Application of Heterogeneous Catalysts in the First Steps of the Oseltamivir Synthesis



Abstract
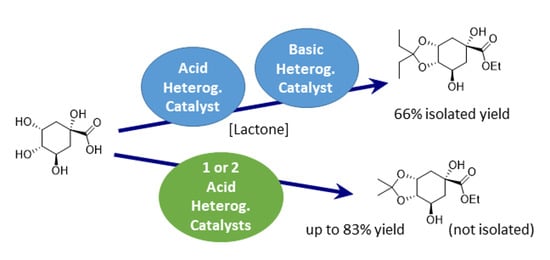
:
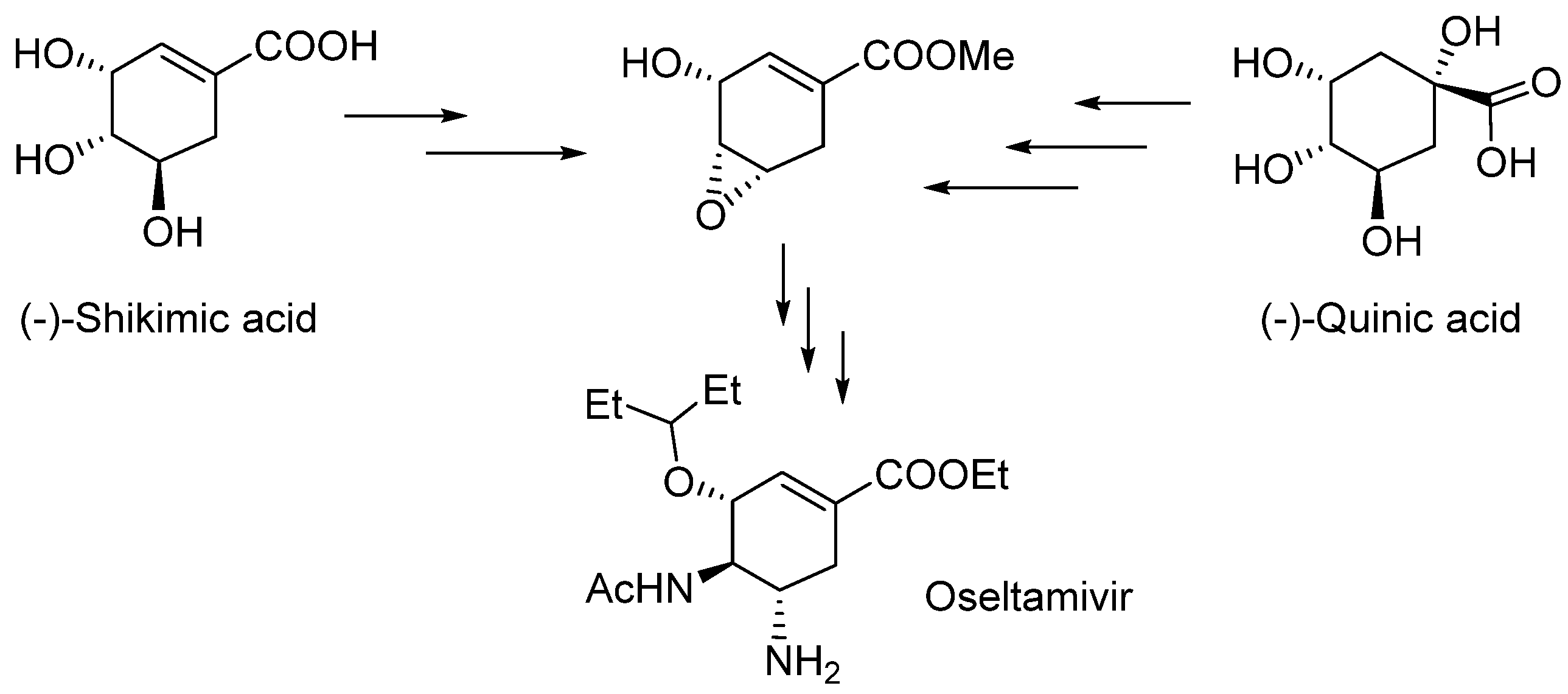
1. Introduction
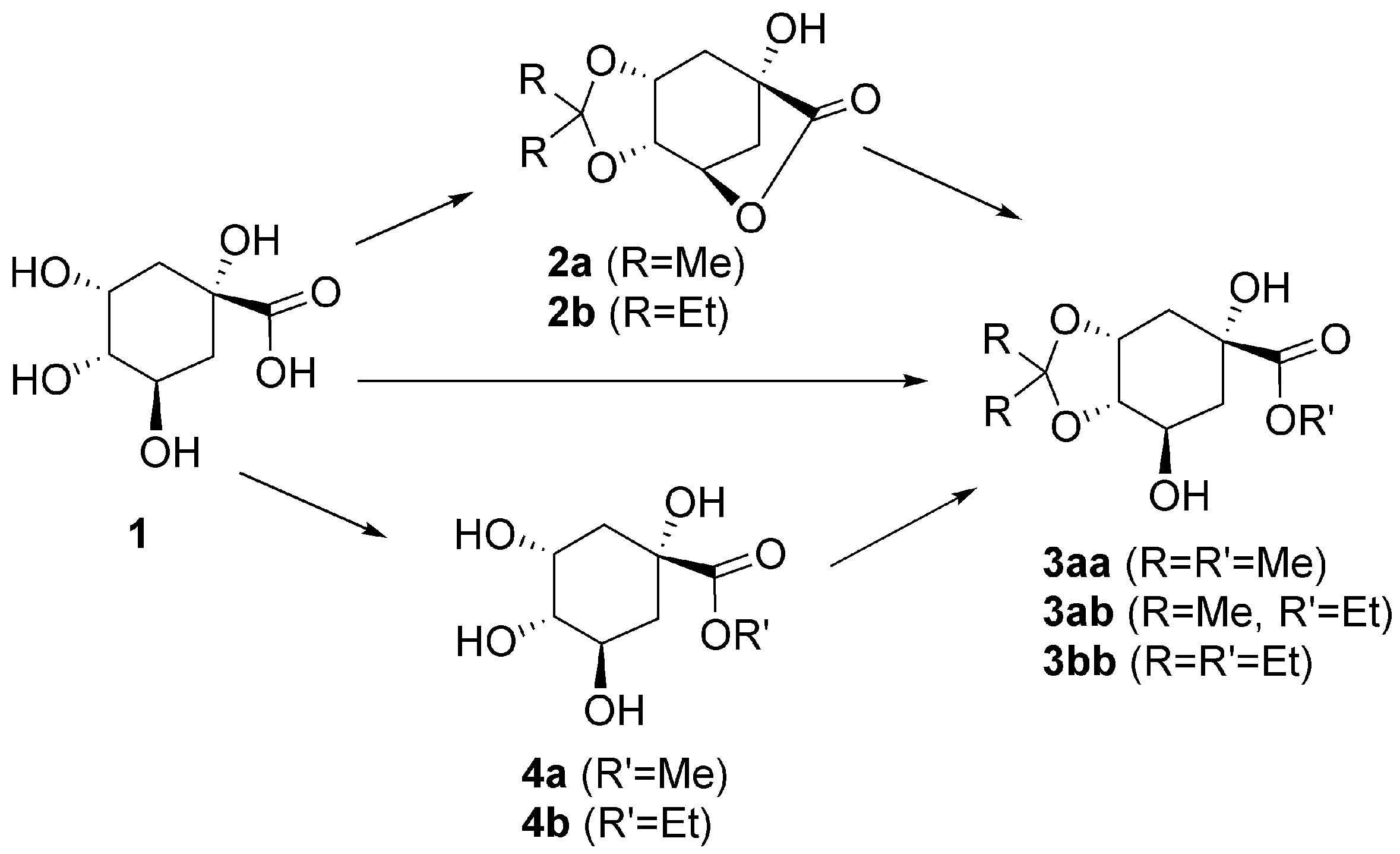
2. Results and Discussion
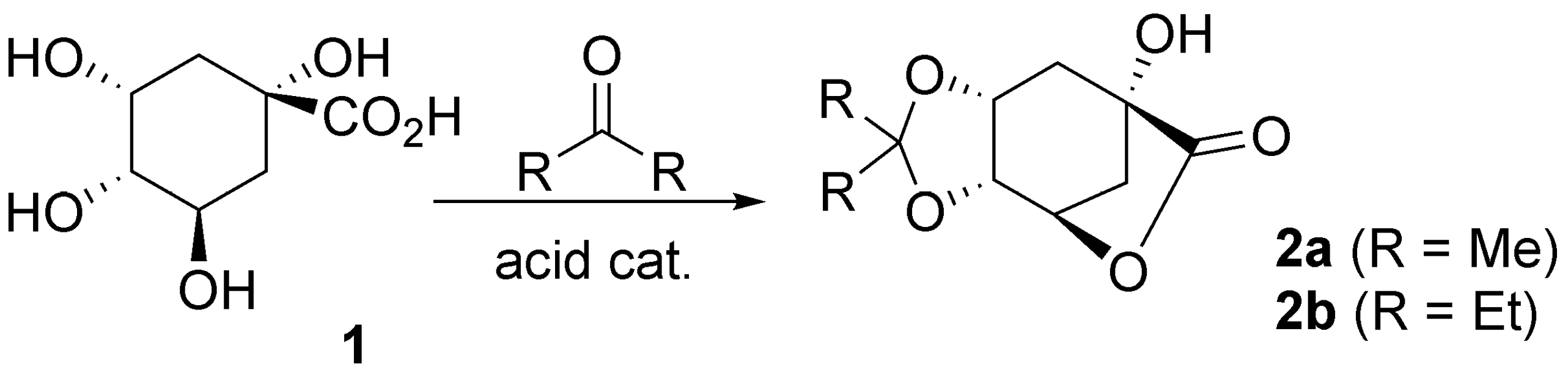
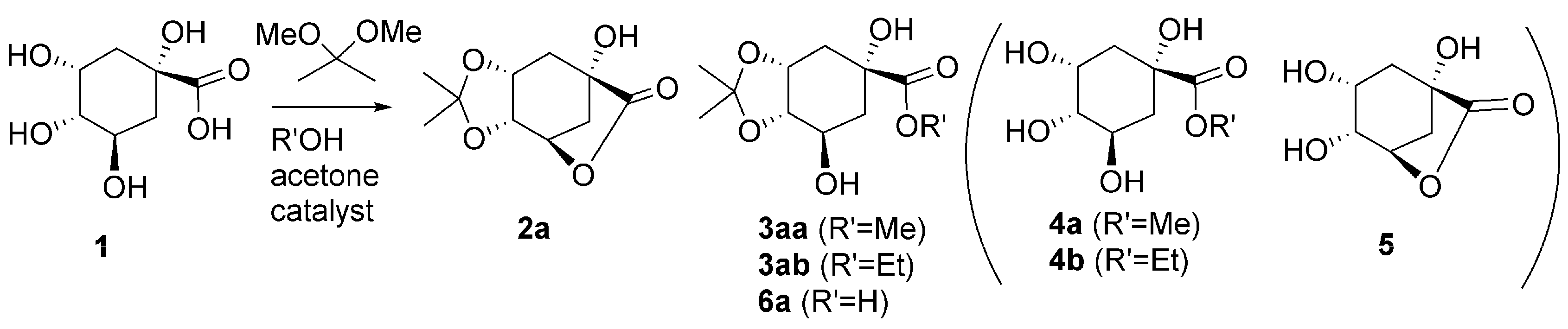
2.1. Single Step One: Acetal Formation from Quinic Acid
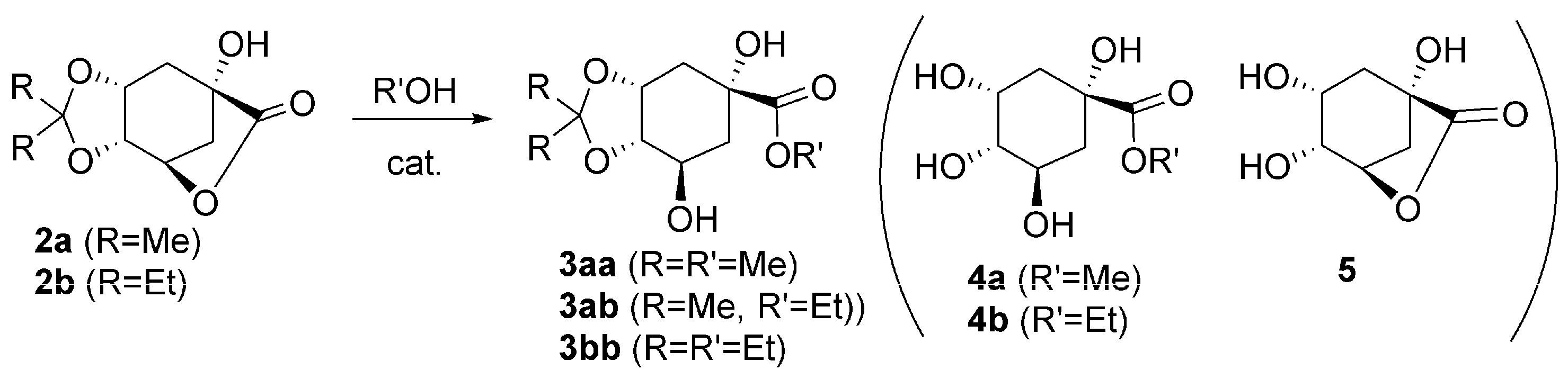
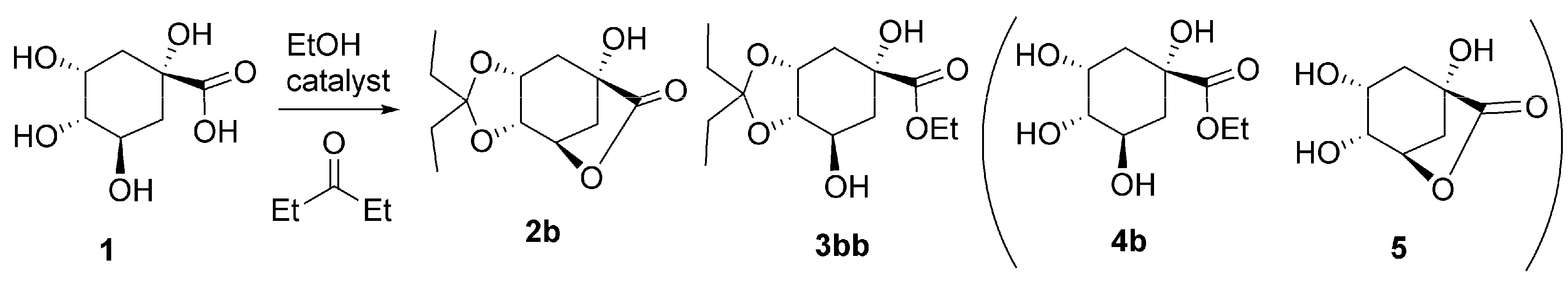
2.2. Single Step Two: Lactone Alcoholysis
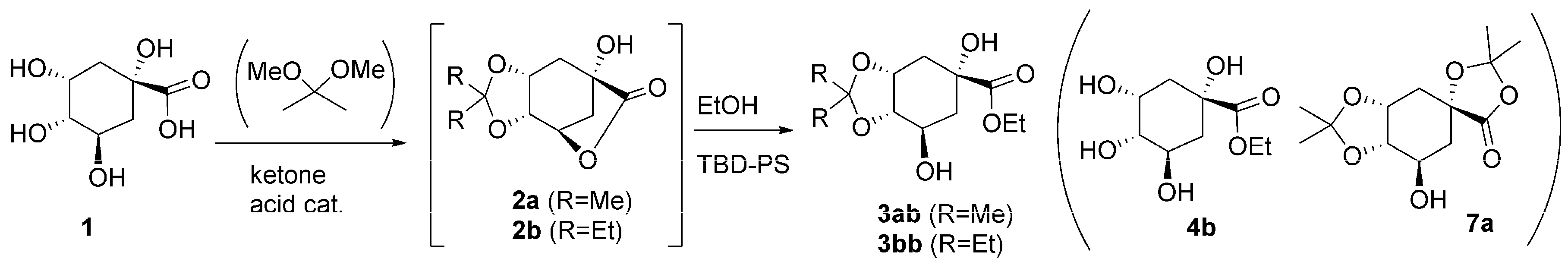
2.3. One-Pot Acetalization and Lactone Alcoholysis: Single Acid Catalyst
2.4. One-Pot Sequential Process with One Acid and One Base Catalyst: Acetalization and Lactone Alcoholysis
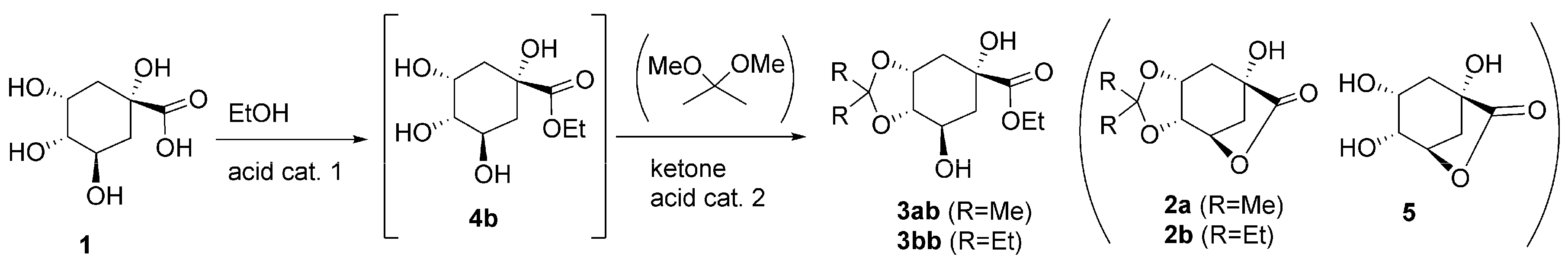
2.5. Single Step Three: Esterification of Quinic Acid
2.6. Sequential Process with One or Two Acid Catalysts: Esterification and Acetalization
2.7. Process at Gram Scale
3. Materials and Methods
3.1. Acetalization of Quinic Acid (1)
3.2. Alcoholysis of Acetal–Lactones (2)
3.3. One-Pot Acetalization and Lactone Alcoholysis with a Single Acid Catalyst
3.4. One-Pot Acetalization and Lactone Alcoholysis with One Acid and One Basic Catalyst
3.5. Esterification of Quinic Acid
3.6. Sequential Esterification and Acetalization with Two Acid Catalysts
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Federsel, H.-J. Chemical process research and development in the 21st century: Challenges, strategies, and solutions from a pharmaceutical industry perspective. Acc. Chem. Res. 2009, 42, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Verma, V.A.; Paxton, T.J.; Pillow, T.H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.A.; Santos, S.; Martin, W.H.C. Combining pot, atom and step economy (PASE) in organic synthesis. Synthesis of tetrahydropyran-4-ones. Green Chem. 2007, 9, 438–440. [Google Scholar] [CrossRef] [Green Version]
- Vaxelaire, C.; Winter, P.; Christmann, M. One-pot reactions accelerate the synthesis of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2011, 50, 3605–3607. [Google Scholar] [CrossRef] [PubMed]
- Climent, M.J.; Corma, A.; Iborra, S.; Sabater, M.J. Heterogeneous catalysis for tandem reactions. ACS Catal. 2014, 4, 870–891. [Google Scholar] [CrossRef]
- Fraile, J.M.; Mallada, R.; Mayoral, J.A.; Menéndez, M.; Roldán, L. Shift of multiple incompatible equilibriums by a combination of heterogeneous catalysis and membranes. Chem. Eur. J. 2010, 16, 3296–3299. [Google Scholar] [CrossRef] [PubMed]
- Magano, J. Synthetic approaches to the neuraminidase inhibitors zanamivir (relenza) and oseltamivir phosphate (tamiflu) for the treatment of influenza. Chem. Rev. 2009, 109, 4398–4438. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, M.; Fischer, R.; Hennig, M.; Mair, H.J.; Oberhauser, T.; Rimmler, G.; Albiez, T.; Bruhin, J.; Estermann, H.; Gandert, C.; et al. Industrial synthesis of the key precursor in the synthesis of the anti-influenza drug oseltamivir phosphate (Ro 64-0796/002, GS-4104-02): Ethyl (3R,4S,5S)-4,5-epoxy-3-(1-ethyl-propoxy)-cyclohex-1-ene-1-carboxylate. Org. Proc. Res. Dev. 1999, 3, 266–274. [Google Scholar] [CrossRef]
- Mita, T.; Fukuda, N.; Roca, F.X.; Kanai, M.; Shibasaki, M. Second generation catalytic asymmetric synthesis of Tamiflu: Allylic substitution route. Org. Lett. 2007, 9, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Zutter, U.; Iding, H.; Spurr, P.; Wirz, B. New, efficient synthesis of Oseltamivir phosphate (Tamiflu) via enzymatic desymmetrization of a meso-1,3-cyclohexanedicarboxylic acid diester. J. Org. Chem. 2008, 73, 4895–4902. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Zhang, T. A concise synthesis of (−)-Oseltamivir. Angew. Chem. Int. Ed. 2008, 47, 3759–3761. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.; Hong, S.; Corey, E.J. A short enantioselective pathway for the synthesis of the anti-influenza neuramidase inhibitor Oseltamivir from 1,3-butadiene and acrylic acid. J. Am. Chem. Soc. 2006, 128, 6310–6311. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Akiba, R.; Yokoshima, S.; Fukuyama, T. A practical synthesis of (−)-Oseltamivir. Angew. Chem. Int. Ed. 2007, 46, 5734–5736. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-A.; Fang, J.-M. Synthesis of oseltamivir and tamiphosphor from N-acetyl-d-glucosamine. Org. Biomol. Chem. 2013, 11, 7687–7699. [Google Scholar] [CrossRef] [PubMed]
- Kongkathip, B.; Akkarasamiyo, S.; Kongkathip, N. A new and efficient asymmetric synthesis of oseltamivir phosphate (Tamiflu) from D-glucose. Tetrahedron 2015, 71, 2393–2399. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ogasawara, S. Time economical total synthesis of (−)-Oseltamivir. Org. Lett. 2016, 18, 3426–3429. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.C.; Kent, K.M.; Postich, M.J.; Becker, M.W.; Chapman, H.H.; Kelly, D.E.; Lew, W.; Louie, M.S.; McGee, L.R.; Prisbe, E.J.; et al. Practical total synthesis of the anti-influenza drug GS-4104. J. Org. Chem. 1998, 63, 4545–4550. [Google Scholar] [CrossRef]
- Karpf, M.; Trussardi, R. New, azide-free transformation of epoxides into 1,2-diamino compounds: Synthesis of the anti-influenza neuraminidase inhibitor oseltamivir phosphate (tamiflu). J. Org. Chem. 2001, 66, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Andraos, J. Global green chemistry metrics analysis algorithm and spreadsheets: Evaluation of the material efficiency performances of synthesis plans for oseltamivir phosphate (tamiflu) as a test case. Org. Proc. Res. Dev. 2009, 13, 161–185. [Google Scholar] [CrossRef]
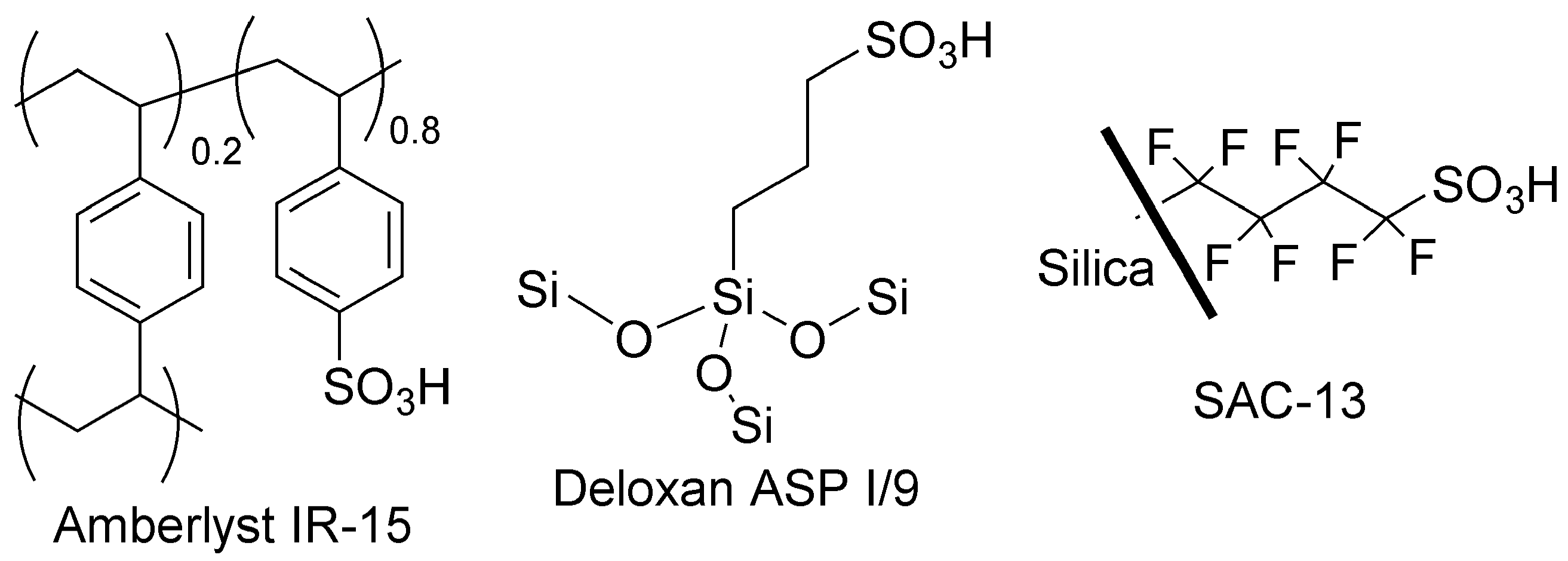
- Harmer, M.A.; Sun, Q. Solid acid catalysis using ion-exchange resins. Appl. Catal. A 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Gelbard, G. Organic synthesis by catalysis with ion-exchange resins. Ind. Eng. Chem. Res. 2005, 44, 8468–8498. [Google Scholar] [CrossRef]
- Melero, J.A.; van Grieken, R.; Morales, G. Advances in the synthesis and catalytic applications of organosulfonic-functionalized mesostructured materials. Chem. Rev. 2006, 106, 3790–3812. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Sharma, M.M. Cationic ion exchange resins as catalyst. React. Polym. 1993, 20, 1–45. [Google Scholar] [CrossRef]
- Wieland, S.; Panster, P. Replacing liquid acids in fine chemical synthesis by sulfonated polysiloxanes as solid acids and as supports for precious metal catalysts. Stud. Surf. Sci. Catal. 1997, 108, 67–74. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q.; Farneth, W.E. High surface area nafion resin/silica nanocomposites: A new class of solid acid catalyst. J. Am. Chem. Soc. 1996, 118, 7708–7715. [Google Scholar] [CrossRef]
- Fraile, J.M.; García-Bordejé, E.; Pires, E.; Roldán, L. New insights into the strength and accessibility of acid sites of sulfonated hydrothermal carbon. Carbon 2014, 77, 1157–1167. [Google Scholar] [CrossRef]
- Fraile, J.M.; García-Bordejé, E.; Pires, E.; Roldán, L. Catalytic performance and deactivation of sulfonated hydrothermal carbon in the esterification of fatty acids: Comparison with sulfonic solids of different nature. J. Catal. 2015, 324, 107–118. [Google Scholar] [CrossRef]
- De la Calle, C.; Fraile, J.M.; García-Bordejé, E.; Pires, E.; Roldán, L. Biobased catalyst in biorefinery processes: Sulphonated hydrothermal carbon for glycerol esterification. Catal. Sci. Technol. 2015, 5, 2897–2903. [Google Scholar] [CrossRef]
- Sinisi, V.; Boronov, K.; Colomban, S.; Navarini, L.; Berti, F.; Forzato, C. Synthesis of mono-, di-, and tri-3,4-dimethoxycinnamoyl-1,5-γ-quinides. Eur. J. Org. Chem. 2014, 1321–1326. [Google Scholar] [CrossRef]
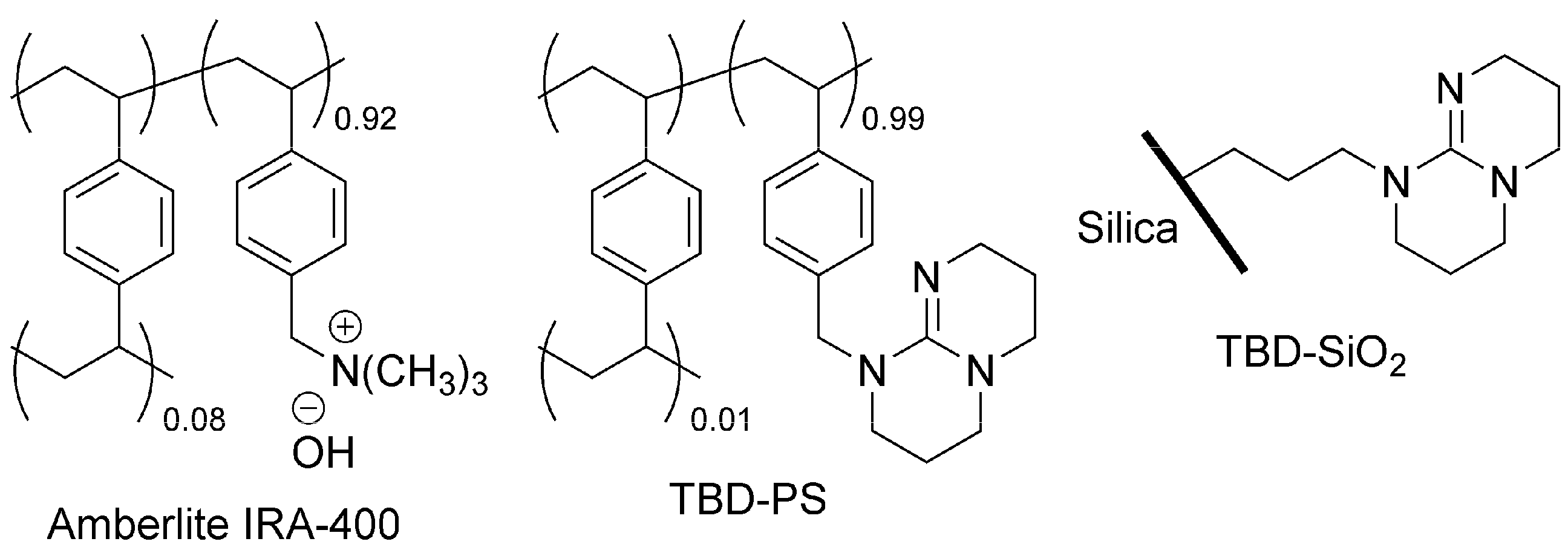
- Solabannavar, S.B.; Desai, U.V.; Mane, R.B. Heck reaction in aqueous medium using Amberlite IRA-400 (basic). Green Chem. 2002, 4, 347–348. [Google Scholar] [CrossRef]
- Simoni, D.; Rondanin, R.; Morini, M.; Baruchello, R.; Invidiata, F.P. 1,5,7-Triazabicyclo[4.4.0]dec-1-ene (TBD), 7-methyl-TBD (MTBD) and the polymer-supported TBD (P-TBD): Three efficient catalysts for the nitroaldol (Henry) reaction and for the addition of dialkyl phosphites to unsaturated systems. Tetrahedron Lett. 2000, 41, 1607–1610. [Google Scholar] [CrossRef]
- Soldi, L.; Ferstl, W.; Loebbecke, S.; Maggi, R.; Malmassari, C.; Sartori, G.; Yada, S. Use of immobilized organic base catalysts for continuous-flow fine chemical synthesis. J. Catal. 2008, 258, 289–295. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, N.; Herrerías, C.I.; Mayoral, J.A. Heterogeneous catalysis for tandem Mukaiyama-Michael and hydrogenation reactions: One-pot vs sequential processes. ACS Catal. 2012, 2, 56–64. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, N.; Herrerías, C.I.; Mayoral, J.A. Integration of heterogeneous catalysts into complex synthetic routes: Sequential vs.one-pot reactions in a (Knoevenagel + Mukaiyama–Michael + hydrogenation + transesterification) sequence. Catal. Sci. Technol. 2013, 3, 436–443. [Google Scholar] [CrossRef]
- Sánchez-Abella, L.; Férnandez, S.; Armesto, N.; Ferrero, M.; Gotor, V. Novel and efficient syntheses of (−)-methyl 4-epi-shikimate and 4,5-epoxy-quinic and -shikimic acid derivatives as key precursors to prepare new analogues. J. Org. Chem. 2006, 71, 5396–5399. [Google Scholar] [CrossRef] [PubMed]
- Baptistella, L.H.B.; Cerchiaro, G. Studies for the transformation of carbocycles into carbohydrates: Approach toward the synthesis of higher sugar derivatives. Carbohydr. Res. 2004, 339, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Lange, G.L.; Humber, C.C.; Manthorpe, J.M. [2+2] Photoadditions with chiral 2,5-cyclohexadienone synthons. Tetrahedron Asymmetry 2002, 13, 1355–1362. [Google Scholar] [CrossRef]
- Aucktor, J.; Brückner, R. Total Synthesis of quercitols: (+)-allo-, (–)-proto-, (+)-talo-, (–)-gala-, (+)-gala-, neo-, and (–)-epi-quercitol. Synlett 2015, 26, 250–258. [Google Scholar] [CrossRef]
- Banwell, M.G.; Hungerford, N.L.; Jolliffe, K.A. Synthesis of the sialic acid (−)-KDN and certain epimers from (−)-3-dehydroshikimic acid or (−)-quinic acid. Org. Lett. 2004, 6, 2737–2740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, X.-L.; Ding, W.; Shi, X.-X. A novel stereoselective synthesis of (−)-quinic acid starting from the naturally abundant (−)-shikimic acid. Tetrahedron Asymmetry 2015, 26, 1375–1381. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ketone | Catalyst 2 | T (°C) | DMP 3 | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Acetone | PTSA | 56 | Yes | 3 | 83 |
| 2 | (R = Me) | IR-15 | 56 | Yes | 3 | 48 |
| 48 | 73 | |||||
| 3 | Deloxan | 56 | Yes | 3 | 91 | |
| 4 | 56 | No | 3 | 10 | ||
| 5 | 25 | Yes | 68 | 63 | ||
| 6 | 66 | Yes 4 | 3 | 11 | ||
| 7 | SAC-13 | 56 | Yes | 3 | 86 | |
| 8 | 56 | No | 3 | 15 | ||
| 24 | 40 | |||||
| 72 | 64 | |||||
| 9 | SHTC | 56 | Yes | 3 | 90 5 | |
| 10 | 56 | No | 3 | 9 | ||
| 24 | 64 | |||||
| 48 | 80 | |||||
| 11 | 25 | Yes | 24 | 82 | ||
| 12 | 66 | Yes 4 | 3 | 61 | ||
| 13 | Pentan-3-one | Deloxan | 101 | No | 24 | 100 |
| 14 | (R = Et) | SHTC | 101 | No | 24 | 91 |
| 48 | 100 |
| Entry | Reaction | Catalyst | T | Time | Yield (%) | |||
|---|---|---|---|---|---|---|---|---|
| (°C) | (h) | 2 | 3 | 4 | 5 | |||
| 1 | 2a + EtOH | SAC-13 | 78 | 24 | 13 | 1 | 76 | 10 |
| 2 | 25 | 24 | 87 | 0 | 6 | 7 | ||
| 3 | SHTC | 78 | 24 | 17 | 2 | 72 | 9 | |
| 4 | 25 | 24 | 82 | 0 | 9 | 9 | ||
| 5 | IRA-400 | 78 | 5 | 42 | 58 | - | - | |
| 6 | TBD-SiO2 | 78 | 4 | 40 | 60 | - | - | |
| 7 | 25 | 4 | 69 | 31 | - | - | ||
| 19 | 45 | 55 | - | - | ||||
| 8 | −20 | 48 | 21 | 79 | - | - | ||
| 9 | TBD-PS | 78 | 4 | 36 | 64 | - | - | |
| 10 | 0 | 4 | 60 | 40 | - | - | ||
| 24 | 14 | 86 | - | - | ||||
| 48 | 10 | 90 2 | - | - | ||||
| 11 | Me4NOH | 78 | 24 | 39 | 61 | - | - | |
| 12 | 0 | 24 | 30 | 70 | - | - | ||
| 48 | 30 | 70 | - | - | ||||
| 13 | TBD | 78 | 24 | 42 | 58 | - | - | |
| 14 | 0 | 24 | 42 | 58 | - | - | ||
| 48 | 43 | 57 | - | - | ||||
| 15 | 2b + EtOH | TBD-SiO2 | 78 | 4 | 48 | 52 | - | - |
| 16 | TBD-PS | 78 | 4 | 44 | 56 | - | - | |
| 17 | 0 | 4 | 29 | 71 | - | - | ||
| 24 | 16 | 84 | - | - | ||||
| 48 | 14 | 86 2 | - | - | ||||
| 18 | 2a + MeOH | TBD-PS | 0 | 24 | 31 | 69 | - | - |
| 48 | 4 | 96 2 | - | - | ||||
| Entry | Alcohol | Catalyst | Yield (%) | ||||
|---|---|---|---|---|---|---|---|
| 2a | 3 | 4 | 5 | 6 | |||
| 1 | EtOH | Deloxan 2 | 56 | 8 | 9 | 7 | 20 |
| 2 | IR-15 | 54 | 0 | 32 | 6 | 8 | |
| 3 | SAC-13 | 38 | 3 | 38 | 10 | 11 | |
| 4 | SHTC | 46 | 0 | 24 | 12 | 3 3 | |
| 5 | MeOH | Deloxan | 41 | 25 | 23 | 7 | 4 |
| 6 | IR-15 | 35 | 9 | 45 | 10 | 1 | |
| 7 | SAC-13 | 38 | 26 | 26 | 7 | 3 | |
| 8 | SHTC | 39 | 0 | 49 | 12 | 0 | |
| Catalyst | Yield (%) | |||
|---|---|---|---|---|
| 2b | 3bb | 4b | 5 | |
| Deloxan | 27 | 3 | 43 | 24 |
| IR-15 | 34 | 2 | 47 | 17 |
| SAC-13 | 20 | 10 | 53 | 17 |
| SHTC | 67 | 0 | 29 | 4 |
| Entry | Ketone | Acid Catalyst | Time (h) | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 2 | 3 | 4b | 7a | |||
| 1 | Acetone | Deloxan | 4 | 24 | 54 | 21 | - | 19 |
| 48 | 53 | 26 | - | 16 | ||||
| 2 | SHTC | 4 | 24 | 41 | 28 | - | 31 2 | |
| 48 | 38 | 32 | - | 30 | ||||
| 3 | SHTC 3 | 4 | 24 | 48 | 23 | - | 28 | |
| 120 | 25 | 46 | - | 29 | ||||
| 4 | SHTC 4 | 4 | 24 | 19 | 65 | 16 | - | |
| 48 | 11 | 49 | 40 | - | ||||
| 5 | Pentan-3-one | Deloxan | 24 | 24 | 100 | - | - | - |
| 48 | 85 | - | 7 5 | - | ||||
| 6 | Deloxan 4 | 24 | 24 | 53 | 23 | 24 | - | |
| 7 | Deloxan 6 | 24 | 24 | 47 | 53 | - | - | |
| 48 | 35 | 65 | - | - | ||||
| 8 | SHTC | 48 | 24 | 87 | 13 | - | - | |
| 120 | 87 | 13 | - | - | ||||
| 9 | SHTC 5 | 48 | 24 | 46 | 54 | - | - | |
| 48 | 29 | 71 | - | - | ||||
| 96 | 24 | 76 | - | - | ||||
| Entry | Alcohol | Catalyst | Time (h) | Yield (%) | |
|---|---|---|---|---|---|
| 4 | 5 | ||||
| 1 | EtOH | Deloxan | 3 | 19 | 12 |
| 24 | 72 | 25 2 | |||
| 48 | 81 | 19 | |||
| 68 | 87 | 13 | |||
| 2 | Deloxan 3 | 24 | 83 | 17 | |
| 3 | SAC-13 | 24 | 65 | 22 | |
| 4 | SHTC | 24 | 82 | 9 | |
| 48 | 90 | 10 | |||
| 72 | 93 | 7 | |||
| 5 | MeOH | Deloxan | 48 | 94 | 6 |
| 6 | SHTC | 24 | 93 | 7 | |
| Entry | Acid catalyst 1 | Esterification Time (h) | Acid catalyst 2 | Ketone | Acetalization | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | Time (h) | 4b | 3 | 2 | 5 | |||||
| 1 | Deloxan | 70 | SHTC | Acetone | 25 | 15 | 29 | 56 | 15 | 0 |
| 24 | 13 | 63 | 24 | 0 | ||||||
| 2 | Deloxan | 70 | SHTC | Acetone 2 | 25 | 40 | 27 | 57 | 16 | 0 |
| 3 | Deloxan 3 | 70 | - | Acetone | 25 | 40 | 3 | 83 | 14 | 0 |
| 4 | SHTC 3 | 96 | - | Acetone | 25 | 40 | 11 | 74 | 15 | 0 |
| 5 | Deloxan 3 | 70 | - | Acetone | 56 | 40 | 7 | 49 | 44 | 0 |
| 6 | SHTC 3 | 96 | - | Acetone | 56 | 40 | 0 | 9 | 91 | 0 |
| 7 | Deloxan | 70 | SHTC | Acetone | 0 | 70 | 0 | 83 | 17 | 0 |
| 8 | Deloxan 3 | 70 | - | Acetone | 0 | 40 | 10 | 76 | 14 | 0 |
| 9 | Deloxan | 70 | SHTC | Pentan-3-one | 25 | 70 | 64 | 24 | 3 | 9 |
| 10 | Deloxan 3 | 70 | - | Pentan-3-one | 25 | 78 | 79 | 9 | 2 | 10 |
| 11 | SHTC | 48 | Deloxan | Pentan-3-one | 25 | 78 | 41 | 39 | 4 | 16 |
| 12 | SHTC | 48 | SHTC | Pentan-3-one | 25 | 78 | 44 | 35 | 13 | 8 |
| 13 | SHTC | 48 | Deloxan | Pentan-3-one | 40 | 68 | 47 | 14 | 33 | 6 |
| 14 | SHTC | 48 | SHTC | Pentan-3-one | 40 | 20 | 62 | 12 | 20 | 6 |
| 68 | 41 | 14 | 38 | 7 | ||||||
| 15 | SHTC | 48 | SHTC | Pentan-3-one | 101 | 24 | 2 | 0 | 82 | 16 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraile, J.M.; Saavedra, C.J. Application of Heterogeneous Catalysts in the First Steps of the Oseltamivir Synthesis. Catalysts 2017, 7, 393. https://doi.org/10.3390/catal7120393
Fraile JM, Saavedra CJ. Application of Heterogeneous Catalysts in the First Steps of the Oseltamivir Synthesis. Catalysts. 2017; 7(12):393. https://doi.org/10.3390/catal7120393
Chicago/Turabian StyleFraile, José M., and Carlos J. Saavedra. 2017. "Application of Heterogeneous Catalysts in the First Steps of the Oseltamivir Synthesis" Catalysts 7, no. 12: 393. https://doi.org/10.3390/catal7120393
APA StyleFraile, J. M., & Saavedra, C. J. (2017). Application of Heterogeneous Catalysts in the First Steps of the Oseltamivir Synthesis. Catalysts, 7(12), 393. https://doi.org/10.3390/catal7120393