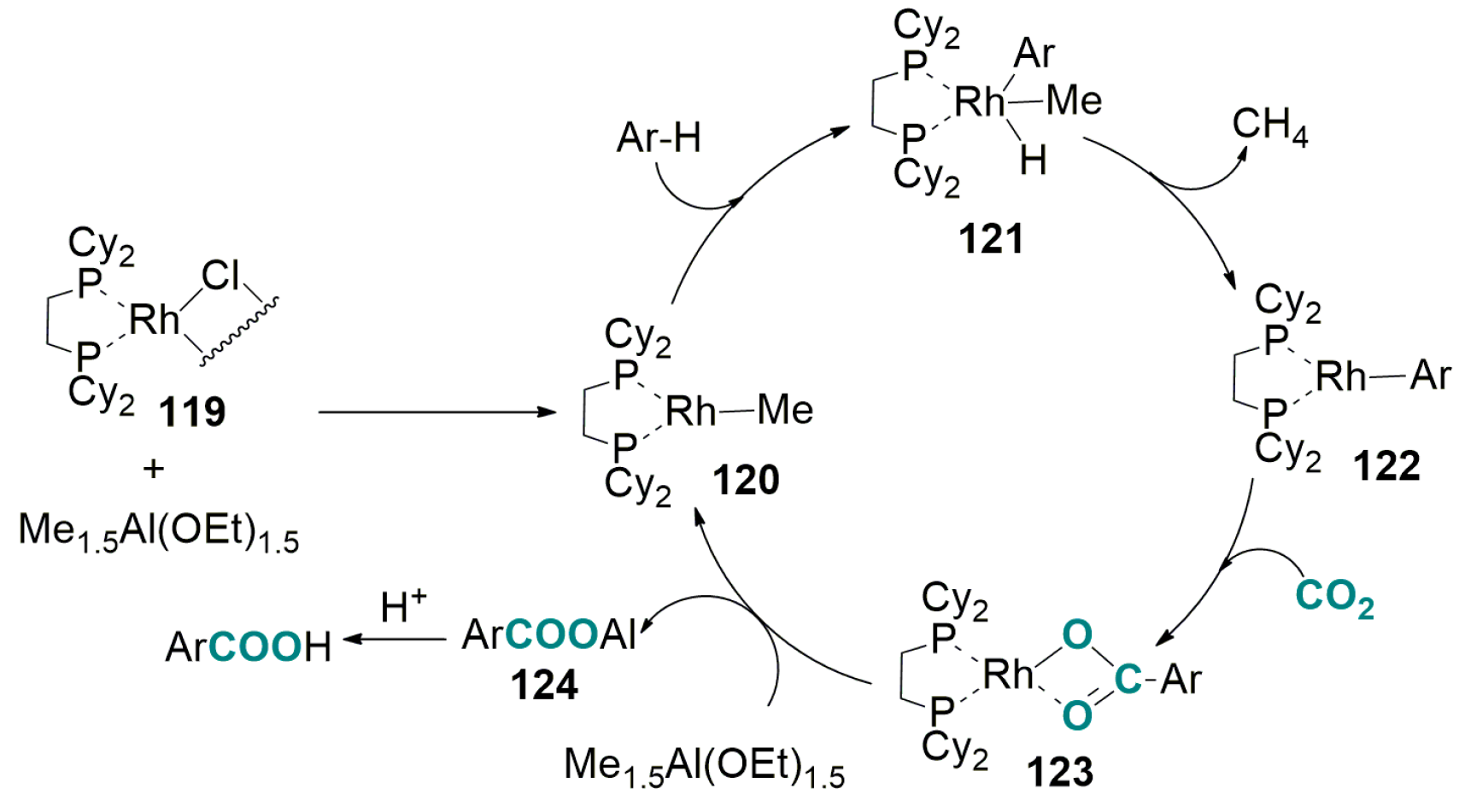
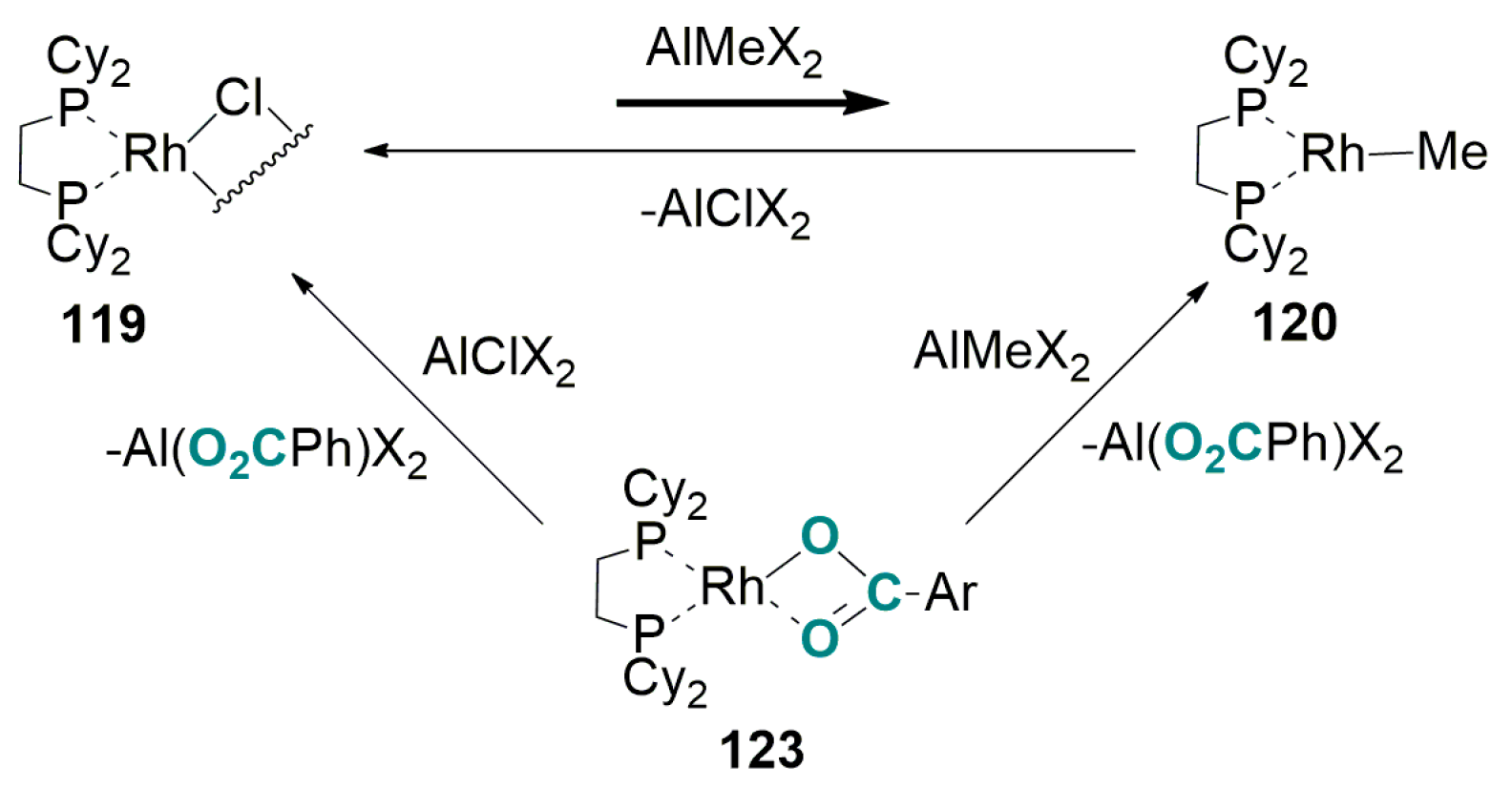
5.2. Carboxylation of Aromatic and Heteroaromatic Compounds
6. Brønsted Base-Mediated Carboxylation Reactions of C(sp2)-H Bonds with CO2
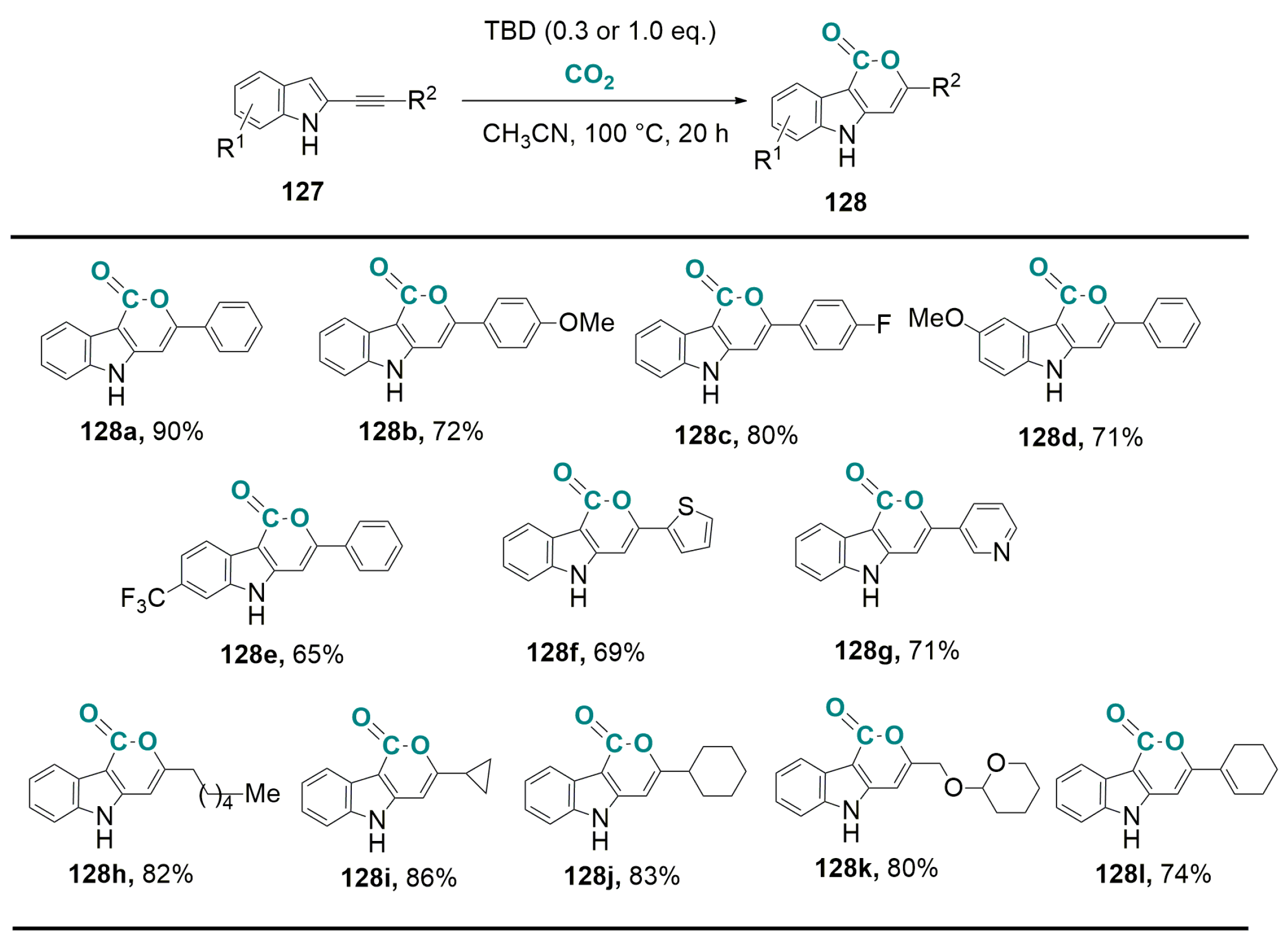
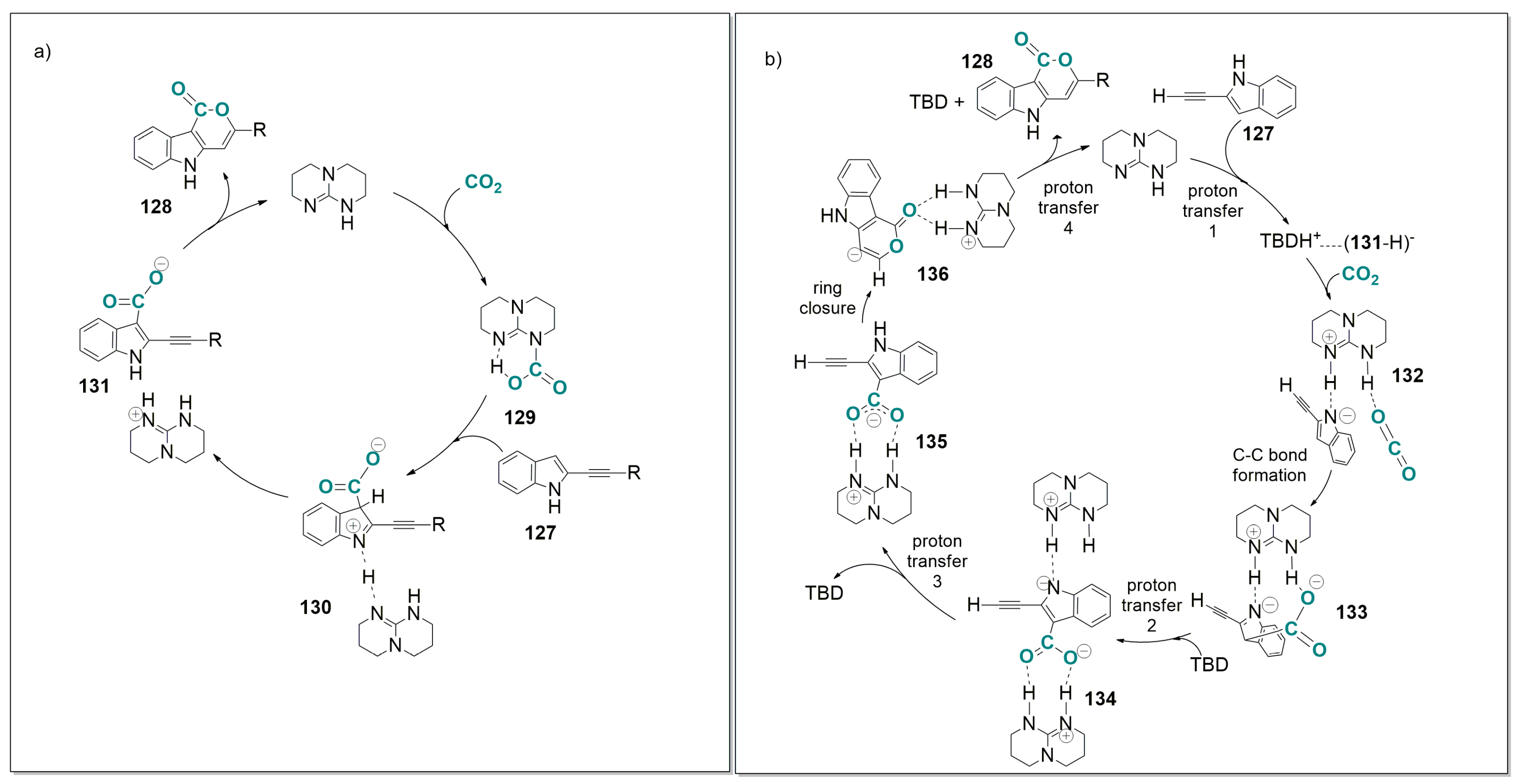
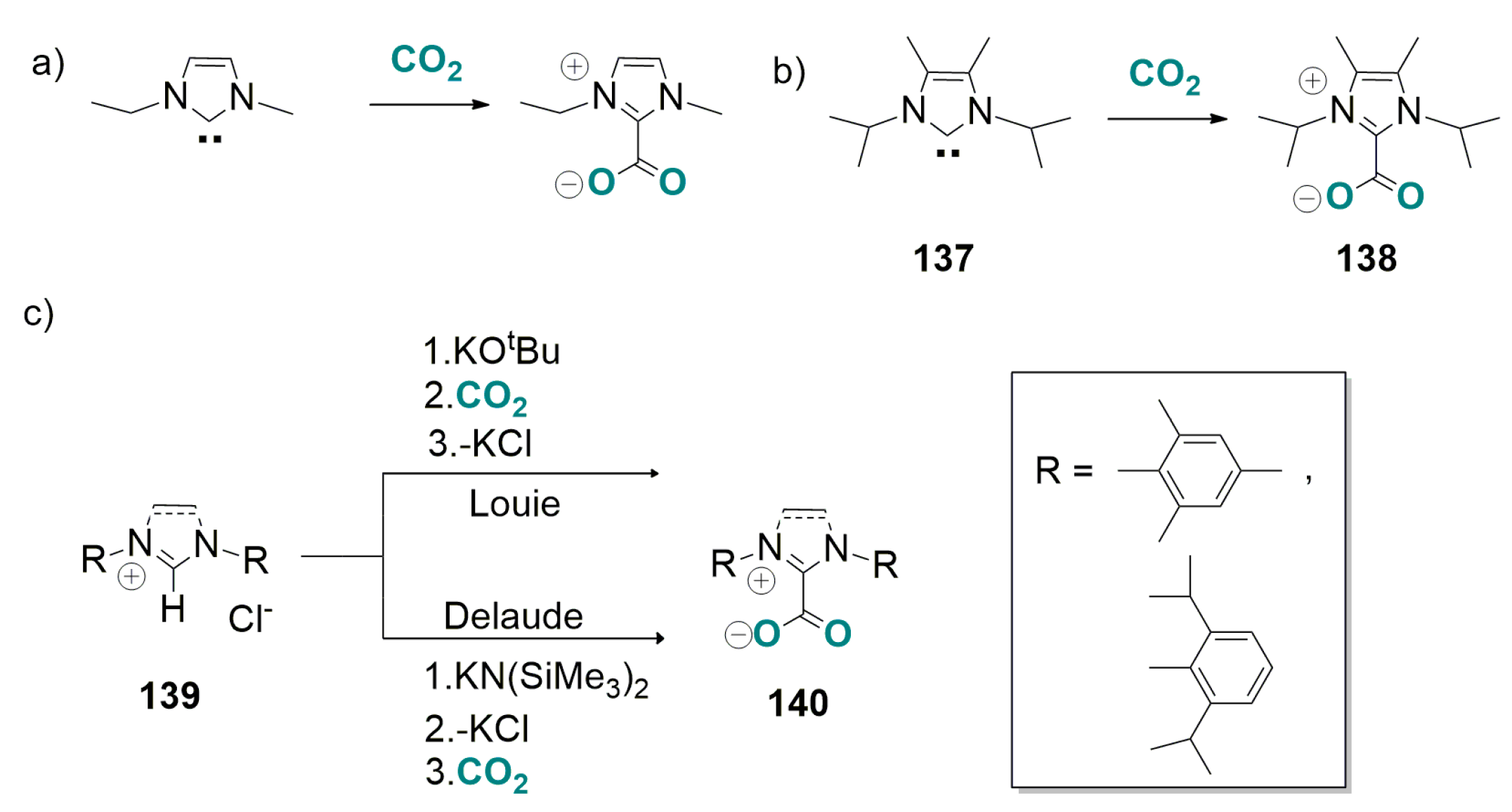
6.1. Base-Catalyzed Carboxylation of 2-Alkynyl Indoles with CO2
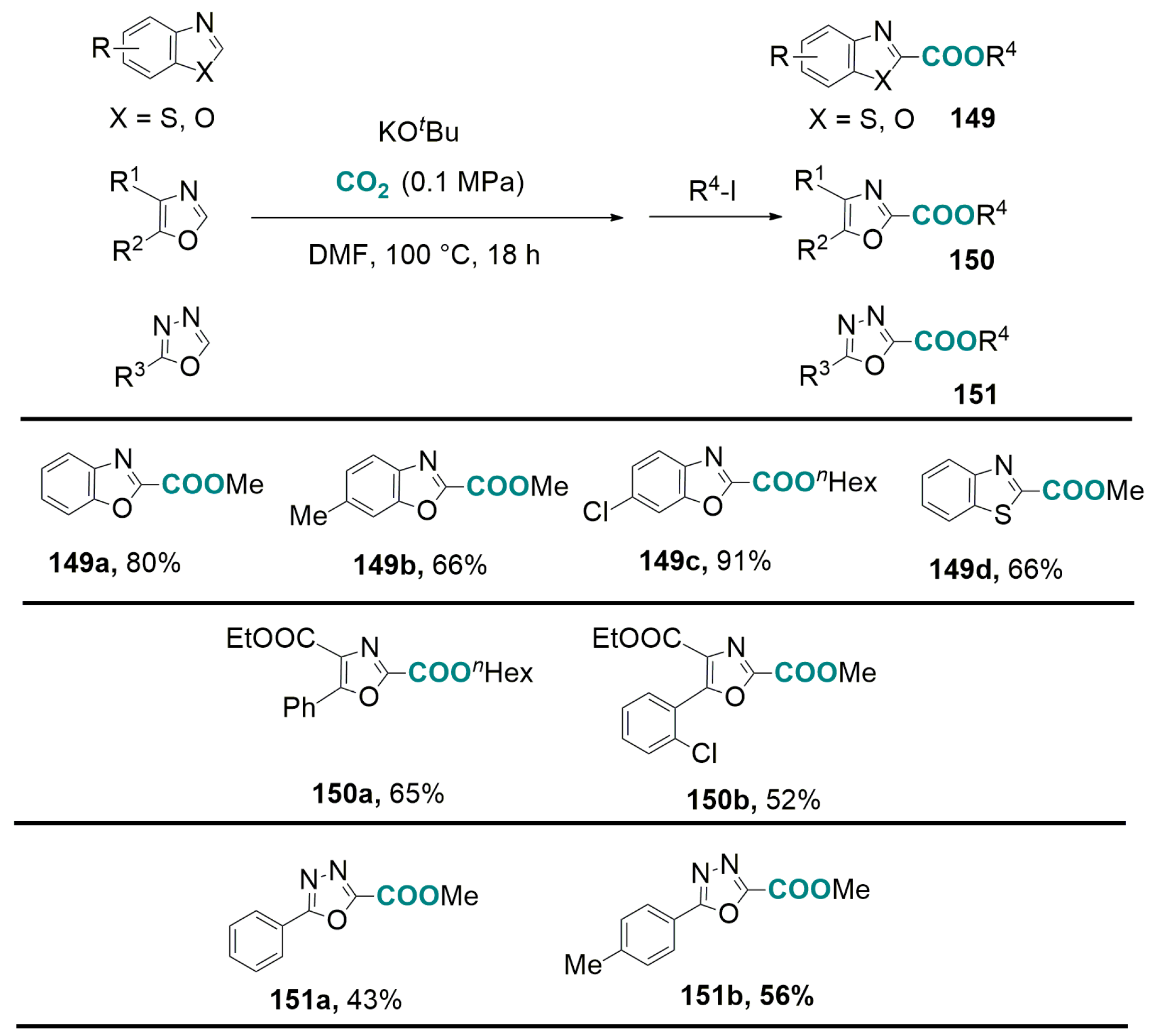
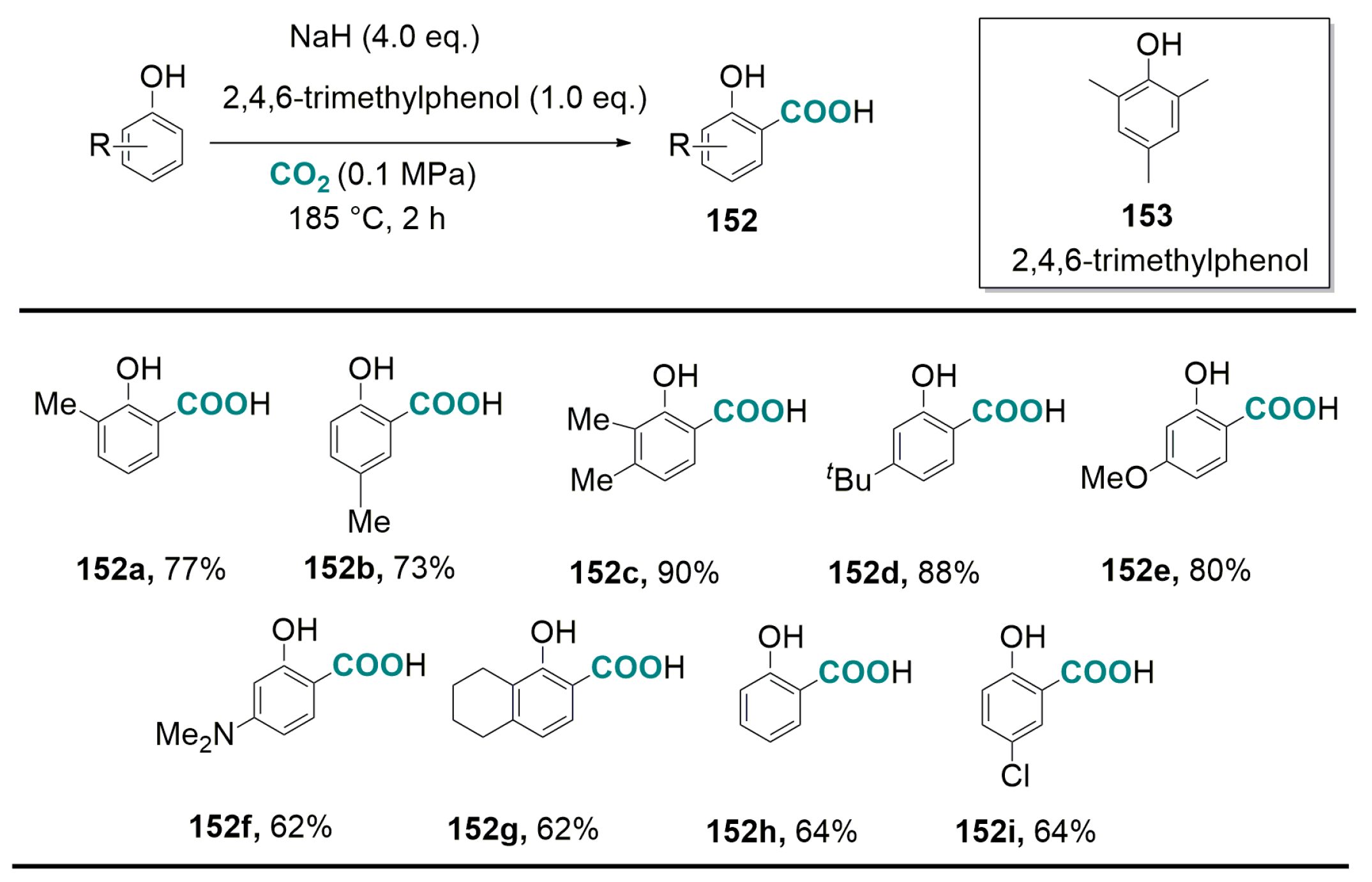
6.2. Base-Promoted Carboxylation of Aromatic and Heteroaromatic Compounds with CO2
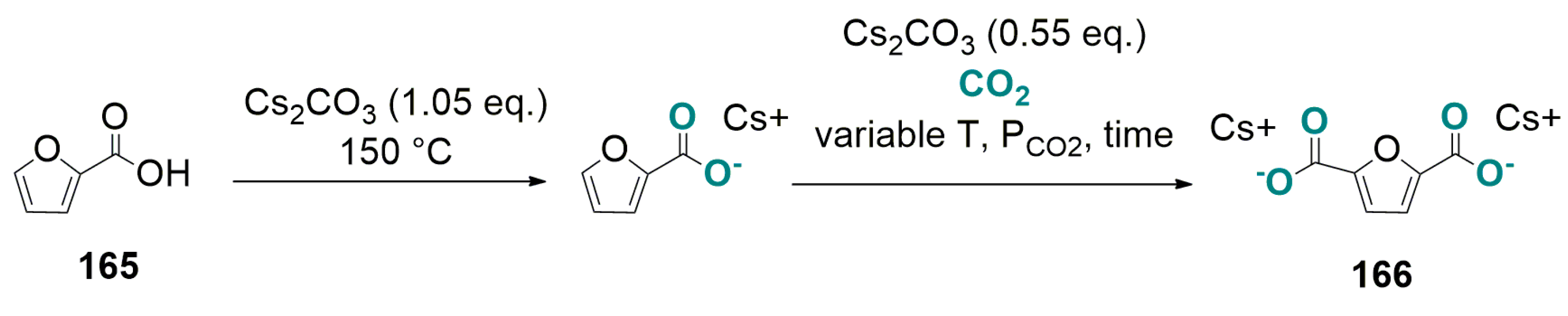
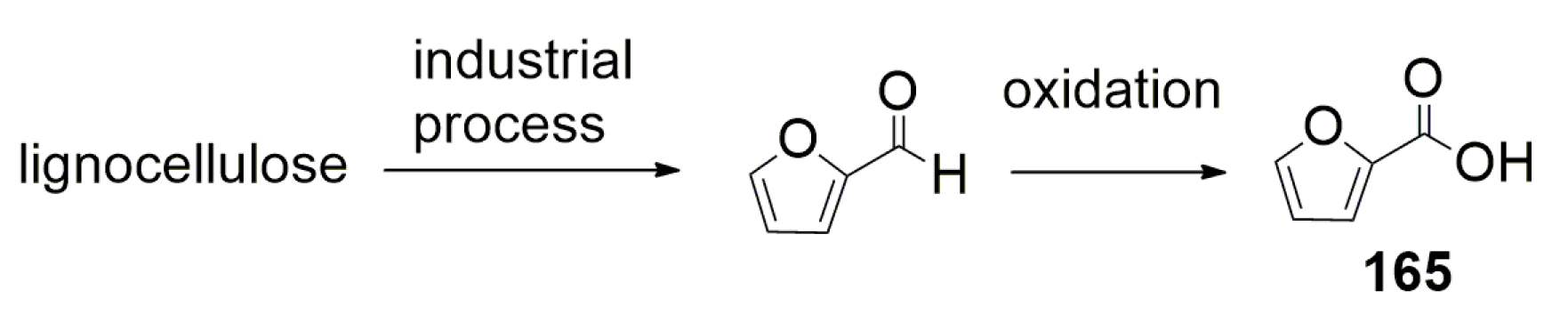
6.3. Base-Promoted Carboxylation of Furan-2-Carboxylic Acid to Furan-2,5-Dicarboxylate
- (i)
- carboxylation of furan-2-carboxylic to furan-2,5-carboxylate;
- (ii)
- protonation of furan-2,5-carboxylate with HCl (to give furan-2,5-carboxylic acid);
- (iii)
- regeneration of HCl and Cs2CO3 from CsCl and CO2.
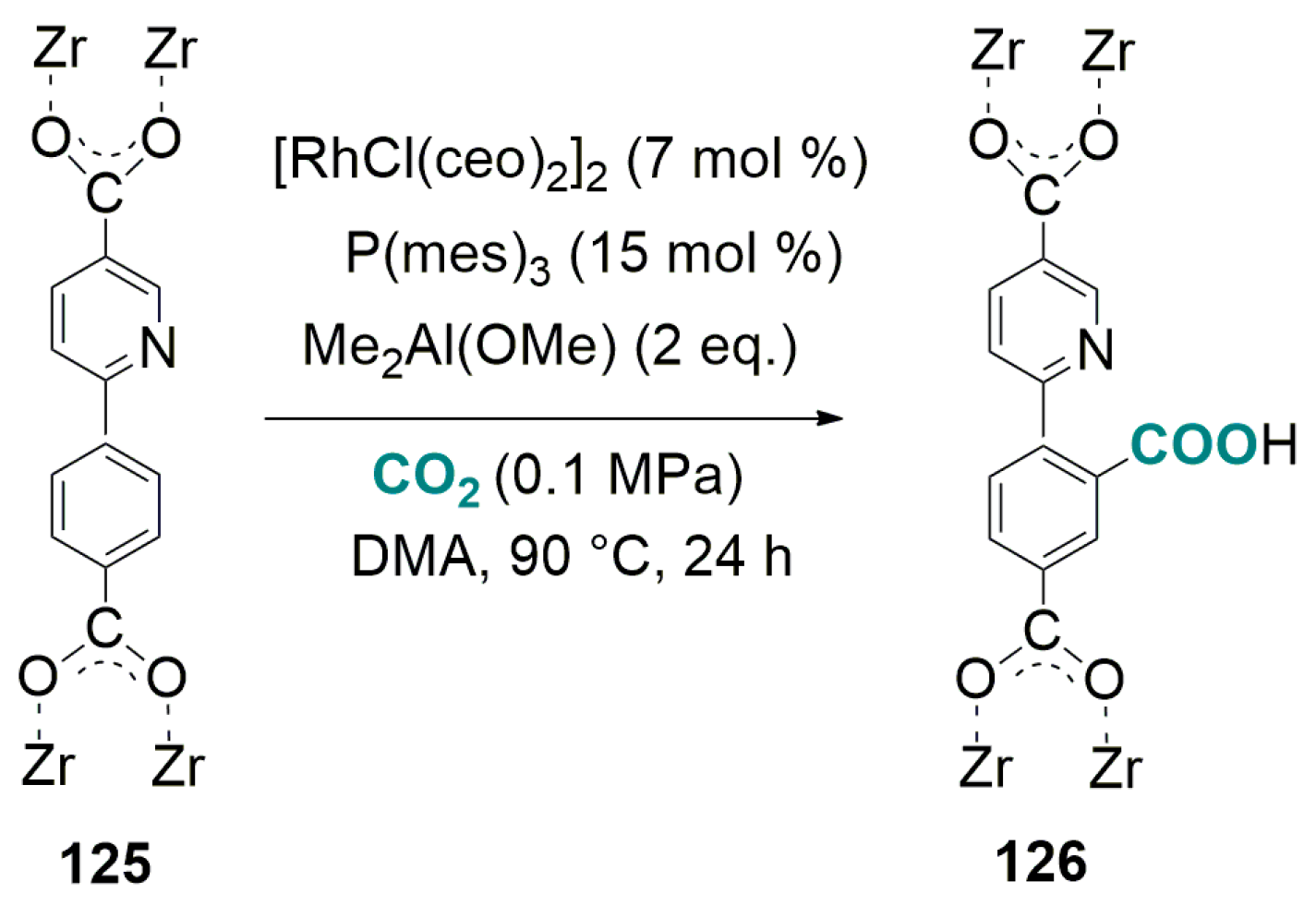
7. C-H Carboxylation with CO2 via Heterogeneous Catalysis
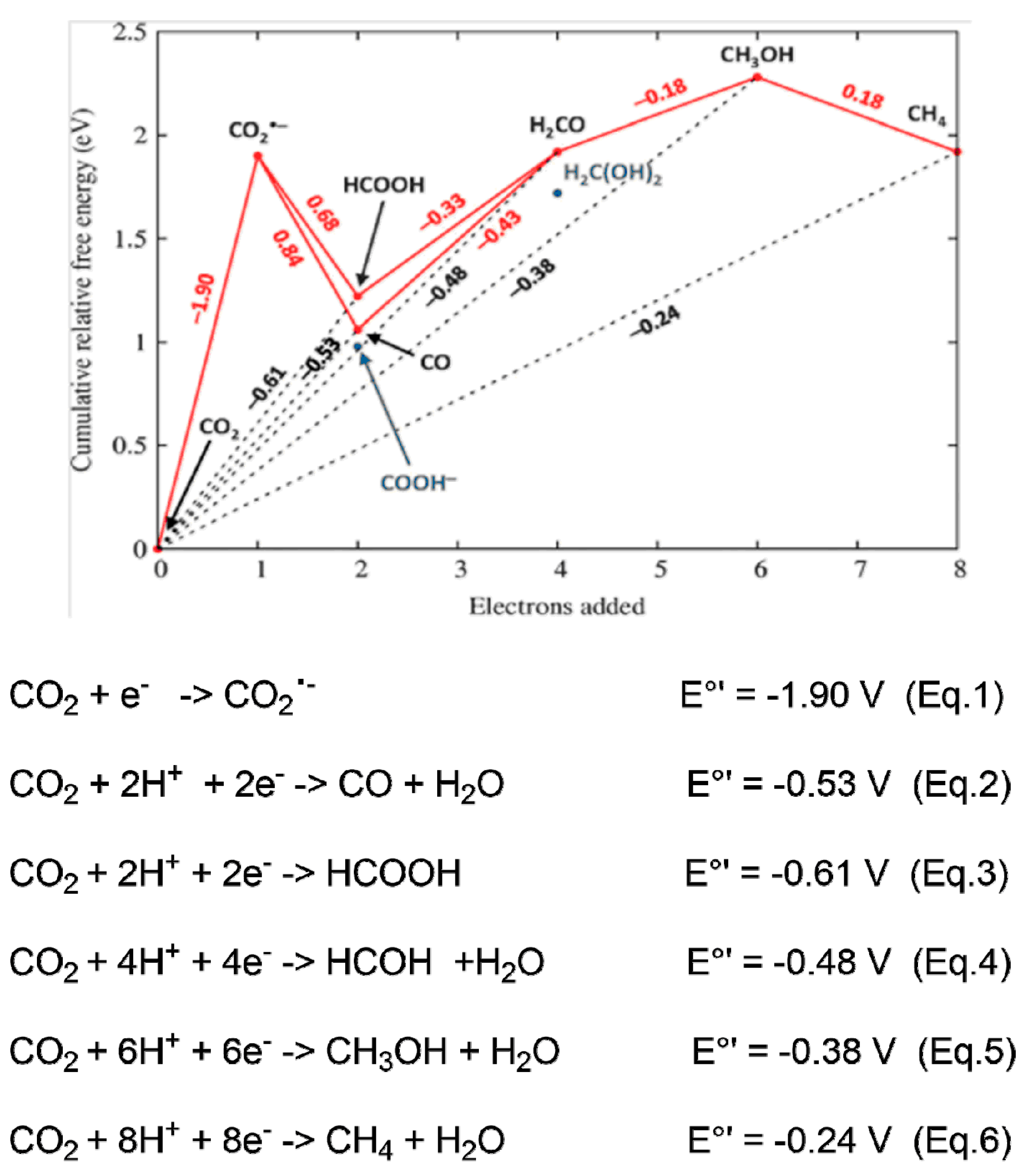
8. Thermodynamics of C-H Carboxylation Reactions with CO2
9. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviation
| AgOBz | silver benzoate |
| BINAP | 2,2′-bis(difenilfosfino)-1,1′-binaftile |
| coe | cyclooctene |
| DBU | 1,8-diazabicyclo(5.4.0)-7-undecene |
| Dcppy | 2-phenylpyridine-5-4′-dicarboxylic acid |
| DFT | density functional theory |
| DMA | dimethylacetamide |
| DMF | dimethylformamide |
| DMSO | dimethyl sulfoxide |
| DRITS | diffuse reflectance infrared Fourier transform spectroscopy |
| EMIM BF4 | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| EMIM Tf2N | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| HMS | methylhydrosiloxane dimethyl siloxane copolymer |
| HMPT | hexamethylphosphoric acid triamide |
| ItBuCO2 | 1,3-bis(tert-butyl)imidazolium-2-carboxylate |
| ICP | Inductively coupled plasma |
| IMesCO2 | 1,3-bis(2,4,6-trimethylphenyl)imidazolium-2-carboxylate |
| IPrCO2 | 1,3-bis(2,6-diisopropylphenyl)imidazolium-2-carboxylate |
| LDA | Lithium diisopropylamide |
| MMC | Magnesium methylcarbonate |
| MOF | Metal-organic framework |
| MTBD | 7-Methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| NHC | N-Heterocyclic carbene |
| NPs | Nanoparticles |
| PXRD | Powder X-ray diffraction |
| RCC | reversible CO2-carrier |
| TBD | 1,5,7-Triazabicyclo[4.4.0]dec-5-ene |
| TGA | thermogravimetric analysis |
| TMP | 2,4,6-trimethylphenol |
| TMU | 1,1,3,3-tetramethylurea |
| TMSCHN2 | trimethylsilyldiazomethane |
| TON | Turn Over Number |
| TOF | Turn Over Frequency |
| TPD | Temperature-programmed desorption |
| TPR | Temperature programmed reaction |
| XPS | X-ray photoelectron spectroscopy |
References
- Lim, X. How to make the most of carbon dioxide. Nature 2015, 526, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and photelectrochemical reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xie, B. Direct carboxylative reactions for the transformation of carbon dioxide into carboxylic acids and derivatives. Synthesis 2013, 45, 3305–3324. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials and fuels. Technological use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef] [PubMed]
- Lia, K.; Ana, X.; Parka, K.H.; Khraishehb, M.; Tanga, J. A critical review of CO2 photoconversion: Catalysts and reactors. Catal. Today 2014, 224, 3–12. [Google Scholar] [CrossRef]
- Liu, A.-H.; Yu, B.; He, L.-N. Catalytic conversion of carbon dioxide to carboxylic acid derivatives. Greenh. Gas. Sci. Technol. 2015, 5, 17–33. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Luca, O.R.; Fenwick, A.Q. Organic reactions for the electrochemical and photochemical production of chemical fuels from CO2—The reduction chemistry of carboxylic acids and derivatives as bent CO2 surrogates. J. Photochem. Photobiol. B 2015, 152, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. Reaction Mechanisms of Carbon Dioxide Conversion; Springer: Berlin, Germany, 2016. [Google Scholar]
- Wu, X.-F.; Zheng, F. Synthesis of carboxylic acids and esters from CO2. Top. Curr. Chem. 2017, 375, 4. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Teong, S.P.; Zhang, Y. Transition metal complex catalyzed carboxylation reactions with CO2. Coord. Chem. Rev. 2015, 293–294, 279–291. [Google Scholar] [CrossRef]
- Kho, E.T.; Tan, T.H.; Lovell, E.; Wong, R.J.; Scott, J.; Amal, R. A review on photo-thermal catalytic conversion of carbon dioxide. Green Energy Environ. 2017, 2, 204–217. [Google Scholar] [CrossRef]
- Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. Photochemical carboxylation of activated C(sp3)-H bonds with CO2. ChemSusChem 2017, 10, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Pinaka, A.; Vougioukalakis, G.C. Using sustainable metals to carry out “green” transformations: Fe- and Cu-catalyzed monetization. Coord. Chem. Rev. 2015, 288, 69–97. [Google Scholar] [CrossRef]
- Omae, I. Recent developments in carbon dioxide utilization for the production of organic chemicals. Coord. Chem. Rev. 2012, 256, 1384–1405. [Google Scholar]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L. Transition-metal-catalyzed carboxylation of C-H bonds. Angew. Chem. Int. Ed. 2011, 50, 3842–3844. [Google Scholar] [CrossRef] [PubMed]
- Bew, S.P. Comprehensive Organic Functional Groups Transformation II; Katritzky, A.R., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2005; p. 19. [Google Scholar]
- Goosen, L.J.; Rodriguez, N.; Goosen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martin, R. Metal-catalyzed carboxylation of organometallic reagents with carbon dioxide. Angew. Chem. Int. Ed. 2009, 48, 6201–6204. [Google Scholar] [CrossRef] [PubMed]
- Maag, H. Prodrugs of Carboxylic Acids; Springer: New York, NY, USA, 2007; pp. 703–729. [Google Scholar]
- Shi, M.; Nicholas, K.M. Palladium-catalyzed carboxylation of allyl stannanes. J. Am. Chem. Soc. 1997, 119, 5057–5058. [Google Scholar] [CrossRef]
- Johansson, R.; Wendt, O.F. Insertion of CO2 into a palladium allyl bond and a Pd(II) catalysed carboxylation of allyl stannanes. Dalton Trans. 2007, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hazari, N. Palladium catalyzed carboxylation of allylstannanes and boranes using CO2. Chem. Commun. 2011, 47, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Sugawara, M.; Hasegawa, H.; Sato, Y. Synthesis of arylglycine and mandelic acid derivatives through carboxylations of α-amido and α-acetoxy stannanes with carbon dioxide. J. Org. Chem. 2012, 77, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Ukai, K.; Aoki, M.; Takaya, J.; Iwasawa, N. Rhodium(I)-catalyzed carboxylation of aryl- and alkenylboronic esters with CO2. J. Am. Chem. Soc. 2006, 128, 8706–8707. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Tadami, S.; Ukai, K.; Iwasawa, N. Copper(I)-catalyzed carboxylation of aryl- and alkenylboronic esters. Org. Lett. 2008, 10, 2697–2700. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Nishiura, M.; Hou, Z. Carboxylation of organoboronic esters catalyzed by N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Ed. 2008, 47, 5792–5795. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Zhang, L.; Nishiura, M.; Hou, Z. Carboxylation of alkylboranes by N-heterocyclic carbene copper catalysts: Synthesis of carboxylic acids from terminal alkenes and carbon dioxide. Angew. Chem. Int. Ed. 2011, 50, 8114–8117. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, H.; Tanabe, M.; Sawamura, M. Copper-catalyzed carboxylation of alkylboranes with carbon dioxide: Formal reductive carboxylation of terminal alkenes. Org. Lett. 2011, 13, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Jang, M.; Hirano, K.; Yorimitsu, H.; Oshima, K. Nickel-catalyzed carboxylation of organozinc reagents with CO2. Org. Lett. 2008, 10, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Beyond Aresta’s complex: Ni- and Pd-catalyzed organozinc coupling with CO2. J. Am. Chem. Soc. 2008, 130, 7826–7827. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martin, R. Palladium-catalyzed direct carboxylation of aryl bromides with carbon dioxide. J. Am. Chem. Soc. 2009, 131, 15974–15975. [Google Scholar] [CrossRef] [PubMed]
- Börjesson, M.; Moragas, T.; Gallego, D.; Martin, R. Metal-catalyzed carboxylation of organic (pseudo)halides with CO2. ACS Catal. 2016, 6, 6739–6749. [Google Scholar] [CrossRef] [PubMed]
- Juliá-Hernández, F.; Moragas, T.; Cornella, J.; Martin, R. Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 2017, 545, 84–88. [Google Scholar] [CrossRef] [PubMed]
- North, M. Synthesis of β,γ-unsaturated acids from allenes and carbon dioxide. Angew. Chem. Int. Ed. 2009, 48, 4104–4105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Riduan, S.N. Catalytic hydrocarboxylation of alkenes and alkynes with CO2. Angew. Chem. Int. Ed. 2011, 50, 6210–6212. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Johnson, J.B.; Rovis, T. Nickel-catalyzed reductive carboxylation of styrenes using CO2. J. Am. Chem. Soc. 2008, 130, 14936–14937. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Iwasawa, N. Hydrocarboxylation of allenes with CO2 catalyzed by silyl pincer-type palladium complex. J. Am. Chem. Soc. 2008, 130, 15254–15255. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Sasano, K.; Iwasawa, N. Efficient one-to-one coupling of easily available 1,3-dienes with carbon dioxide. Org. Lett. 2011, 13, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Gaydou, M.; Morgas, T.; Juliá-Hernández, F.; Martin, R. Site-selective catalytic carboxylation of unsaturaded hydrocarbons with CO2 and water. J. Am. Chem. Soc. 2017, 139, 12161–12164. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Liu, A.; Jamisom, T.F. Direct b-selective hydrocarboxylation of styrenes with CO2 enabled by contiuous flow photoredox catalysis. J. Am. Chem. Soc. 2017, 139, 13969–13972. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhou, F.; Lim, D.S.W.; Su, H.; Zhang, Y. NHC-Ag/Pd-catalyzed reductive carboxylation of terminal alkynes with CO2 and H2: A combined experimental and computational study for fine-tuned selectivity. ChemSusChem 2017, 10, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, G.; Hoberg, H. Oxanickelacyclopentene derivatives from Nickel(0), carbon dioxide, and alkynes. Angew. Chem. Int. Ed. 1982, 21, 76. [Google Scholar] [CrossRef]
- Takimoto, M.; Mori, M. Cross-coupling reaction of oxo-π-allylnickel complex generated from 1,3-diene under an atmosphere of carbon dioxide. J. Am. Chem. Soc. 2001, 123, 2895–2896. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.; Gibby, J.E.; Farnworth, M.V.; Tekavec, T.N. Efficient nickel-catalyzed [2 + 2 + 2] cycloaddition of CO2 and diynes. J. Am. Chem. Soc. 2002, 124, 15188–15189. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, M.; Nakamura, Y.; Kimura, K.; Mori, M. Highly enantioselective catalytic carbon dioxide incorporation reaction: Nickel-catalyzed asymmetric carboxylative cyclization of bis-1,3-dienes. J. Am. Chem. Soc. 2004, 126, 5956–5957. [Google Scholar] [CrossRef] [PubMed]
- Tekavec, T.N.; Arif, A.M.; Louie, J. Regioselectivity in nickel(0) catalyzed cycloadditions of carbon dioxide with diynes. Tetrahedron 2004, 60, 7431–7437. [Google Scholar] [CrossRef]
- Shimizu, K.; Takimoto, M.; Sato, Y.; Mori, M. Nickel-Catalyzed regioselective synthesis of tetrasubstituted alkene using alkylative carboxylation of disubstituted alkyne. Org. Lett. 2005, 7, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; Jackstell, R.; Dastgir, S.; Al-Shihi, B.; Beller, M. Efficient and selective palladium-catalyzed telomerization of 1,3-Butadiene with carbon dioxide. ChemCatChem 2017, 9, 542–546. [Google Scholar] [CrossRef]
- Michigami, K.; Mita, T.; Sato, Y. Cobalt-catalyzed allylic C(sp3)-H carboxylation with CO2. J. Am. Chem. Soc. 2017, 139, 6094–6097. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Nolan, S.P. Direct C-H carboxylation with complexes of the coinage metals. Chem. Commun. 2011, 47, 3021–3024. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Igor Larrosa, I. C-H Carboxylation of aromatic compounds through CO2 fixation. ChemSusChem 2017, 10, 3317–3332. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N-H/C-H bonds using N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Ed. 2010, 49, 8674–8677. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C-H bonds using N-heterocyclic carbene Gold(I) complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef] [PubMed]
- Flowers, B.J.; Gautreau-Service, R.; Jessop, P.G. β-Hydroxycarboxylic acids from simple ketones by carboxylation and asymmetric hydrogenation. Adv. Synth. Catal. 2008, 350, 2947–2958. [Google Scholar] [CrossRef]
- Beckman, E.J.; Munshi, P. Ambient carboxylation on a supported reversible CO2 carrier: Ketone to keto ester. Green Chem. 2011, 13, 376–383. [Google Scholar] [CrossRef]
- Banarjee, A.; Dick, G.R.; Yoshino, T.; Kanan, M.W. Carbon dioxide utilization via carbonate-promoted C-H carboxylation. Nature 2016, 531, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, J.; Ohishi, T.; Hou, Z. Copper-catalyzed direct carboxylation of C-H bonds with carbon dioxide. Angew. Chem. Int. Ed. 2010, 49, 8670–8673. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. C-C bond formation with carbon dioxide promoted by a silver catalyst. Angew. Chem. Int. Ed. 2012, 51, 6989–6992. [Google Scholar] [CrossRef] [PubMed]
- Joumier, J.M.; Fournier, J.; Bruneau, C.; Dixneuf, P.H. Functional carbonates: Cyclic α-methylene and β-oxopropyl carbonates from prop-2-ynyl alcohol derivatives and CO2. J. Chem. Perkin Trans. 1991, 3271–3274. [Google Scholar] [CrossRef]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. Stereoselective formation of α-alkylidene cyclic carbonates via carboxylative cyclization of propargyl alcohols in supercritical carbon dioxide. J. Org. Chem. 2007, 72, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Kawaguchi, T.; Takayama, H.; Nakamura, A.; Inoue, Y. Preparation of alkylidene cyclic carbonates via cyclization of propargylic carbonates. J. Mol. Catal. A: Chem. 1999, 139, 1–9. [Google Scholar] [CrossRef]
- Tommasi, I.; Sorrentino, F. 1,3-Dialkylimidazolium-2-carboxylates as versatile N-heterocyclic carbene–CO2 adducts employed in the synthesis of carboxylates and α-alkylidene cyclic carbonates. Tetrahedron Lett. 2009, 50, 104–107. [Google Scholar] [CrossRef]
- Della Ca, N.; Gabriele, B.; Ruffolo, G.; Veltri, L.; Zanetta, T.; Costa, M. Effective guanidine-catalyzed synthesis of carbonate and carbamate derivatives from propargyl alcohols in supercritical carbon dioxide. Adv. Synth. Catal. 2011, 353, 133–146. [Google Scholar] [CrossRef]
- Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. Carbon-dioxide mediated catalytic rearrangment of propargyl alcohols into α,β-unsaturated ketones. J. Am. Chem. Soc. 2007, 129, 12902–12903. [Google Scholar] [CrossRef] [PubMed]
- Yamada, W.; Sugawara, Y.; Cheng, H.M.; Ikeno, T.; Yamada, T. Silver-Catalyzed Incorporation of carbon dioxide into propargylic alcohols. Eur. J. Org. Chem. 2007, 2604–2607. [Google Scholar] [CrossRef]
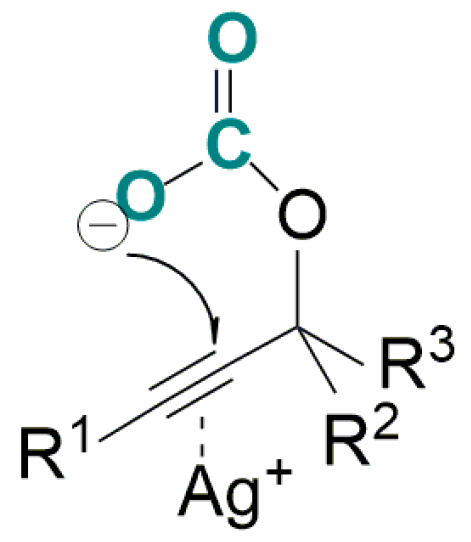
- Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Silver-catalyzed C-C bond formation with carbon dioxide: Significant synthesis of dihydroisobenzofurans. Chem. Commun. 2013, 49, 11320–11322. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Salerno, G.; Fazio, A.; Pittelli, R. Versatile synthesis of (Z)-1-alkylidene-1,3 dihydroisobenzofurans and 1H-isochromenes by palladium-catalyzed cycloisomerization of 2-alkynylbenzyl alcohols. Tetrahedron 2003, 59, 6251–6259. [Google Scholar] [CrossRef]
- Peng, P.; Tang, B.-X.; Pi, S.-F.; Liang, Y.; Li, J.-H. Synthesis of (E)-3-(Isobenzofuran-3(1H)-ylidene)indolin-2-ones by the palladium-catalyzed intramolecular C-H functionalization process. J. Org. Chem. 2009, 74, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Silver versus gold catalysis in tandem reactions of carbonyl functions onto alkynes: A versatile access to furoquinoline and pyranoquinoline cores. Chem. Eur. J. 2007, 13, 5632–5641. [Google Scholar] [CrossRef] [PubMed]
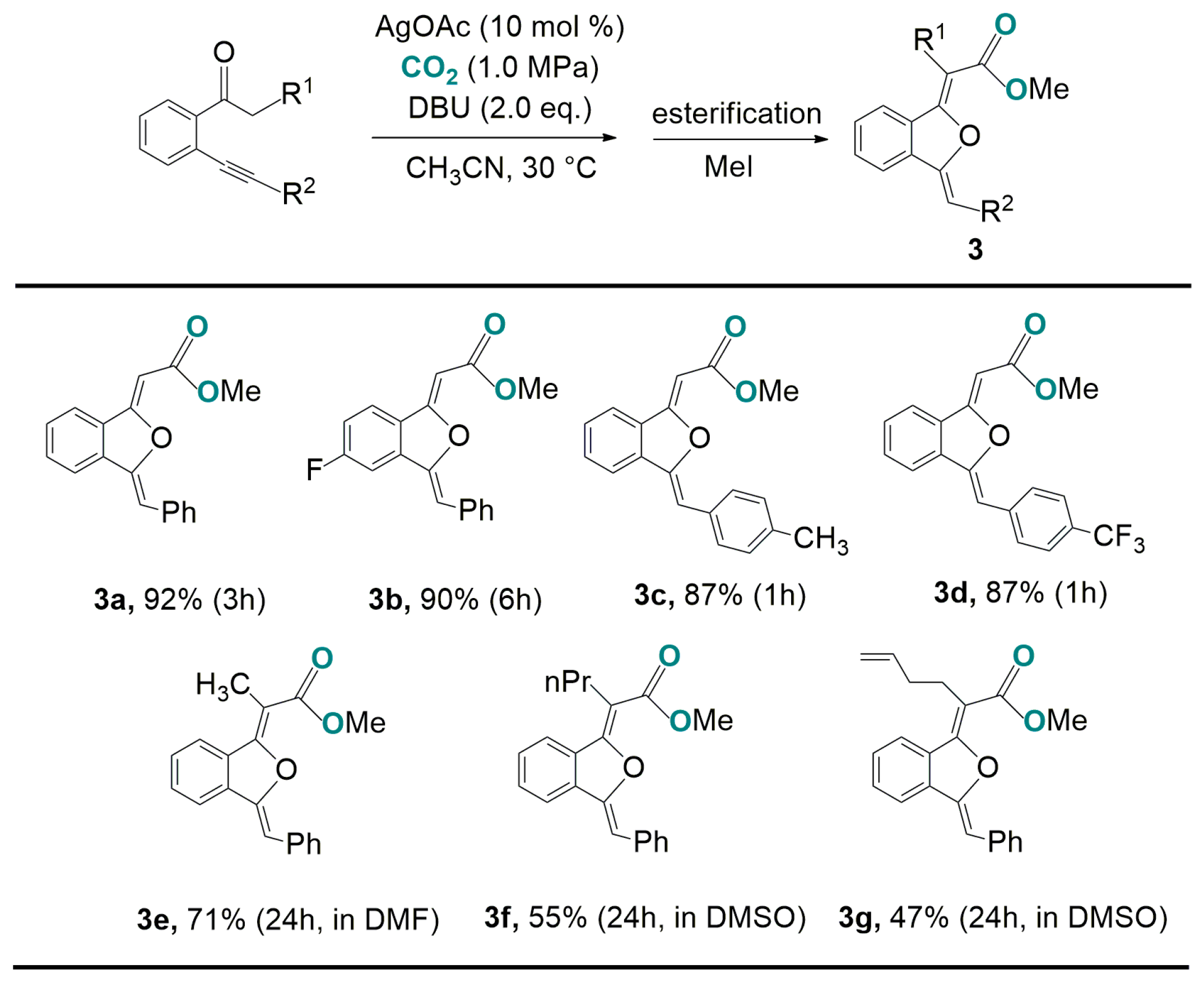
- Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Sequential carboxylation /intramolecular cyclization reaction of a o-alkynyl acetophenone with CO2. Org. Chem. Front. 2014, 1, 275–283. [Google Scholar] [CrossRef]
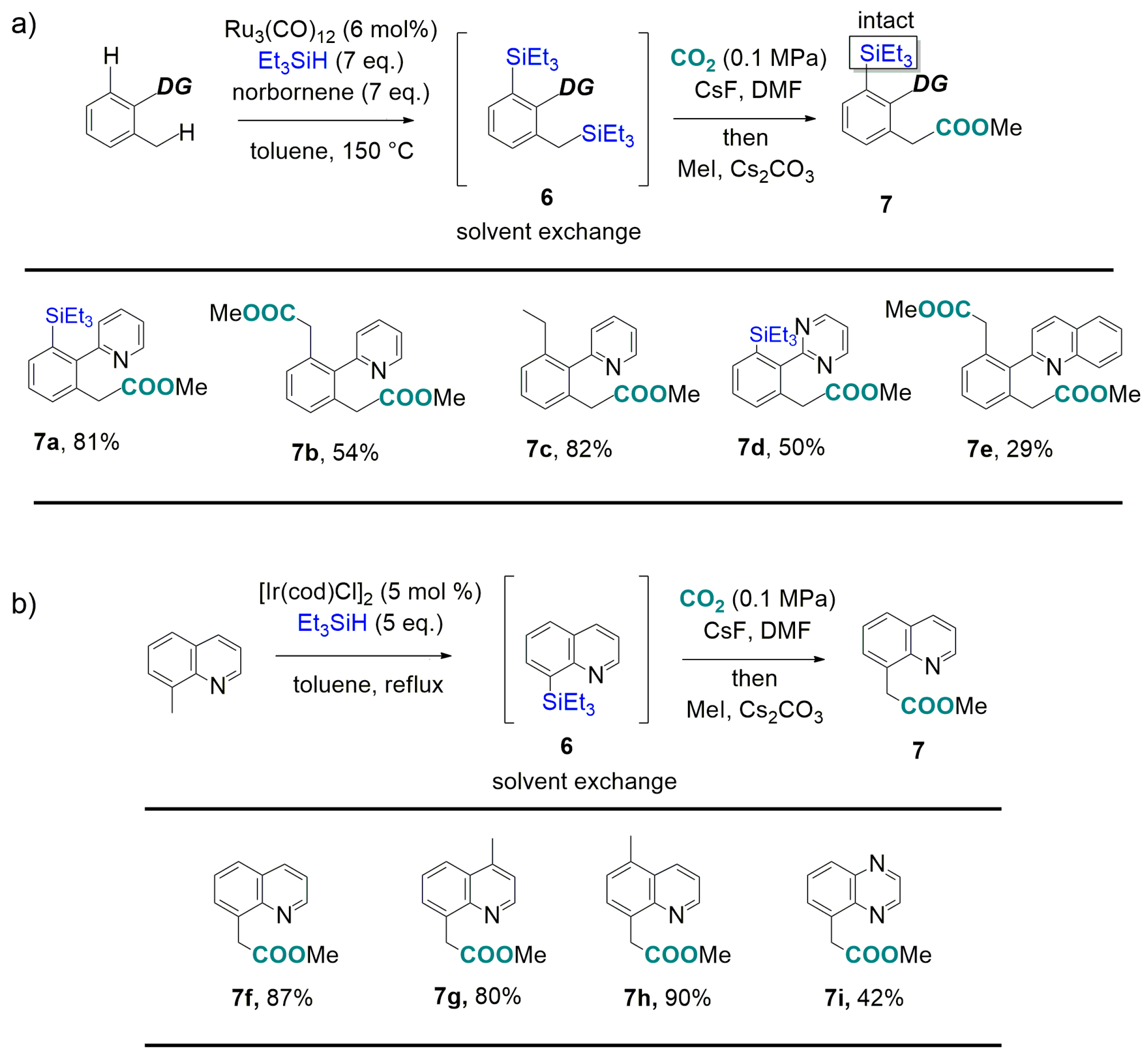
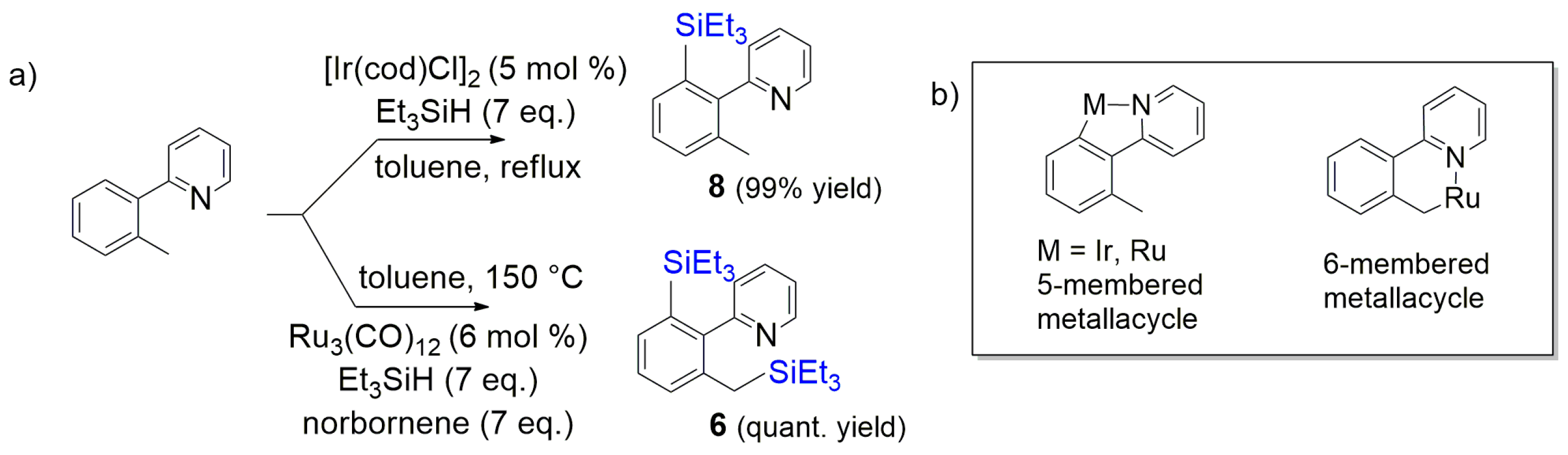
- Mita, T.; Michigami, K.; Sato, Y. Sequential protocol for C(sp3)-H carboxylation with CO2: Transition-metal-catalyzed benzylic C-H silylation and fluoride-mediated carboxylation. Org. Lett. 2012, 14, 3462–3465. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Tsuchiya, K.; Matsumoto, M.; Mizushima, E.; Chatani, N. Ru3(CO)12-catalyzed silylation of benzylic C-H bonds in arylpyridines and arylpyrazoles with hydrosilanes via C-H bond cleavage. J. Am. Chem. Soc. 2004, 126, 12792–12793. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Takimoto, M.; Hou, Z. Synthesis of 2-aryloxy butenoates by copper catalysed allylic C–H carboxylation of allyl aryl ethers with carbon dioxide. Org. Biomol. Chem. 2017, 15, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Naka, H.; Moreu, J.V.; Haywood, J.; Eisler, D.J.; McPartlin, M.; Garcia, F.; Kudo, H.; Kondo, Y.; Uchiyama, M.; Wheatley, E.H. Mixed alkylamido aluminate as a kinetically controlled base. J. Am. Chem. Soc. 2008, 130, 16193–16200. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, E.M.; Roberts, G.W.; Spivey, J.J. Direct catalytic formation of acetic acid from CO2 and methane. Catal. Today 2003, 88, 83–90. [Google Scholar] [CrossRef]
- Spivei, J.J.; Wilcox, E.M.; Roberts, G.W. Direct utilization of carbon dioxide in chemical synthesis: Vinyl acetate via methane carboxylation. Catal. Commun. 2008, 9, 685–689. [Google Scholar] [CrossRef]
- Seo, H.; Katcher, M.H.; Jamison, T.F. Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow. Nat. Chem. 2017, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Baran, T.; Dibenedetto, A.; Aresta, M.; Kcuczala, K.; Macyk, W. Photocatalytic carboxylation of organic substrates with carbon dioxide at zinc sulfide with deposited ruthenium nanoparticles. ChemPlusChem 2014, 79, 708–715. [Google Scholar] [CrossRef]
- Masuda, Y.; Ishida, N.; Murakami, M. Light-driven carboxylation of o-alkylphenyl ketones with CO2. J. Am. Chem. Soc. 2015, 137, 14063–14066. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Masuda, Y.; Uemoto, S.; Murakami, M. A Light/ketone/copper system for carboxylation of allylic C-H bonds of alkenes with CO2. Chem. Eur. J. 2016, 22, 6524–6527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Z.; Yang, M.-W.; Lu, X.-B. Carboxylative cyclization of substituted propenyl ketones using CO2: Transition-metal-free synthesis of α-pyrones. Green Chem. 2016, 18, 4181–4184. [Google Scholar] [CrossRef]
- Benetti, S.; Romagnoli, R.; De Risi, C.; Spalluto, G.; Zanirato, V. Mastering β-keto esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]
- Abdel-Rahman, H.M.; Hussein, M.A. Synthesis of β-hydroxypropanoic acid derivatives as potential anti-inflammatory, analgesic and antimicrobial agents. Arch. Pharm. Chem. Life Sci. 2006, 339, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Thaisrivongs, S.; Schostarez, H.J.; Pals, D.T.; Turner, S.R. alpha,alpha-Difluoro-beta-aminodeoxystatine-containing renin inhibitory peptides. J. Med. Chem. 1987, 30, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Schostarez, H.J. A stereospecific synthesis of 3-aminodeoxystatine. J. Org. Chem. 1988, 53, 3628–3631. [Google Scholar] [CrossRef]
- Capozzi, G.; Roelens, S.; Talami, S. A protocol for the efficient synthesis of enantiopure β-substituted β-lactones. J. Org. Chem. 1993, 58, 7932–7936. [Google Scholar] [CrossRef]
- Oertle, K.; Beyeler, H.; Duthaler, R.O.; Lottenbach, W.; Riediker, M.; Steiner, E. A facile synthesis of optically pure (−)-(S)-ipsenol using a chiral titanium complex. Helv. Chim. Acta 1990, 73, 353–358. [Google Scholar] [CrossRef]
- Genêt, J.P.; Pinel, C.; Ratovelomanana-Vidal, V.; Mallart, S.; Pfister, X.; Bischoff, L.; Andrade, M.C.C.D.; Darses, S.; Galopin, C.; Laffitte, J.A. Enantioselective hydrogenation reactions with a full set of preformed and prepared in situ chiral diphosphine-ruthenium(II) catalysts. Tetrahedron 1994, 5, 675–690. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulphoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry. Part B: Reactions and Synthesis, 4th ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001; p. 4. [Google Scholar]
- Ishikawa, T. (Ed.) Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalyst; John Wiley & Sons: Chichester, UK, 2009; p. 2. [Google Scholar]
- Tables. Available online: https://www.chem.wisc.edu/areas/reich/pkatable/ (accessed on 30 November 2017).
- Callear, S. Preparation, Characterization and Structural Assessment of Salts and Co-Crystals of Organic Compounds. Ph.D. Thesis, University of Southampton, Southampton, UK, 2008. [Google Scholar]
- Magill, A.M.; Cavell, K.J.; Yates, B.F. Basicity of nucleophilic carbenes in acqueous and non acqueous solvents-theoretical predictions. J. Am. Chem. Soc. 2004, 126, 8717–8724. [Google Scholar] [CrossRef] [PubMed]
- Bottaccio, G.; Chiusoli, G.P.; Felicioli, M.G. Organic syntheses on solvent-differentiated ion pairs. Carboxylation with carbon dioxide in aprotic dipolar solvents. Gazz. Chim. Ital. 1973, 103, 105–116. [Google Scholar] [CrossRef]
- Tirpak, R.E.; Olsen, R.S.; Rathke, M.W. Carboxylation of ketones using triethylamine and magnesium halides. J. Org. Chem. 1985, 50, 4877–4879. [Google Scholar] [CrossRef]
- Haruki, E.; Arakawa, M.; Matsumura, N.; Ostuji, Y.; Imoto, E. Carboxylation of active methylene compounds using the reagent of 1,8-diazabiciclo(5.4.0)-7-undecene and carbon dioxide. Chem. Lett. 1974, 427–428. [Google Scholar] [CrossRef]
- Patmore, E.L.; Fishkill, N.Y. Carboxylation Process. U.S. Patent 3,694,496, 26 September 1972. [Google Scholar]
- Stiles, M. Chelation as a driving force in synthesis. II Use of magnesium methylcarbonate in the carboxylation and alkylation of ketones. J. Am. Chem. Soc. 1959, 81, 2598–2599. [Google Scholar] [CrossRef]
- Tsuda, T.; Chujo, Y.; Hayasaki, T.; Saegusa, T. Preparation and transcarboxylation of magnesium(II) and manganese(II) 2-oxoimidazoline-1-carboxylato-complexes. J. Chem. Soc. Chem. Commun. 1979, 797–798. [Google Scholar] [CrossRef]
- Chiba, K.; Tagaya, H.; Karasu, M.; Ishizuka, M.; Sugo, T. Carboxylation of active methylene compounds using anilide, potassium carbonate and carbon dioxide. Bull. Chem. Soc. Jpn. 1994, 67, 452–454. [Google Scholar] [CrossRef]
- Tommasi, I.; Sorrentino, F. Utilisation of 1,3-dialkylimidazolium-2-carboxylates as CO2-carriers in the presence of Na+ and K+: Application in the synthesis of carboxylates, monomethylcarbonate anions and halogen-free ionic liquids. Tetrahedron Lett. 2005, 46, 2141–2145. [Google Scholar] [CrossRef]
- Rose, B.A.; Salehi-Khojin, A.; Thorson, M.R.; Zhu, W.; Whipple, D.T.; Kenis, P.J.A.; Masel, R.I. Ionic-liquid mediated selective conversion of CO2 to CO at lower overpotentials. Science 2011, 334, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ding, Z.; Hou, Y.; Wang, X. Ionic liquid co-catalyzed artificial photosynthesis of CO2. Sci. Rep. 2013, 3, 1056. [Google Scholar] [CrossRef]
- Denning, D.M.; Thum, M.D.; Falvey, D.E. Photochemical reduction of CO2 using 1,3-dimethyimidazolidene. Org. Lett. 2005, 17, 4152–4155. [Google Scholar] [CrossRef] [PubMed]
- Mastumura, N.; Asai, N.; Yoneda, S. α-carboxylation reactions of ketones with a bromomagnesium thioureide-carbon dioxide complex. J. Chem. Soc. Chem. Commun. 1983, 1487–1488. [Google Scholar] [CrossRef]
- Tommasi, I.; Sorrentino, F. Synthesis of 1,3-dialkylimidazolium-2-carboxylates by direct carboxylation of 1,3-dialkylimidazolium chlorides with CO2. Tetrahedron Lett. 2006, 47, 6453–6456. [Google Scholar] [CrossRef]
- Van Ausdall, B.-R.; Poth, N.F.; Kincaid, V.A.; Arif, A.M.; Louie, J. Imidazolidene Carboxylate Bound MBPh4 complexes (M = Li, Na) and their relevance in transcarboxylation reactions. J. Org. Chem. 2011, 76, 8413–8420. [Google Scholar] [CrossRef] [PubMed]
- Ratusky, J. Transcarboxylation reactions of salts of organic acids. XIX. The effect of various cations on the course of the transcarboxylation and the catalytic effect of some of these cations. Collect. Czechoslov. Chem. Commun. 1973, 38, 74–99. [Google Scholar] [CrossRef]
- Van Ausdall, B.-R.; Glass, G.E.; Wiggins, K.M.; Arif, A.M.; Louie, J. A systematic investigation of factors influencing the decarboxylation of imidazolium carboxylates. J. Org. Chem. 2009, 74, 7935–7942. [Google Scholar] [CrossRef] [PubMed]
- Dalton, D.M.; Rovis, T. Organometallic chemistry: C-H carboxylation takes gold. Nat. Chem. 2010, 2, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Sasano, K.; Takaya, J.; Iwasawa, N. Palladium(II)-catalyzed direct carboxylation of alkenyl C-H bonds with CO2. J. Am. Chem. Soc. 2013, 135, 10954–10957. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Boorman, T.C.; Slawin, A.Z.; Larrosa, I. Gold(I)-mediated C-H activation of arenes. J. Am. Chem. Soc. 2010, 132, 5580–5581. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. What are the pKa values of C-H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar] [CrossRef]
- Dang, L.; Lin, Z. DFT Studies on the carboxylation of arylboronate esters with CO2 catalyzed by copper(I) complexes. Organometallics 2010, 29, 917–927. [Google Scholar] [CrossRef]
- Inomata, H.; Ogata, K.; Fukuzawa, S.; Hou, Z. Direct C-H carboxylation with carbon dioxide using 1,2,3-triazol-5-ylidene copper(I)-complexes. Org. Lett. 2012, 14, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Takimoto, M.; Wylie, W.N.O.; Nishiura, M.; Ikariya, T.; Hou, Z. Copper-catalyzed formal C-H carboxylation of aromatic compounds with carbon dioxide through arylalluminium intermediates. Chem. Asian J. 2015, 10, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, L.J.; Rodriguez, N.; Manjolinho, F.; Langea, P. Synthesis of propiolic acids via copper-catalyzed insertion of carbon dioxide into the C-H bond of terminal alkynes. Adv. Synth. Catal. 2010, 352, 2913–2917. [Google Scholar] [CrossRef]
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-catalyzed direct carboxylation of arenes with CO2 via chelation-assisted C-H bond activation. J. Am. Chem. Soc. 2011, 133, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Mizuno, H.; Takaya, J.; Iwasawa, N. Direct carboxylation of simple arenes with CO2 through a rhodium-catalyzed C-H bond activation. Chem. Commun. 2014, 50, 14360–14363. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Saitou, T.; Takaya, J.; Iwasawa, N. Mechanistic study of the rhodium-catalyzed carboxylation of simple aromatic compounds with carbon dioxide. Chem. Sci. 2017, 8, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-Y.; Wu, H.; Leng, K.; Sun, Y.; Ma, S. Inserting CO2 into aryl C-H bonds of metal–organic frameworks: CO2 utilization for direct heterogeneous C-H activation. Angew. Chem. Int. Ed. 2016, 55, 5472–5476. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass. In Results of Screening for Potential Candidates from Sugars and Synthesis Gas Vol. 1; USDOE, 2004; pp. 26–28. Available online: http://www.nrel.gov/docs/fy04osti/35523.pdf (accessed on 5 December 2017).
- Xin, Z.; Lescot, C.; Friis, S.D.; Daasbjerg, K.; Skrydstrup, T. Organocatalyzed CO2 trapping using alkynyl indoles. Angew. Chem. Int. Ed. 2015, 54, 6862–6866. [Google Scholar] [CrossRef] [PubMed]
- Kee, C.W.; Peh, K.Q.E.; Wong, M.W. Coupling reactions of alkynyl indoles and CO2 by bicyclic guanidine: Origin of catalytic activity? Chem. Asian J. 2017, 12, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. A combined kinetic—Quantum mechanical model for assessment of catalytic cycles: Application to cross-coupling and Heck reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.A.; Haan, J.L.; Mukherjee, P.; Braunschweig, B.; Zhu, W.; Salehi-Khojin, A.; Dlott, D.D.; Masel, R.I. In situ spectroscopic examination of the low overpotential pathway for carbon dioxide conversion to carbon monoxide. J. Phys. Chem. C 2012, 116, 15307–15312. [Google Scholar] [CrossRef]
- Sun, L.; Ramesha, G.K.; Kamat, P.V.; Brennecke, J.F. Switching the reaction course of electrochemical CO2 reduction with ionic liquids. Langmuir 2014, 30, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Kuhn, N.; Steimann, M.; Weyers, G. Synthesis and properties of 1,3-diisopropyl-4,5-dimethyimidazolium-2-carboxylate. A stable carbene adduct of carbon dioxide. Z. Naturforsch. 1999, 54b, 427–433. [Google Scholar]
- Duong, H.A.; Tekavec, T.N.; Arif, A.M.; Louie, J. Reversible carboxylation of N-heterocyclic carbenes. Chem. Commun. 2004, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Tudose, A.; Delaude, L.; Andre, B.; Demonceau, A. Imidazol(in)ium carboxylates as N-heterocyclic carbene ligand precursors for Suzuki–Miyaura reactions. Tetrahedron Lett. 2006, 47, 8529–8533. [Google Scholar] [CrossRef]
- Holbrey, J.D.; Reichert, W.M.; Tkatchenko, I.; Bouajila, E.; Walter, O.; Tommasi, I.; Rogers, R.D. 1,3-Dimethylimidazolium-2-carboxylate: The planned and unexpected synthesis of an ionic liquid precursor and carbene-CO2 adduct. Chem. Commun. 2003, 28–29. [Google Scholar] [CrossRef]
- Voutchkova, A.M.; Feliz, M.; Clot, E.; Eisenstein, O.; Crabtree, R.H. Imidazolium carboxylates as versatile and selective N-heterocyclic carbene transfer agents: Synthesis, mechanism and applications. J. Am. Chem. Soc. 2007, 129, 12834–12846. [Google Scholar] [CrossRef] [PubMed]
- Vechorkin, O.; Hirt, N.; Hu, X. Carbon dioxide as the C1 source for direct C-H functionalization of aromatic heterocycles. Org. Lett. 2010, 12, 3567–3569. [Google Scholar] [CrossRef] [PubMed]
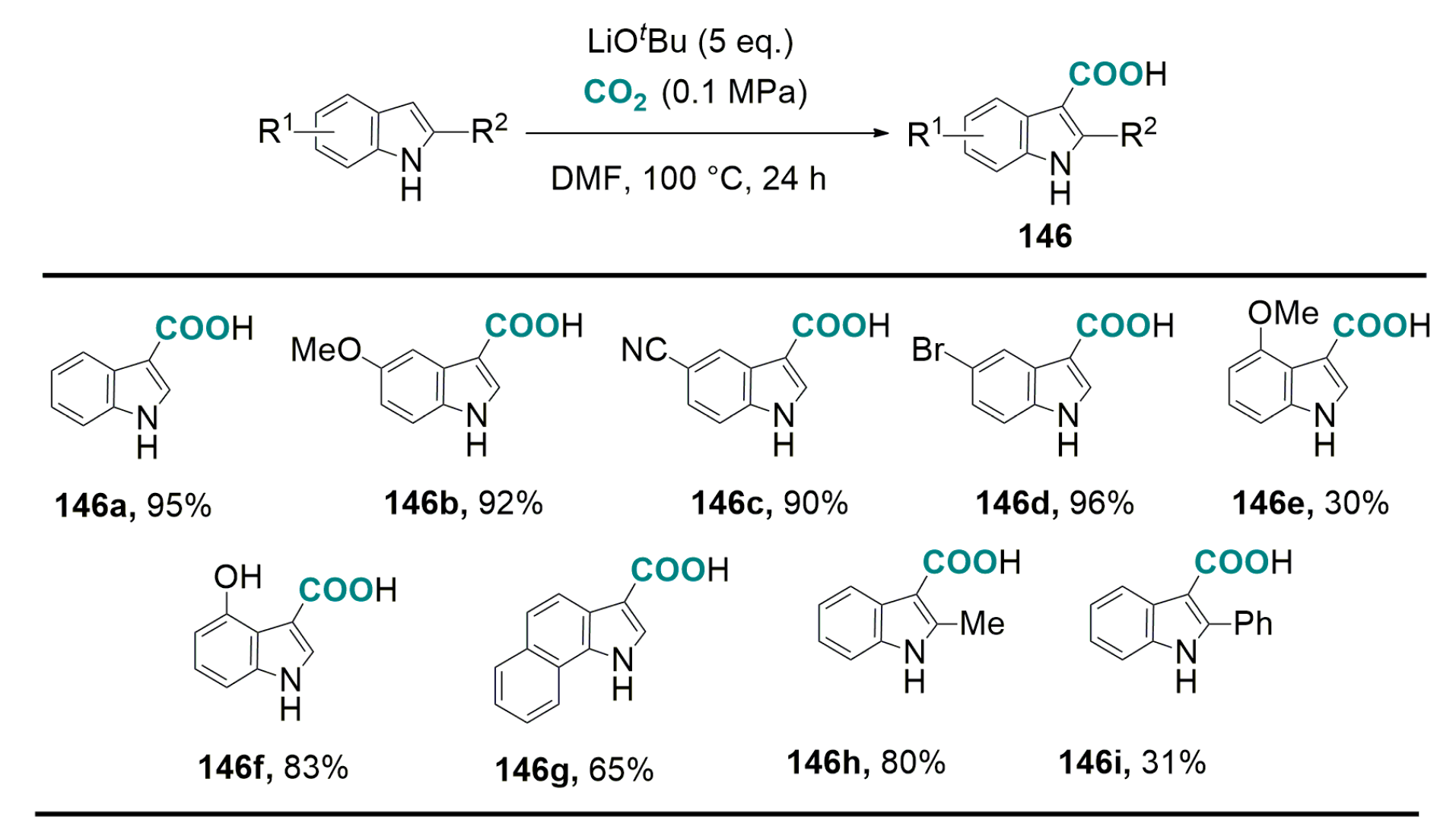
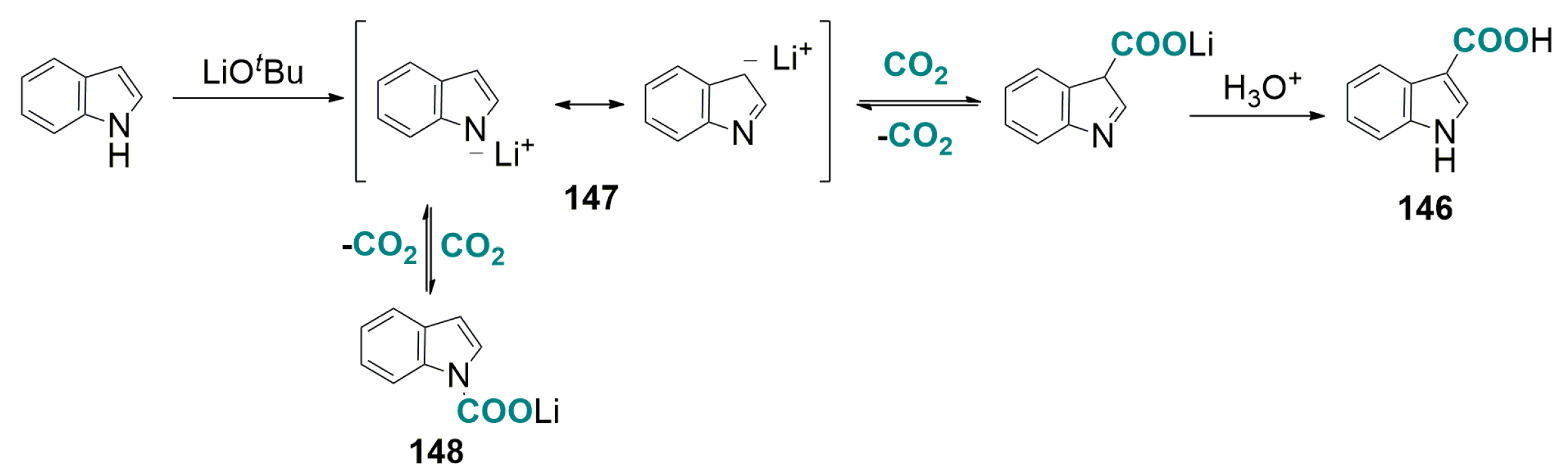
- Yoo, W.-J.; Capdevila, M.G.; Du, X.; Kobayashi, S. Base-mediated carboxylation of unprotected indole derivatives with carbon dioxide. Org. Lett. 2012, 14, 5326–5329. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.-J.; Nguyen, T.V.Q.; Capdevila, M.G.; Kobayashi, S. Lithium tert-butoxide-mediated carboxylation reactions of unprotected indoles and pyrroles with carbon dioxide. Heterocycles 2015, 90, 1196–1204. [Google Scholar]
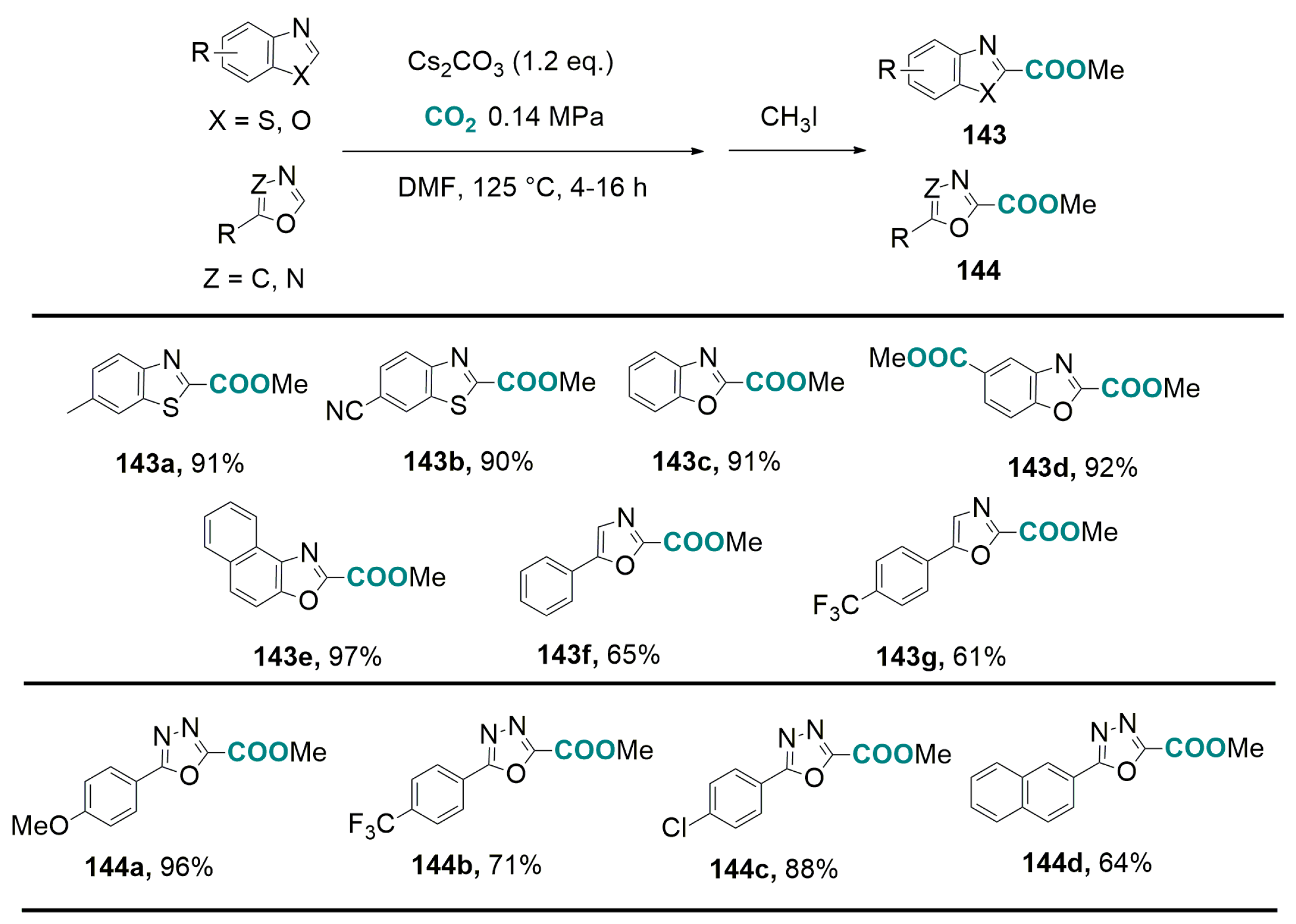
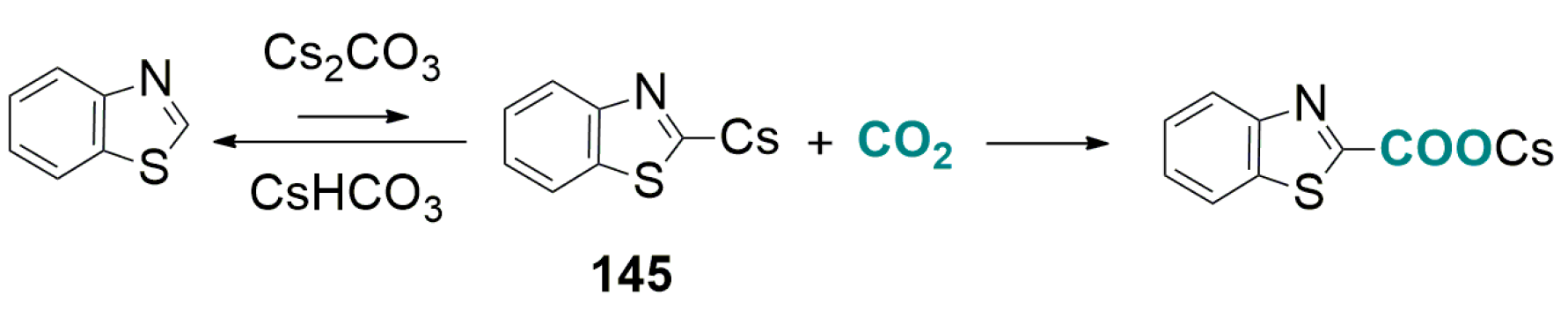
- Fenner, S.; Ackermann, L. C-H carboxylation of heteroarenes with ambient CO2. Green Chem. 2016, 18, 3804–3807. [Google Scholar] [CrossRef]
- Luo, J.; Preciado, S.; Xie, P.; Larrosa, I. Gold(I)-mediated C-H activation of arenes. Chem. Eur. J. 2016, 22, 5580–5581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ju, T.; Miao, M.; Han, J.-L.; Zhang, Y.-H.; Zhu, X.-Y.; Ye, J.-H.; Yu, D.-G.; Zhi, Y.-G. Transition-metal-free lactonization of sp2 C-H bonds with CO2. Org. Lett. 2017, 19, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Wang, L.; He, Y.-Q.; Ye, J.-H.; Li, J.; Zhi, Y.-G.; Yu, D.-G. Lactamization of sp2 C-H bonds with CO2: Transition-metal-free and redox-neutral. Angew. Chem. Int. Ed. 2016, 55, 7068–7072. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Furukawa, K.; Nagai, Y.; Uno, H. 4-Phenyl-2-(1-piperazinyl)quinolines with potent antidepressant activity. Chem. Pharm. Bull. 1980, 28, 2618–2622. [Google Scholar] [CrossRef] [PubMed]
- Labaudinière, R.; Hendel, W.; Terlain, B.; Cavy, F.; Marquis, O.; Dereu, N. omega-[(4-Phenyl-2-quinolyl)oxy]alkanoic acid derivatives: A new family of potent LTB4 antagonists. J. Med. Chem. 1992, 35, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Angibaud, P.R.; Venet, M.G.; Filliers, W.; Broeckx, R.; Ligny, Y.A.; Muller, P.; Poncelet, V.S.; End, D.W. Synthesis routes towards the farnesyl protein transferase inhibitor ZARNESTRATM. Eur. J. Org. Chem. 2004, 479–486. [Google Scholar] [CrossRef]
- Ferguson, J.; Zeng, F.; Alwis, N.; Alper, H. Synthesis of 2(1H)-Quinolinones via Pd-catalyzed oxidative cyclocarbonylation of 2-vinylanilines. Org. Lett. 2013, 15, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Hu, Z.; Peng, J.; Huang, J.; Zhu, Q. Synthesis of phenanthridinones via palladium-catalyzed C(sp2)–H aminocarbonylation of unprotected o-arylanilines. Chem. Commun. 2013, 49, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, V.; Lee, T.-H.; Chuang, S.-C. Palladium-catalyzed oxidative insertion of carbon monoxide to N-sulfonyl-2-aminobiaryls through C–H bond activation: Access to bioactive phenanthridinone derivatives in one pot. Org. Lett. 2013, 15, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, L.; Liu, Q.; Yang, X.; Ye, X.; Hub, Y.; Huang, Y. A Schiff base-modified silver catalyst for efficient fixation of CO2 as carboxylic acid at ambient pressure. Green Chem. 2017, 19, 2080–2085. [Google Scholar] [CrossRef]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Liu, X.-H.; Ma, J.-G.; Niu, Z.; Yang, G.-M.; Cheng, P. An efficient nanoscale heterogeneous catalyst for the capture and conversion of carbon dioxide at ambient pressure. Angew. Chem. Int. Ed. 2015, 54, 988–991. [Google Scholar] [CrossRef] [PubMed]
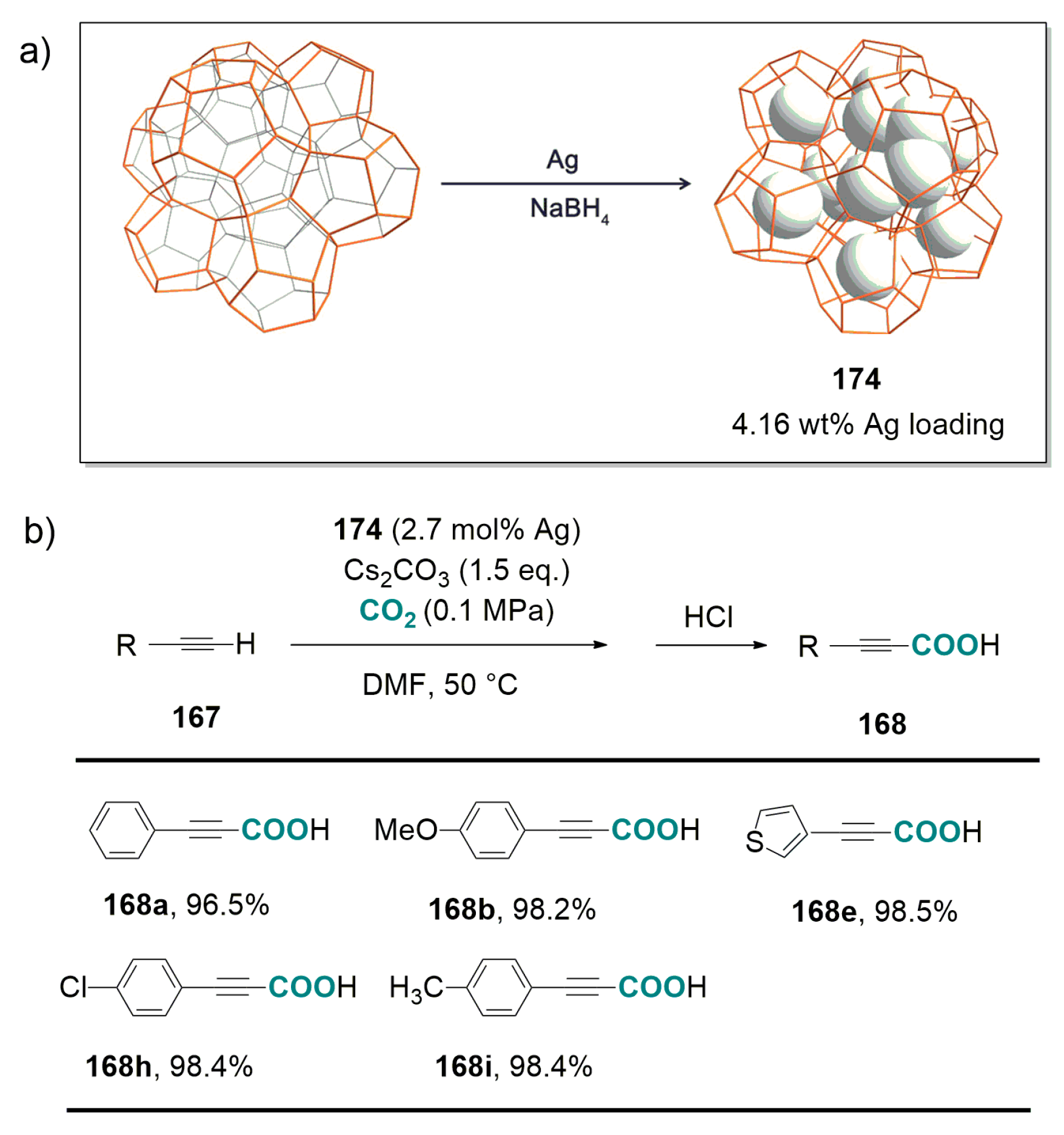
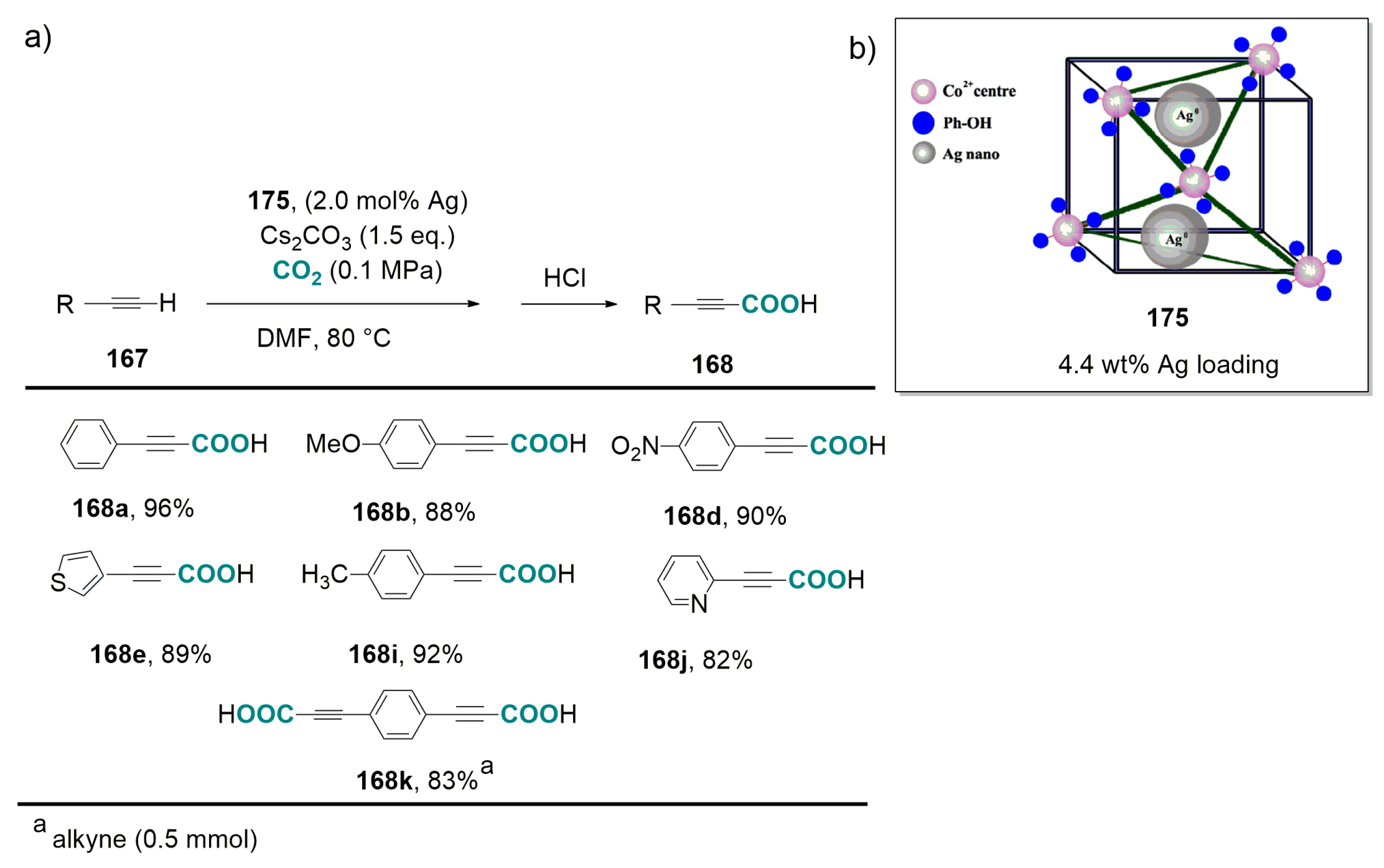
- Molla, R.A.; Ghosh, K.; Banerjee, B.; Iqubal, M.I.; Kundu, S.K.; Islam, S.M.; Bhaumik, A. Silver nanoparticles embedded over porous metal organic frameworks for carbon dioxide fixation via carboxylation of terminal alkynes at ambient pressure. J. Colloid Interf. Sci. 2016, 477, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Glueck, S.M.; Gümüs, S.; Fabian, W.M.F.; Faber, K. Biocatalytic Carboxylation. Chem. Soc. Rev. 2010, 39, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Jia, H.; Muckermana, J.T.; Fujita, E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef] [PubMed]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J. Electroanal. Chem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Matsuoka, S.; Kohzuki, T.; Pac, C.; Ishida, A.; Takamuku, S.; Kusaba, M.; Nakashima, N.; Yanagida, S. Photocatalysis of oligo(p-phenylenes): Photochemical reduction of carbon dioxide with triethylamine. J. Phys. Chem. 1992, 96, 4437–4442. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, J.; Zhang, Y.; Li, Q.; Gong, J.R. Visible light photocatalytic H2-production activity of CuS/ZnS porous nanosheets based on photoinduced interfacial charge transfer. Nano Lett. 2011, 11, 4774–4779. [Google Scholar] [CrossRef] [PubMed]
- Kočí, K.; Reli, M.; Kozák, O.; Lacný, Z.; Plachá, D.; Praus, P.; Obalová, L. Influence of reactor geometry on the yield of CO2 photocatalytic reduction. Catal. Today 2011, 176, 212–214. [Google Scholar] [CrossRef]




















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
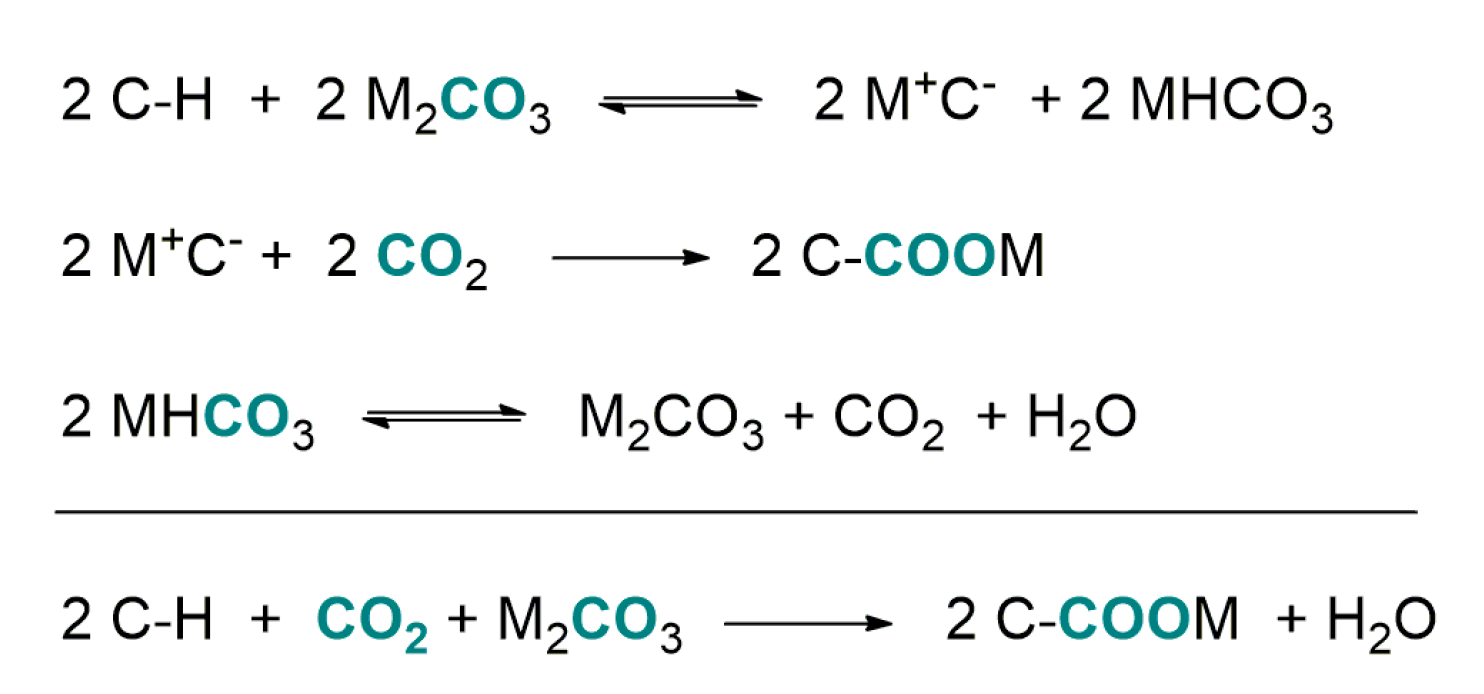{kind=link}
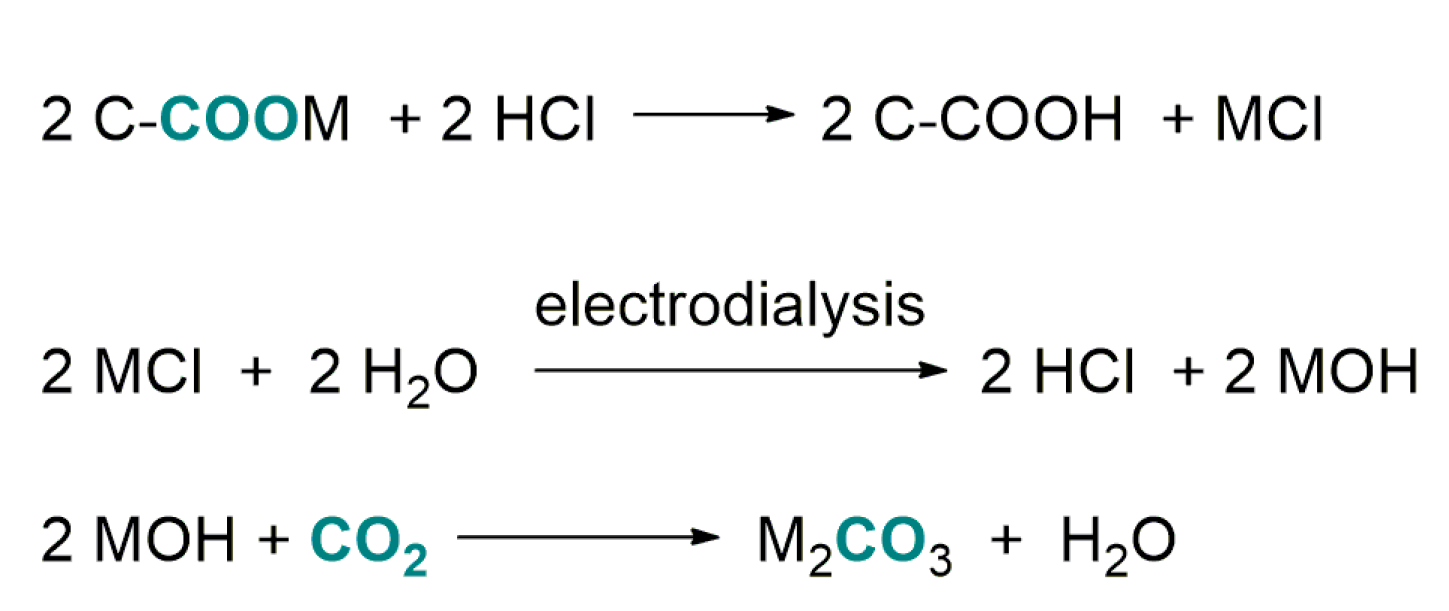{kind=link}
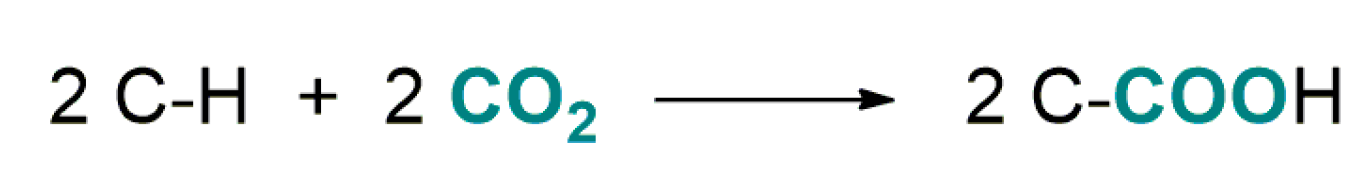{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acid-Base Equilibrium | pKa Value (of the Conjugated Acid) | Reference |
|---|---|---|---|
| 1 |  | 18.0 in DMSO [93] | [99] |
| 2 |  (in conjuction with MgI2) | 10.70 in H2O [94] | [100] |
| 3 |  | 11.82 in MeCN [95] | [101] |
| 4 |  | 28.5 in DMSO [96] | [102] |
| 5 |  | 29.0 in DMSO [93] considering CH3OH | [103] |
| 6 |  | 17.97 in H2O [97] (calculated value) | [104] |
| 7 |  | 21.5 in DMSO [96] | [105] |
| 8 |  | 21.1 in DMSO [98] 32.4 in CH3CN (calculated value) | [106] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tommasi, I. Direct Carboxylation of C(sp3)-H and C(sp2)-H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions. Catalysts 2017, 7, 380. https://doi.org/10.3390/catal7120380
Tommasi I. Direct Carboxylation of C(sp3)-H and C(sp2)-H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions. Catalysts. 2017; 7(12):380. https://doi.org/10.3390/catal7120380
Chicago/Turabian StyleTommasi, Immacolata. 2017. "Direct Carboxylation of C(sp3)-H and C(sp2)-H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions" Catalysts 7, no. 12: 380. https://doi.org/10.3390/catal7120380
APA StyleTommasi, I. (2017). Direct Carboxylation of C(sp3)-H and C(sp2)-H Bonds with CO2 by Transition-Metal-Catalyzed and Base-Mediated Reactions. Catalysts, 7(12), 380. https://doi.org/10.3390/catal7120380