α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
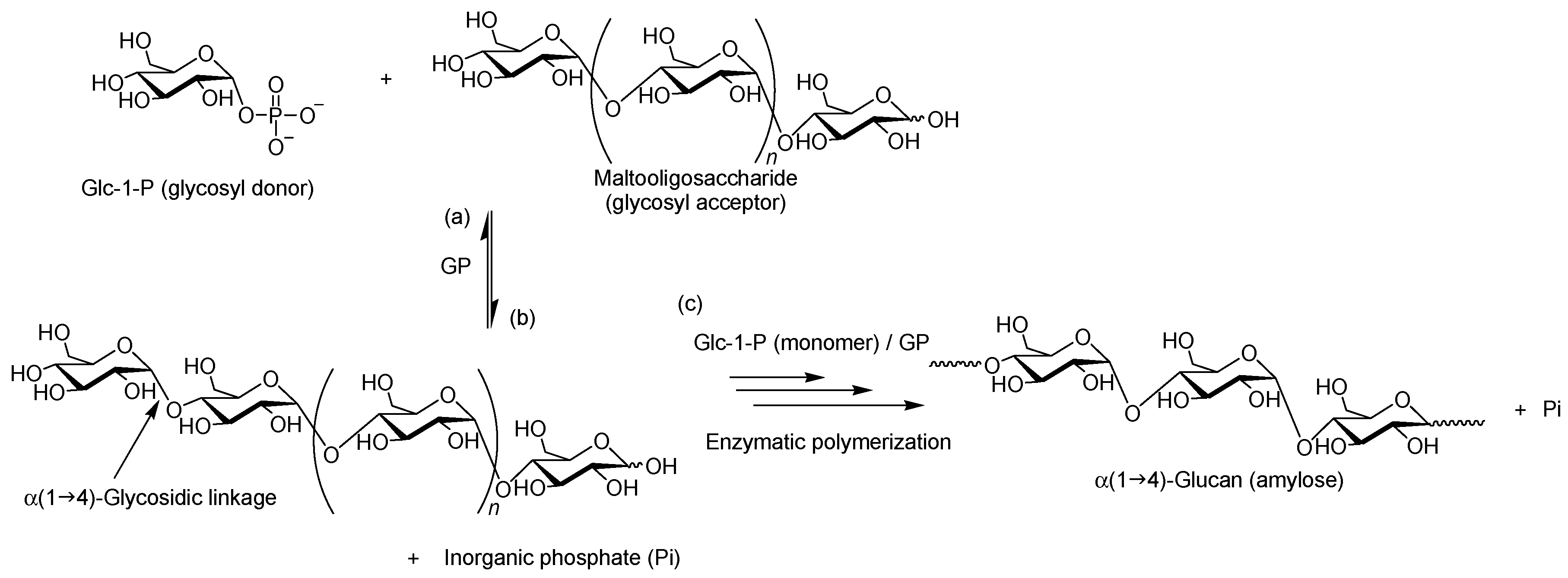
2. Characteristics of GP-Catalyzed Enzymatic Reactions
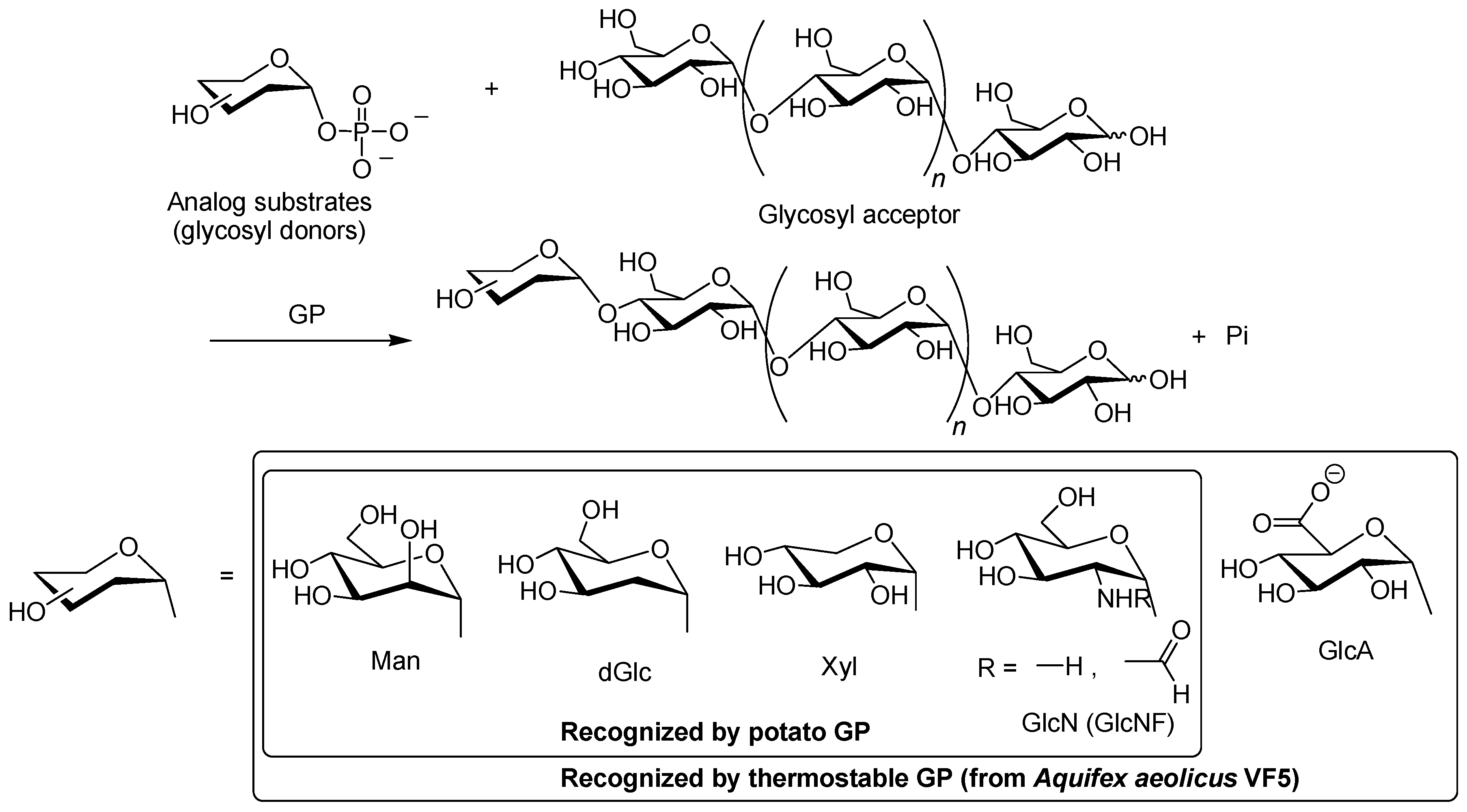
3. Synthesis of Non-Natural Oligosaccharides by Phosphorylase-Catalyzed Enzymatic Glycosylations Using Analog Substrates
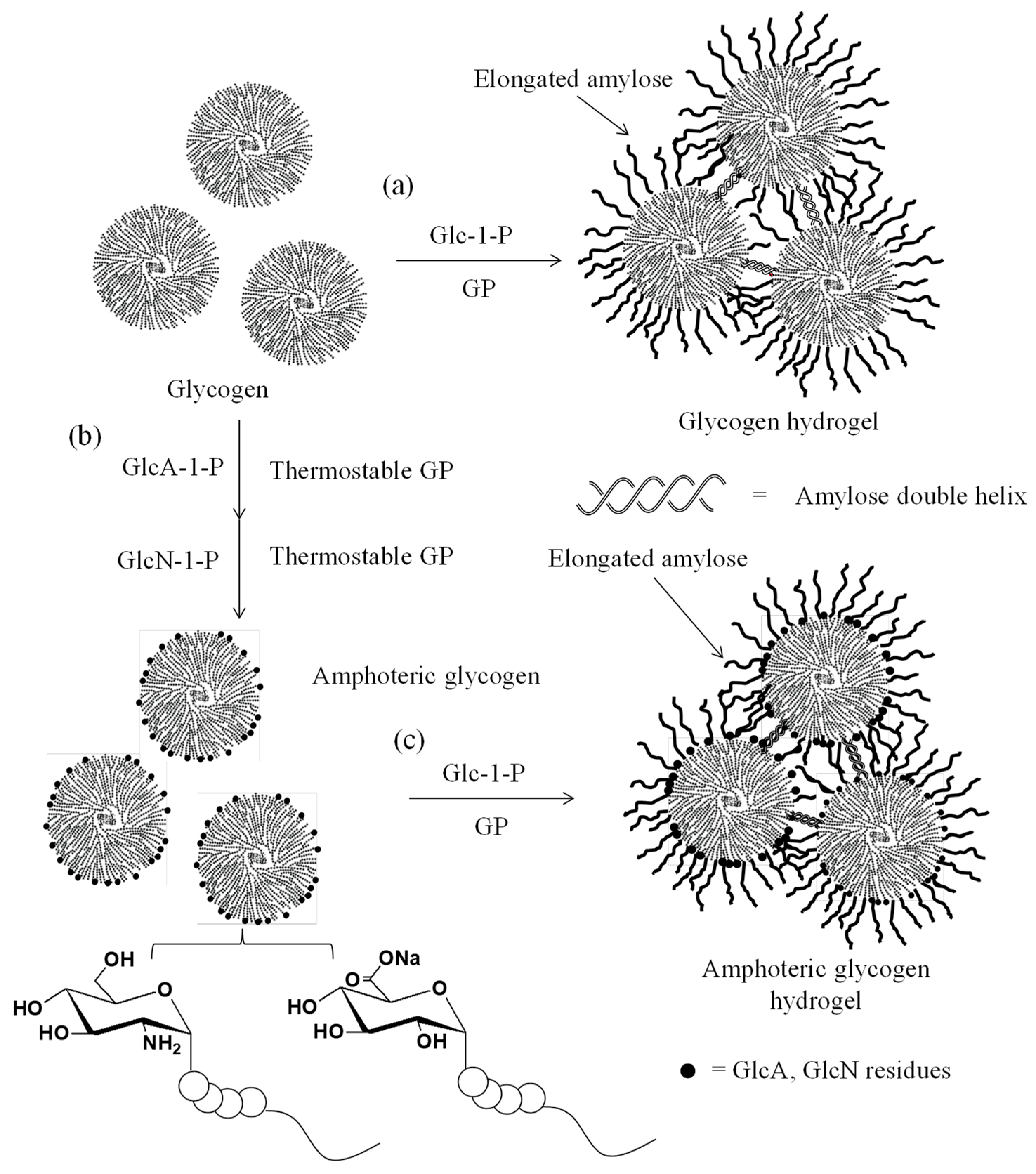
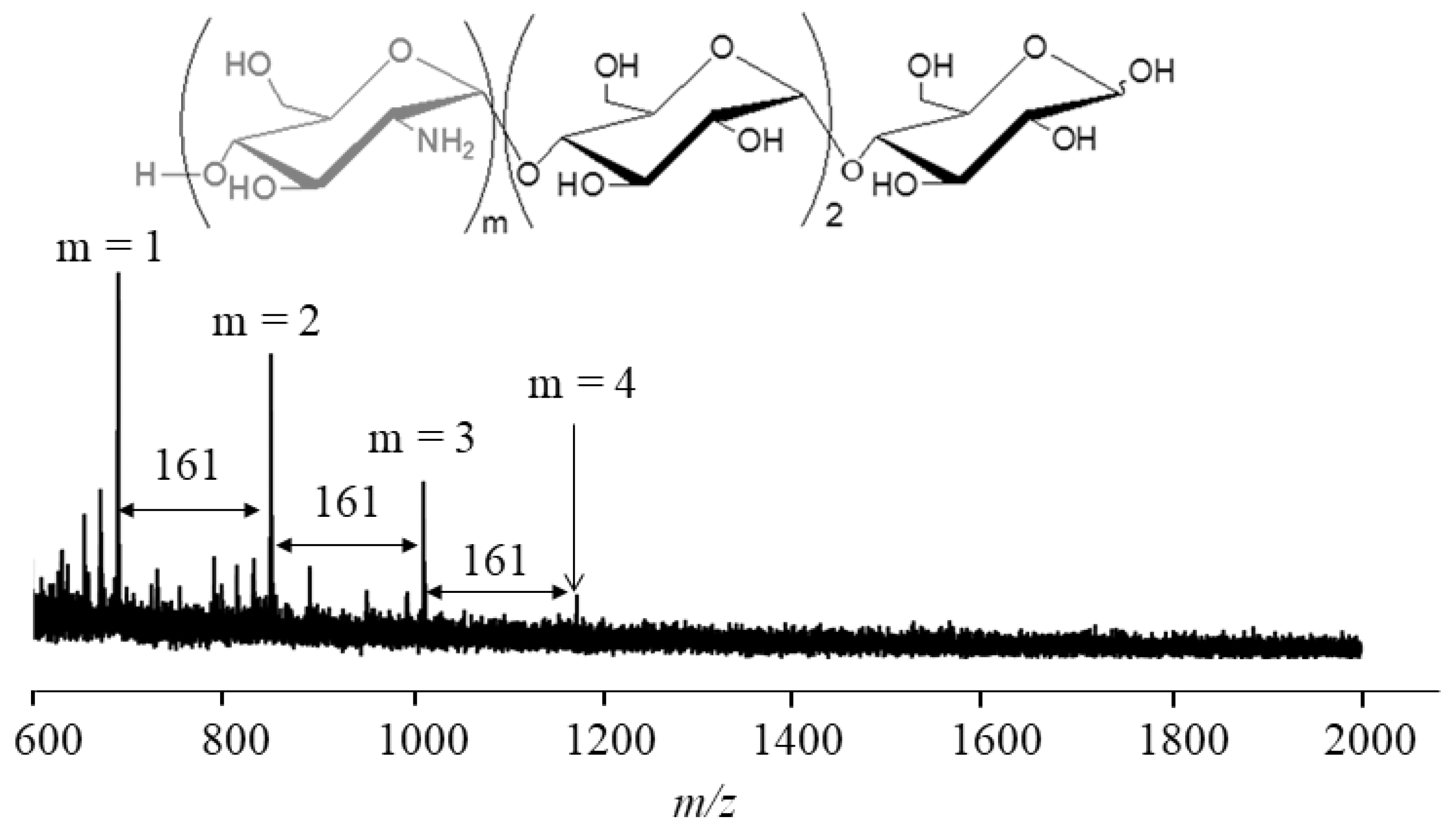
4. Thermostable GP-Catalyzed Enzymatic Oligomerization and Polymerization of Analog Monomers
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Schuerch, C. Polysaccharides. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bilkales, N., Overberger, C.G., Eds.; John Wiley & Sons: New York, NY, USA, 1986; Volume 13, pp. 87–162. [Google Scholar]
- Yalpani, M. Polysaccharides: Syntheses, Modifications, and Structure/Property Relations; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Paulsen, H. Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem. Int. Ed. 1982, 21, 155–173. [Google Scholar] [CrossRef]
- Schmidt, R.R. New methods for the synthesis of glycosides and oligosaccharides—Are there alternatives to the Königs-Knorr method? Angew. Chem. Int. Ed. 1986, 25, 212–235. [Google Scholar] [CrossRef]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Shoda, S. Enzymatic glycosylation. In Glycoscience Chemistry and Chemical Biology; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; Volume II, pp. 1465–1496. [Google Scholar]
- Shoda, S.; Izumi, R.; Fujita, M. Green process in glycotechnology. Bull. Chem. Soc. Jpn. 2003, 76, 1–13. [Google Scholar] [CrossRef]
- Blixt, O.; Razi, N. Enzymatic glycosylation by transferases. In Glycoscience Chemistry and Chemical Biology, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1361–1385. [Google Scholar]
- Thiem, J.; Thimm, J. Enzymatic glycosylation by glycohydrolases and glycosynthases. In Glycoscience Chemistry and Chemical Biology, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1387–1409. [Google Scholar]
- Kitaoka, M.; Hayashi, K. Carbohydrate-processing phosphorolytic enzymes. Trends Glycosci. Glycotechnol. 2002, 14, 35–50. [Google Scholar] [CrossRef]
- Nakai, H.; Kitaoka, M.; Svensson, B.; Ohtsubo, K. Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr. Opin. Chem. Biol. 2013, 17, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Puchart, V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol. Adv. 2015, 33, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Precision polysaccharide synthesis catalyzed by enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.; Kaneko, Y. Engineering of Polysaccharide Materials–by Phosphorylase-Catalyzed Enzymatic Chain-Elongation; Pan Stanford Publishing Pte Ltd.: Singapore, 2013. [Google Scholar]
- O’Neill, E.C.; Field, R.A. Enzymatic synthesis using glycoside phosphorylases. Carbohydr. Res. 2015, 403, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Precision synthesis of functional polysaccharide materials by phosphorylase-catalyzed enzymatic reactions. Polymers 2016, 8, 138. [Google Scholar] [CrossRef]
- Kadokawa, J. α;-Glucan phosphorylase: A useful catalyst for precision enzymatic synthesis of oligo- and polysaccharides. Curr. Org. Chem. 2017, 21, 1192–1204. [Google Scholar] [CrossRef]
- Ubiparip, Z.; Beerens, K.; Franceus, J.; Vercauteren, R.; Desmet, T. Thermostable α-glucan phosphorylases: Characteristics and industrial applications. Appl. Microbiol. Biotechnol. 2018, 102, 8187–8202. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J. Facile synthesis of unnatural oligosaccharides by phosphorylase-catalyzed enzymatic glycosylations using new glycosyl donors. In Oligosaccharides: Sources Properties and Applications; Gordon, N.S., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011; pp. 269–281. [Google Scholar]
- Kadokawa, J. Synthesis of non-natural oligosaccharides by α-glucan phosphorylase-catalyzed enzymatic glycosylations using analogue substrates of α-d-glucose 1-phosphate. Trends Glycosci. Glycotechnol. 2013, 25, 57–69. [Google Scholar] [CrossRef]
- Kadoakwa, J. Enzymatic synthesis of non-natural oligo- and polysaccharides by phosphorylase-catalyzed glycosylations using analogue substrates. In Green Polymer Chemistry: Biobased Materials and Biocatalysis; Cheng, H.N., Gross, R.A., Smith, P.B., Eds.; ACS Symposium Series 1192; American Chemical Society: Washington, DC, USA, 2015; pp. 87–99. [Google Scholar]
- Nishimura, T.; Akiyoshi, K. Amylose engineering: Phosphorylase-catalyzed polymerization of functional saccharide primers for glycobiomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Palm, D.; Klein, H.W.; Schinzel, R.; Buehner, M.; Helmreich, E.J.M. The role of pyridoxal 5′-phosphate in glycogen phosphorylase catalysis. Biochemistry 1990, 29, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Browner, M.F.; Fletterick, R.J. Phosphorylase: A biological transducer. Trends Biochem. Sci. 1992, 17, 66–71. [Google Scholar] [CrossRef]
- Johnson, L.N. Glycogen phosphorylase: Control by phosphorylation and allosteric effectors. FASEB J. 1992, 6, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Schinzel, R.; Nidetzky, B. Bacterial α-glucan phosphorylases. FEMS Microbiol. Lett. 1999, 171, 73–79. [Google Scholar] [CrossRef]
- Boeck, B.; Schinzel, R. Purification and characterisation of an α-glucan phosphorylase from the thermophilic bacterium Thermus thermophilus. Eur. J. Biochem. 1996, 239, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Takaha, T.; Yanase, M.; Takata, H.; Okada, S. Structure and properties of Thermus aquaticus α-glucan phosphorylase expressed in Escherichichia coli. J. Appl. Glycosci. 2001, 48, 71–78. [Google Scholar] [CrossRef]
- Yanase, M.; Takata, H.; Fujii, K.; Takaha, T.; Kuriki, T. Cumulative effect of amino acid replacements results in enhanced thermostability of potato type L α-glucan phosphorylase. Appl. Environ. Microbiol. 2005, 71, 5433–5439. [Google Scholar] [CrossRef] [PubMed]
- Yanase, M.; Takaha, T.; Kuriki, T. α-Glucan phosphorylase and its use in carbohydrate engineering. J. Sci. Food Agric. 2006, 86, 1631–1635. [Google Scholar] [CrossRef]
- Ziegast, G.; Pfannemüller, B. Linear and star-shaped hybrid polymers. Phosphorolytic syntheses with di-functional, oligo-functional and multifunctional primers. Carbohydr. Res. 1987, 160, 185–204. [Google Scholar] [CrossRef]
- Fujii, K.; Takata, H.; Yanase, M.; Terada, Y.; Ohdan, K.; Takaha, T.; Okada, S.; Kuriki, T. Bioengineering and application of novel glucose polymers. Biocatal. Biotransform. 2003, 21, 167–172. [Google Scholar] [CrossRef]
- Ohdan, K.; Fujii, K.; Yanase, M.; Takaha, T.; Kuriki, T. Enzymatic synthesis of amylose. Biocatal. Biotransform. 2006, 24, 77–81. [Google Scholar] [CrossRef]
- Imberty, A.; Chanzy, H.; Perez, S.; Buleon, A.; Tran, V. The double-helical nature of the crystalline part of A-starch. J. Mol. Biol. 1988, 201, 365–378. [Google Scholar] [CrossRef]
- Imberty, A.; Perez, S. A revisit to the three-dimensional structure of B-type starch. Biopolymers 1988, 27, 1205–1221. [Google Scholar] [CrossRef]
- Kitamura, S. Starch polymers, natural and synthetic. In The Polymeric Materials Encyclopedia, Synthesis, Properties and Applications; Salamone, C., Ed.; CRC Press: New York, NY, USA, 1996; Volume 10, pp. 7915–7922. [Google Scholar]
- Kitamura, S.; Yunokawa, H.; Mitsuie, S.; Kuge, T. Study on polysaccharide by the fluorescence method. 2. Micro-brownian motion and conformational change of amylose in aqueous-solution. Polym. J. 1982, 14, 93–99. [Google Scholar] [CrossRef]
- Izawa, H.; Kadokawa, J. Preparation of functional amylosic materials by phosphorylase-catalyzed polymerization. In Interfacial Researches in Fundamental and Material Sciences of Oligo- and Polysaccharides; Kadoakwa, J., Ed.; Transworld Research Network: Trivandrum, Indian, 2009; pp. 69–86. [Google Scholar]
- Kaneko, Y.; Kadokawa, J. Chemoenzymatic synthesis of amylose-grafted polymers. In Handbook of Carbohydrate Polymers: Development, Properties and Applications; Ito, R., Matsuo, Y., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2009; pp. 671–691. [Google Scholar]
- Omagari, Y.; Kadokawa, J. Synthesis of heteropolysaccharides having amylose chains using phosphorylase-catalyzed enzymatic polymerization. Kobunshi Ronbunshu 2011, 68, 242–249. [Google Scholar] [CrossRef]
- Kadokawa, J. Synthesis of amylose-grafted polysaccharide materials by phosphorylase-catalyzed enzymatic polymerization. In Biobased Monomers, Polymers, and Materials; Smith, P.B., Gross, R.A., Eds.; ACS Symposium Series 1105; American Chemical Society: Washington, DC, USA, 2012; pp. 237–255. [Google Scholar]
- Kadokawa, J. Synthesis of new polysaccharide materials by phosphorylase-catalyzed enzymatic α-glycosylations using polymeric glycosyl acceptors. In Green Polymer Chemistry: Biocatalysis and Materials II; Cheng, H.N., Gross, R.A., Smith, P.B., Eds.; ACS Symposium Series 1144; American Chemical Society: Washington, DC, USA, 2013; pp. 141–161. [Google Scholar]
- Kadokawa, J. Chemoenzymatic synthesis of functional amylosic materials. Pure Appl. Chem. 2014, 86, 701–709. [Google Scholar] [CrossRef]
- Izawa, H.; Nawaji, M.; Kaneko, Y.; Kadokawa, J. Preparation of glycogen-based polysaccharide materials by phosphorylase-catalyzed chain elongation of glycogen. Macromol. Biosci. 2009, 9, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Percival, M.D.; Withers, S.G. Applications of enzymes in the synthesis and hydrolytic study of 2-deoxy-α-d-glucopyranosyl phosphate. Can. J. Chem. 1988, 66, 1970–1972. [Google Scholar] [CrossRef]
- Evers, B.; Thiem, J. Further syntheses employing phosphorylase. Bioorg. Med. Chem. 1997, 5, 857–863. [Google Scholar] [CrossRef]
- Nawaji, M.; Izawa, H.; Kaneko, Y.; Kadokawa, J. Enzymatic synthesis of α-d-xylosylated maltooligosaccharides by phosphorylase-catalyzed xylosylation. J. Carbohydr. Chem. 2008, 27, 214–222. [Google Scholar] [CrossRef]
- Nawaji, M.; Izawa, H.; Kaneko, Y.; Kadokawa, J. Enzymatic α-glucosaminylation of maltooligosaccharides catalyzed by phosphorylase. Carbohydr. Res. 2008, 343, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, S.; Izawa, H.; Nawaji, M.; Kaneko, Y.; Kadokawa, J. Phosphorylase-catalyzed N-formyl-α-glucosaminylation of maltooligosaccharides. Carbohydr. Res. 2010, 345, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.; Mischnick, P.; Thiem, J. Synthesis of 2-deoxy-α-d-arabino-hexopyranosyl phosphate and 2-deoxy-maltooligosaccharides with phosphorylase. Carbohydr. Res. 1994, 262, 335–341. [Google Scholar] [CrossRef]
- Umegatani, Y.; Izawa, H.; Nawaji, M.; Yamamoto, K.; Kubo, A.; Yanase, M.; Takaha, T.; Kadokawa, J. Enzymatic α-glucuronylation of maltooligosaccharides using α-glucuronic acid 1-phosphate as glycosyl donor catalyzed by a thermostable phosphorylase from Aquifex aeolicus vf5. Carbohydr. Res. 2012, 350, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Shimohigoshi, R.; Yamamoto, K.; Kadokawa, J. Enzymatic synthesis of dendritic amphoteric α-glucans by thermostable phosphorylase catalysis. Macromol. Biosci. 2014, 14, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Yamamoto, K.; Kadokawa, J. Preparation of pH-responsive amphoteric glycogen hydrogels by α-glucan phosphorylase-catalyzed successive enzymatic reactions. Macromol. Chem. Phys. 2015, 216, 1415–1420. [Google Scholar] [CrossRef]
- Takemoto, Y.; Izawa, H.; Umegatani, Y.; Yamamoto, K.; Kubo, A.; Yanase, M.; Takaha, T.; Kadokawa, J. Synthesis of highly branched anionic α-glucans by thermostable phosphorylase-catalyzed α-glucuronylation. Carbohydr. Res. 2013, 366, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Shimohigoshi, R.; Takemoto, Y.; Yamamoto, K.; Kadokawa, J. Thermostable α-glucan phosphorylase-catalyzed successive α-mannosylations. Chem. Lett. 2013, 42, 822–824. [Google Scholar] [CrossRef]
- Borgerding, J. Phosphate deposits in digestion systems. J. Water Pollut. Control. Fed. 1972, 44, 813–819. [Google Scholar]
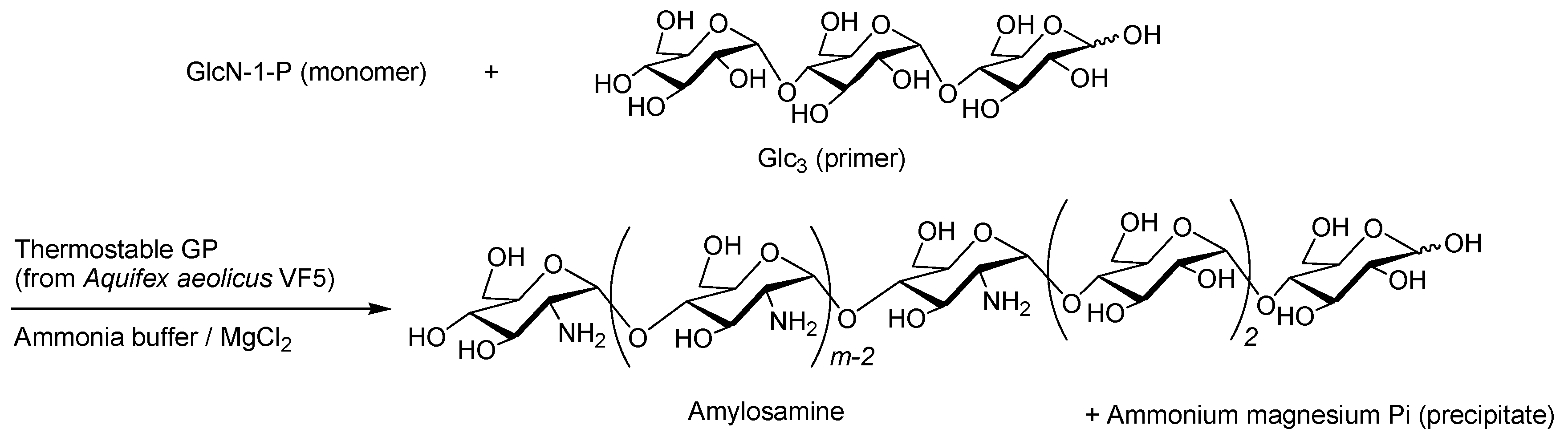
- Kadokawa, J.; Shimohigoshi, R.; Yamashita, K.; Yamamoto, K. Synthesis of chitin and chitosan stereoisomers by thermostable α-glucan phosphorylase-catalyzed enzymatic polymerization of α-d-glucosamine 1-phosphate. Org. Bimol. Chem. 2015, 13, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Yui, T.; Uto, T.; Nakauchida, T.; Yamamoto, K.; Kadokawa, J.I. Double helix formation from non-natural amylose analog polysaccharides. Carbohydr. Polym. 2018, 189, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Nakauchida, T.; Yamamoto, K.; Kadokawa, J. Hierarchically controlled assemblies from amylose analog aminopolysaccharides by reductive amination: From nano- to macrostructures. J. Appl. Polym. Sci. 2018, 135. [Google Scholar] [CrossRef]
- Yamashita, K.; Yamamoto, K.; Kadoakwa, J. Synthesis of non-natural heteroaminopolysaccharides by α-glucan phosphorylase-catalyzed enzymatic copolymerization: α(1→4)-linked glucosaminoglucans. Biomacromolecules 2015, 16, 3989–3994. [Google Scholar] [CrossRef] [PubMed]
- Baba, R.; Yamamoto, K.; Kadokawa, J. Synthesis of α(1→4)-linked non-natural mannoglucans by alphα-glucan phosphorylase-catalyzed enzymatic copolymerization. Carbohydr. Polym. 2016, 151, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
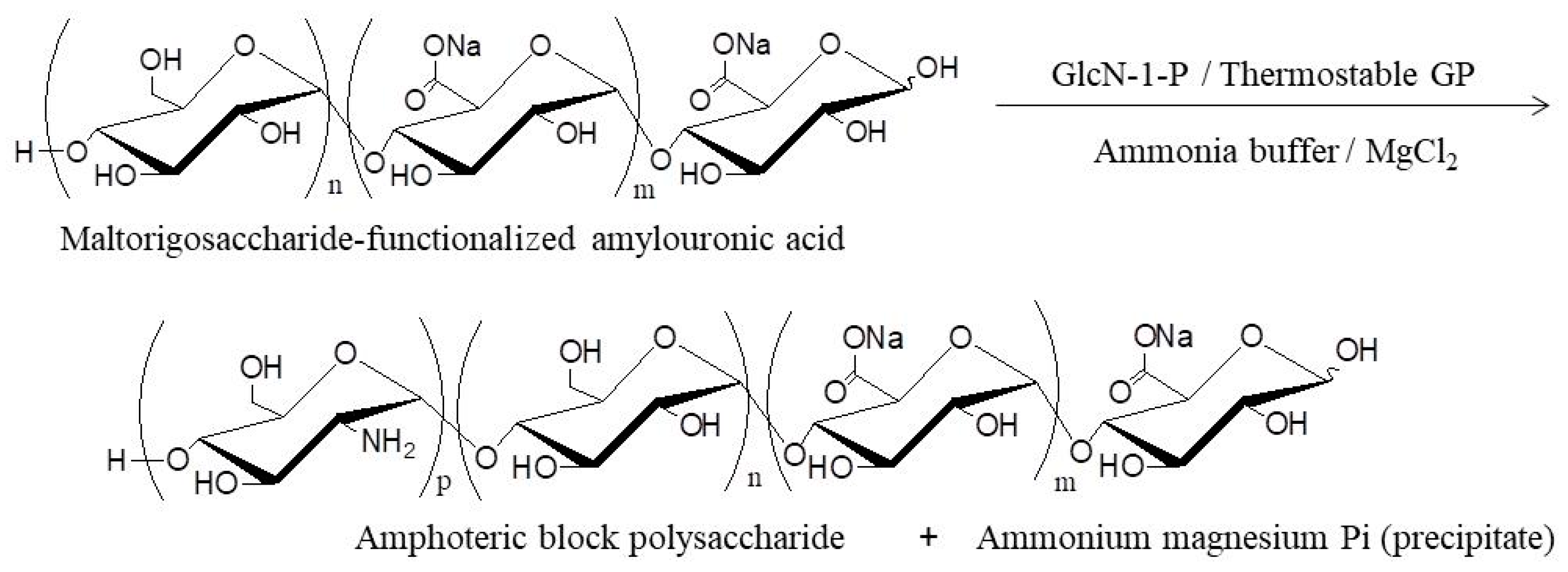
- Nakauchida, T.; Takata, Y.; Yamamoto, K.; Kadokawa, J. Chemoenzymatic synthesis and pH-responsive properties of amphoteric block polysaccharides. Org. Biomol. Chem. 2016, 14, 6449–6456. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadokawa, J.-i. α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides. Catalysts 2018, 8, 473. https://doi.org/10.3390/catal8100473
Kadokawa J-i. α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides. Catalysts. 2018; 8(10):473. https://doi.org/10.3390/catal8100473
Chicago/Turabian StyleKadokawa, Jun-ichi. 2018. "α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides" Catalysts 8, no. 10: 473. https://doi.org/10.3390/catal8100473
APA StyleKadokawa, J. -i. (2018). α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides. Catalysts, 8(10), 473. https://doi.org/10.3390/catal8100473