Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow



Abstract
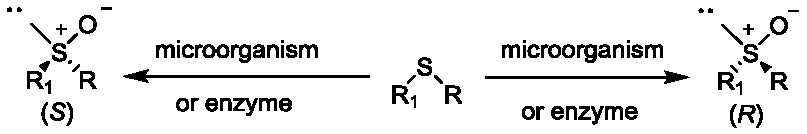
: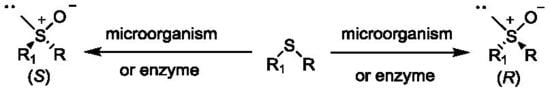
1. Introduction
- nucleophilic substitution of a chiral precursor;
- asymmetric sulphoxidation of prochiral sulfides;
- kinetic separation of racemic sulfoxides.
2. Biotransformations in Cultures of Microorganisms
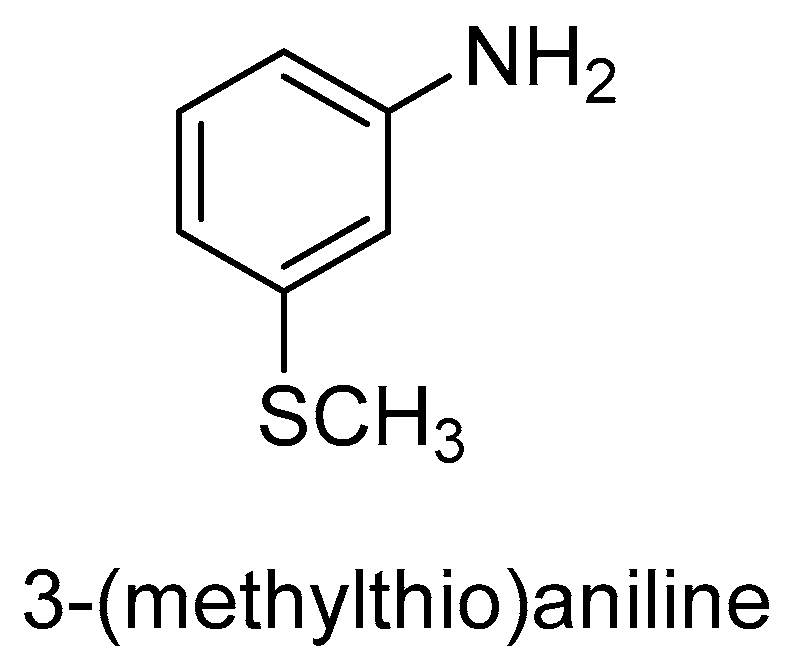
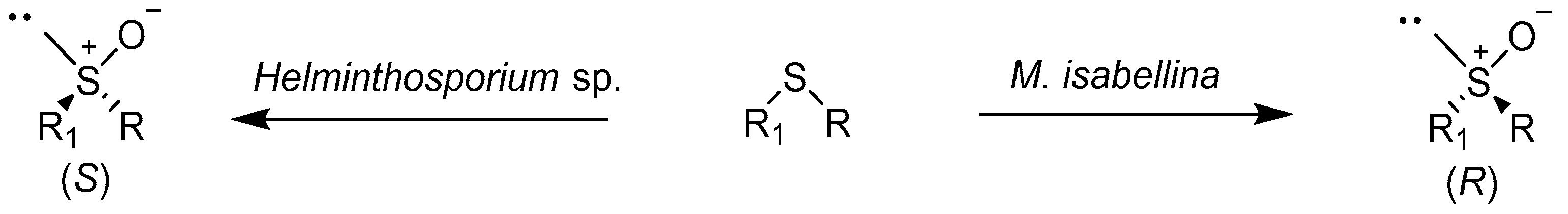
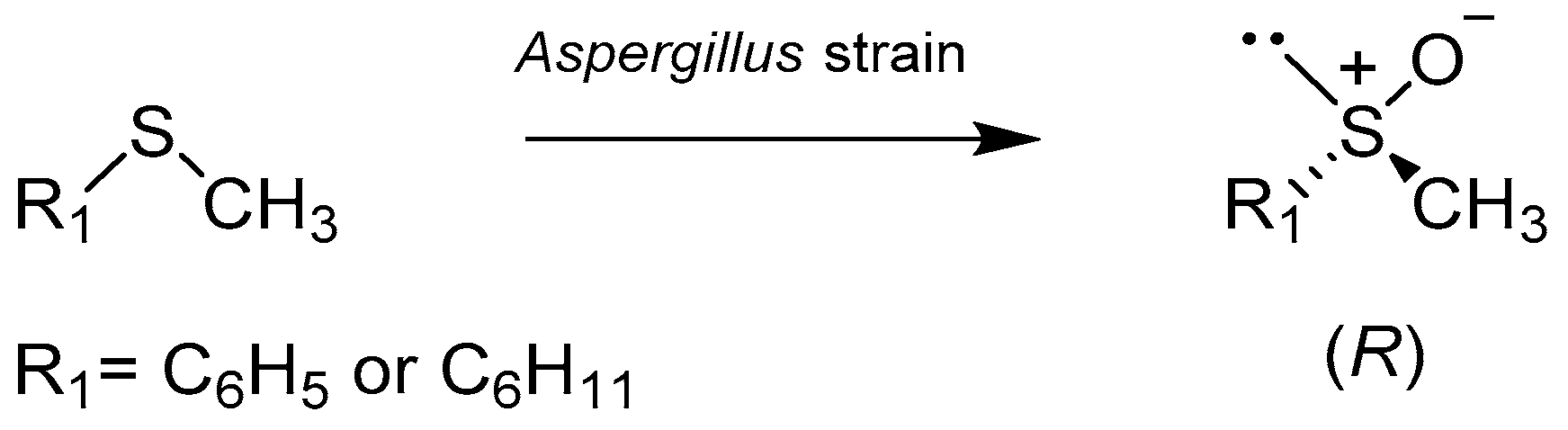
2.1. Fungus and Yeast
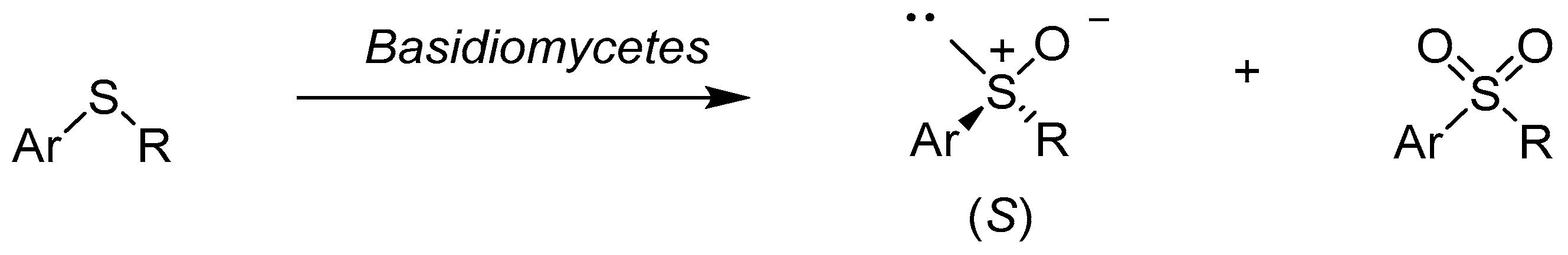
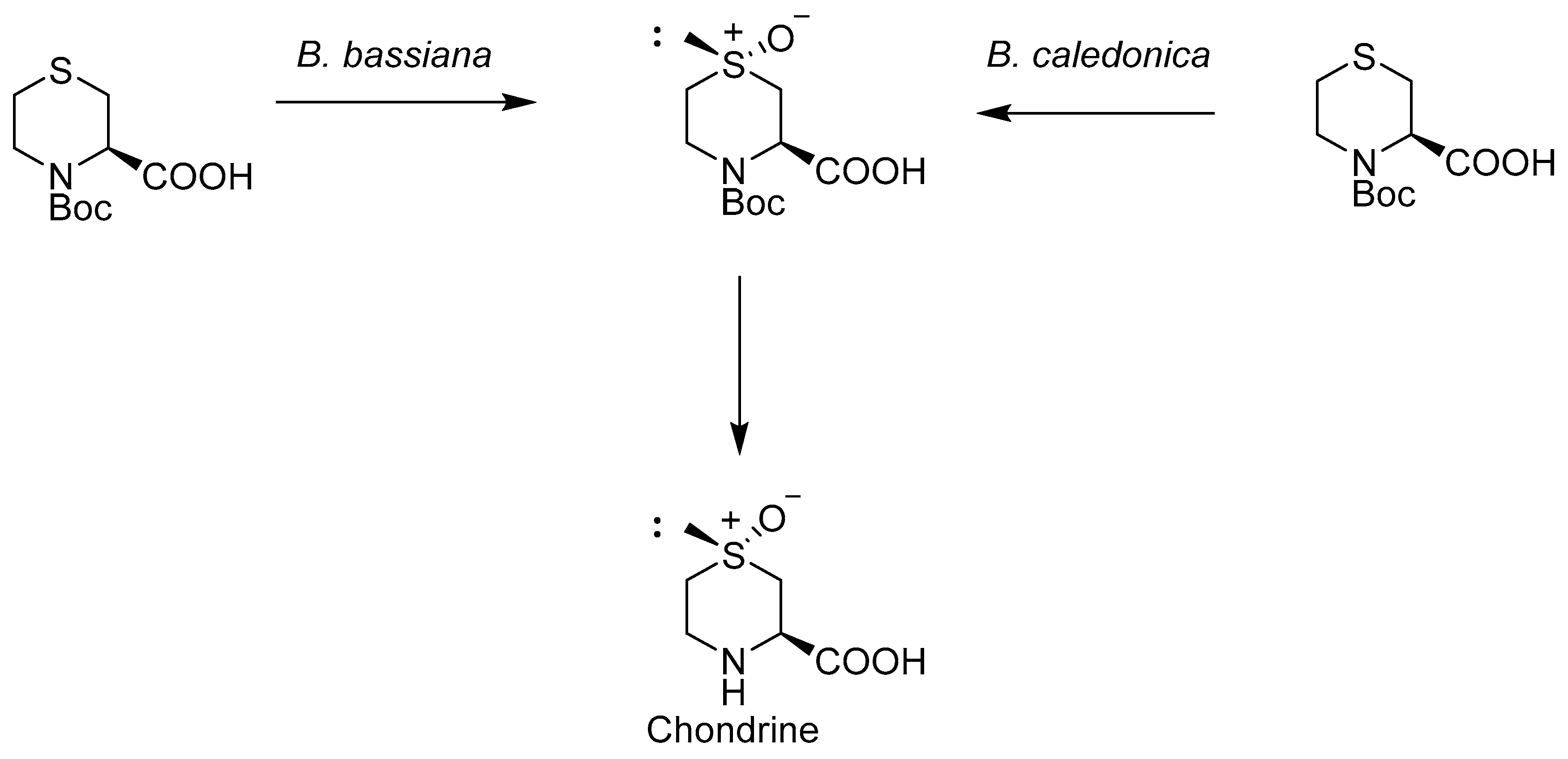
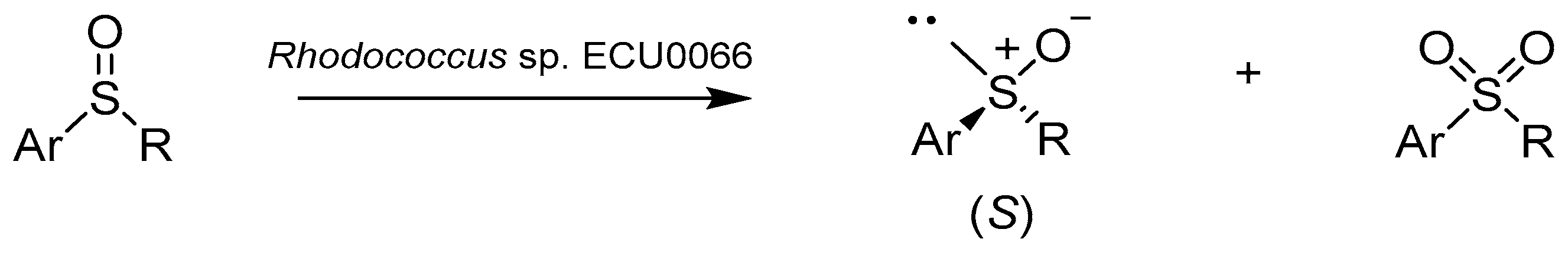
2.2. Bacteria
3. Enzymatic Sulfoxidation
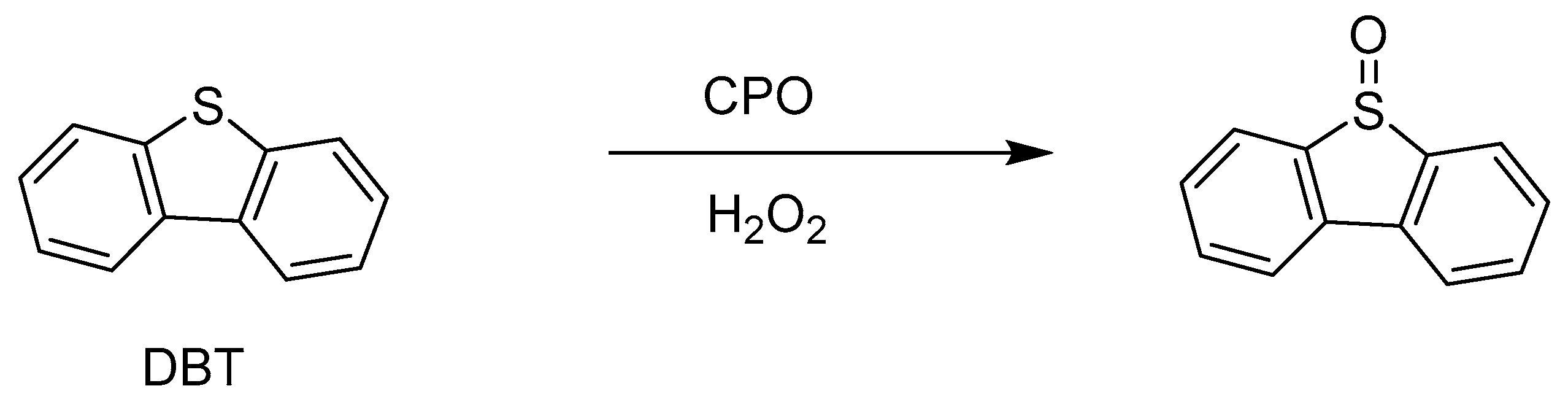
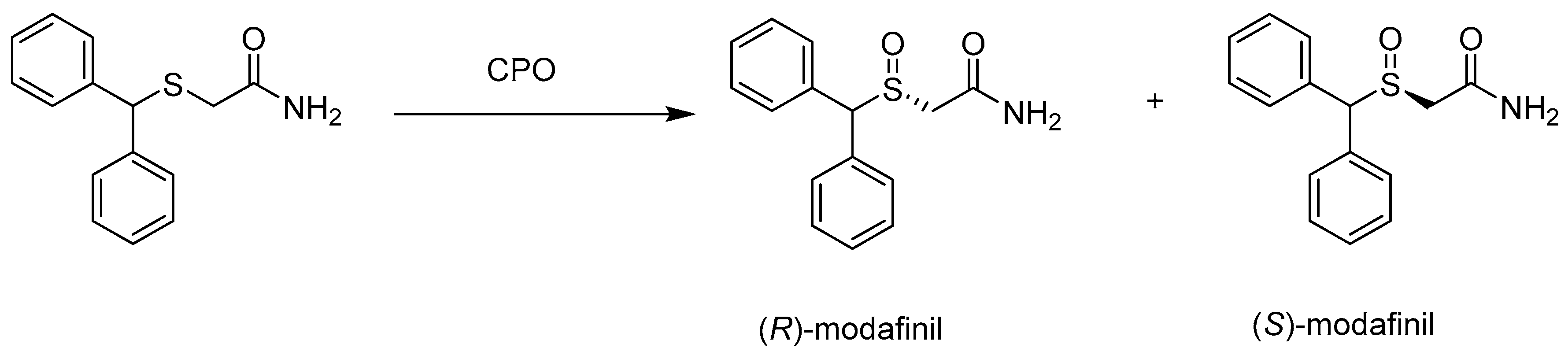
3.1. Chloroperoxidase
3.2. Horseradish Peroxidase
3.3. Dioxygenase
3.3.1. Toluene Dioxygenase
3.3.2. Naphthalene Dioxygenase
3.4. Monooxygenases
3.4.1. Cytochrome P450 Monooxygenases
3.4.2. Flavin-Dependent Monooxygenases
3.4.3. Baeyer-Villiger Monooxygenases (BVMOs)
- cyclohexanone monooxygenase (CHMO, EC 1.14.13.22)
- phenylacetone monooxygenase (PAMO, EC 1.14.13.92)
- 4-hydroxyacetophenone monooxygenase (HAPMO, EC 1.14.13.84)
- styrene monooxygenase (SMO).
3.4.4. Cyclohexanone Monooxygenase
3.4.5. Phenylacetone Monooxygenase
3.4.6. 4-Hydroxyacetophenone Monooxygenase
3.4.7. Styrene Monooxygenase
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bentley, R. Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.S.; Hart, J.; Brems, J.J.; Goldstein, L.; Lewin, K.; Busuttil, R.W. Amanita poisoning: Treatment and the role of liver transplantation. Am. J. Med. 1989, 86, 187–193. [Google Scholar] [CrossRef]
- Thomson, M.; Ali, M. Garlic [Allium sativum]: A review of its potential use as an anti-cancer agent. Curr. Cancer Drug Targets 2003, 3, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.P.; Cooper, D.L.; Gerratt, J.; Karadakov, P.B.; Raimondi, M. Chemical bonding in oxofluorides of hypercoordinate sulfur. J. Chem. Soc. Faraday Trans. 1997, 93, 2247–2254. [Google Scholar] [CrossRef]
- Morris, D.G. Stereochemia; Wydawnictwo Naukowe PWN SA: Warszawa, Poland, 2008. [Google Scholar]
- Andersen, K.K. Synthesis of (+)-ethyl p-tolyl sulfoxide from (−)-menthyl (−)-p-toluensulfinate. Tetrahedron Lett. 1962, 3, 93–95. [Google Scholar] [CrossRef]
- De Sio, V.; Acocella, M.R.; Villano, R.; Scettri, A. New chiral imino- and amino-sulfoxides as activators of allyl trichlorosilane in the asymmetric allylation of aldehydes. Tetrahedron Asymm. 2010, 21, 1432–1435. [Google Scholar] [CrossRef]
- Qi, W.-Y.; Zhu, T.-S.; Xu, M.-H. Design of chiral sulfoxide—Olefins as a new class of sulfur-based olefin ligands for asymmetric catalysis. Org. Lett. 2011, 13, 3410–3413. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, S.; Rachwalski, M.; Kiełbasiński, P. Highly enantioselective conjugate addition of diethylzinc to enones using aziridine-functionalized tridentate sulfinyl ligands. Tetrahedron Asymm. 2010, 21, 1890–1892. [Google Scholar] [CrossRef]
- Cotton, H.; Elebring, T.; Larsson, M.; Li, L.; Sörensen, H.; Unge, S. Asymmetric synthesis of esomeprazole. Tetrahedron Asymm. 2000, 18, 3819–3825. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Janicki, I.; Kiełbasiński, P. Enzyme-promoted kinetic resolution of acetoxymethyl aryl sulfoxides. J. Mol. Catal. B Enzym. 2015, 118, 23–28. [Google Scholar] [CrossRef]
- Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Homogeneous Oxygenase Catalysis. Chem. Rev. 2018, 118, 4912–4945. [Google Scholar] [CrossRef]
- Barth, T.; Hilário, V.C.; Rocha, B.A.; Furtado, N.A.J.C.; Pupo, M.T.; de Oliveira, A.R.M. Asymmetric sulfoxidation of albendazole to ricobendazole by fungi: effect of pH. Quim. Nova 2015, 38, 944–947. [Google Scholar] [CrossRef]
- Borges, W.S.; Borges, K.B.; Bonato, P.S.; Said, S.; Pupo, M.T. Endophytic fungi: Natural products, enzymes and biotransformation reactions. Curr. Org. Chem. 2009, 13, 1137–1163. [Google Scholar] [CrossRef]
- Liu, J.H.; Yu, B.Y. Biotransformation of bioactive natural products for pharmaceutical lead compounds. Curr. Org. Chem. 2010, 14, 1400–1406. [Google Scholar] [CrossRef]
- Parshikov, I.A.; Netrusov, A.I.; Sutherland, J.B. Microbial transformation of antimalarial terpenoids. Biotechnol. Adv. 2012, 30, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Holland, H.L.; Brown, F.M.; Larsen, B.G.; Zabic, M. Biotransformation of organic sulfides. Part 7. Formation of chiral isothiocyanato sulfoxides and related compounds by microbial biotransformation. Tetrahedron Asymm. 1995, 6, 1569–1574. [Google Scholar] [CrossRef]
- Holland, H.L. Chiral Sulfoxidation by Biotransformation of Organic Sulfides. Chem. Rev. 1988, 88, 473–485. [Google Scholar] [CrossRef]
- Holland, H.L.; Carter, I.M. The mechanism of sulfide oxidation by Mortierella isabellina NRRL 1757. Can. J. Chem. 1982, 60, 2420–2426. [Google Scholar] [CrossRef]
- Holland, H.L.; Bergen, E.J.; Chenchaiah, PC.; Khan, S.H.; Munoz, B.; Ninniss, R.W.; Richards, D. Side chain hydroxylation of aromatic compounds by fungi.: 1. Products and stereochemistry. Can. J. Chem. 1987, 65, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Holland, H.L.; Brown, F.M.; Lakshmaiah, G.; Larsen, B.G.; Patel, M. Biotransformation of organic sulfides—VIII. A predictive model for sulfoxidation by Helminthosporiun species NRRL 4671. Tetrahedron Asymm. 1997, 8, 683–697. [Google Scholar] [CrossRef]
- Holland, H.L.; Allen, L.J.; Chernishenko, M.J.; Diez, M.; Kohl, A.; Ozog, J.; Gu, J.-X. Side chain oxidation of aromatic compounds by fungi. A rationale for sulfoxidation, benzylic hydroxylation and olefin oxidation by Mortierella isabellina. J. Mol. Catal. B Enzym. 1997, 3, 311–324. [Google Scholar] [CrossRef]
- Holland, H.L.; Gu, J.-X.; Kerridge, A.; Willetts, A. Biotransformation of organic sulfides. Part 11. Preparation of functionalised phenyl propyl sulfoxides using Helminthosporium species and Mortierella isabellina. Biocatal. Biotransform. 1999, 17, 305–317. [Google Scholar] [CrossRef]
- Holland, H.L. Biotransformation of organic sulfides. Nat. Prod. Rep. 2001, 18, 171–181. [Google Scholar] [CrossRef]
- Ricci, L.C.; Comasseto, J.V.; Andrade, L.H.; Capelari, M.; Cass, Q.B.; Porto, A.L.M. Biotransformations of aryl alkyl sulfides by whole cells of white-rot Basidiomycetes. Enzym. Microb. Technol. 2005, 36, 937–946. [Google Scholar] [CrossRef]
- Pinedo-Rivilla, C.; Aleu, J.; Collado, I.G. Enantiomeric oxidation of organic sulfides by the filamentous fungi Botrytis cinerea, Eutypa lata and Trichoderma viride. J. Mol. Catal. B Enzym. 2007, 49, 18–23. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Ordena, A.A.; Bisogno, F.R.; de Gonzalo, G.; Kurina-Sanz, M. Aspergillus genus as a source of new catalysts for sulfide oxidation. J. Mol. Catal. B Enzym. 2012, 82, 32–36. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Barrett, F.; French, J.; Johnson, V. Biotransformation of beta-ketosulfides to produce chiral beta-hydroxysulfoxides. J. Ind. Microbiol. Biotechnol. 2003, 30, 292–301. [Google Scholar] [CrossRef]
- Holland, H.L.; Andreana, P.R.; Brown, F.M. Biocatalytic and chemical routes to all the stereoisomers of methionine and ethionine sulfoxides. Tetrahedron Asymm. 1999, 10, 2833–2843. [Google Scholar] [CrossRef]
- Mascotti, M.L.; Palazzolo, M.A.; Lewkowicz, E.; Kurina-Sanz, M. Expanding the toolbox for enantioselective sulfide oxidations: Streptomyces strains as biocatalysts. Biocatal. Agric. Biotechnol. 2013, 2, 399–402. [Google Scholar] [CrossRef]
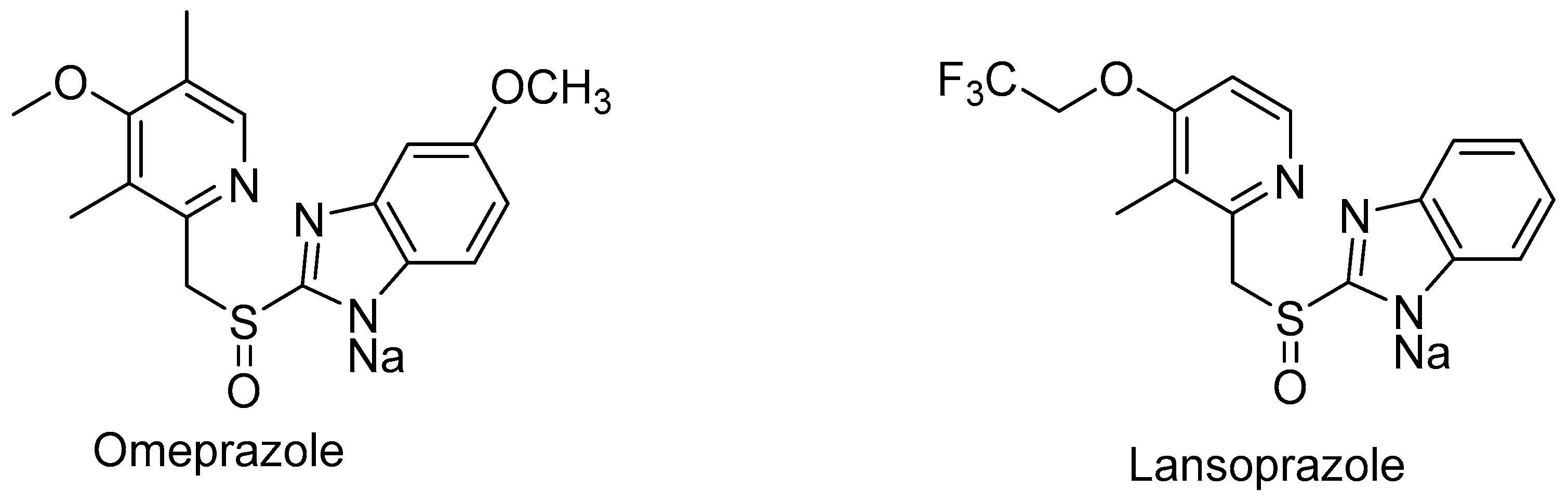
- Yoshida, T.; Kito, M.; Tsujii, M.; Nagasawa, T. Microbial synthesis of a proton pump inhibitor by enantioselective oxidation of a sulfide into its corresponding sulfoxide by Cunninghamella echinulata MK40. Biotechnol. Lett. 2001, 23, 1217–1222. [Google Scholar] [CrossRef]
- French, J.B.; Holland, G.; Holland, H.L.; Gordon, H.L. A comparative molecular field analysis of the biotransformation of sulfides by Rhodococcus erythropolis. J. Mol. Catal. B Enzym. 2004, 31, 87–96. [Google Scholar] [CrossRef]
- Zhang, J.D.; Li, A.T.; Yang, Y.; Xu, J.H. Sequence analysis and heterologous expression of a new cytochrome P450 monooxygenase from Rhodococcus sp. for asymmetric sulfoxidation. Appl. Microbiol. Biotechnol. 2010, 85, 615–624. [Google Scholar] [CrossRef]
- Elkin, A.A.; Kylosovab, T.I.; Grishkoc, V.V.; Ivshinaa, I.B.; Elkin, A.A.; Kylosova, T.I.; Grishko, V.V.; Ivshina, I.B. Enantioselective oxidation of sulfides to sulfoxides by Gordonia terrae IEGM 136 and Rhodococcus rhodochrous IEGM 66. J. Mol. Catal. B Enzym. 2013, 89, 82–85. [Google Scholar] [CrossRef]
- Ohta, H.; Okamoto, Y.; Tsuchihashi, G. Asymmetric synthesis of chiral sulfoxides via microbial oxidation of sulfides. Chem. Lett. 1984, 2, 205–208. [Google Scholar] [CrossRef]
- Ohta, H.; Okamoto, Y.; Tsuchihashi, G. Microbial Oxidation of Alkyl Aryl Sulfides to Corresponding Optically Active Sulfoxides. Agric. Biol. Chem. 1985, 49, 671–676. [Google Scholar] [CrossRef]
- Gray, K.A.; Pogrebinsky, O.; Mrachko, G.T.; Xi, L.; Monticello, D.J.; Squires, C.H. Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat. Biotechnol. 1996, 14, 1705–1709. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Kerridge, A.; Pienkos, P.; Arensdor, J. Biotransformation of sulfides by Rhodocoeccus erythropolis. J. Mol. Catal. B Enzym. 2003, 22, 219–223. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Lozada, D.; Mayne, B.; Szerminski, W.R.; van Vliet, A.J. Chloroperoxidase-catalyzed oxidation of methionine derivatives. Can. J. Chem. 2002, 80, 633–639. [Google Scholar] [CrossRef]
- Holland, H.L.; Brown, F.M.; Johnson, D.V.; Kerridge, A.; Mayne, B.; Turner, C.D.; van Vliet, A.J. Biocatalytic oxidation of S-alkylcysteine derivatives by chloroperoxidase and Beauveria species. J. Mol. Catal. B Enzym. 2002, 17, 249–256. [Google Scholar] [CrossRef]
- Li, A.T.; Yu, H.L.; Pan, J.; Zhang, J.D.; Xu, J.H.; Lin, G.Q. Resolution of racemic sulfoxides with high productivity and enantioselectivity by a Rhodococcus sp. strain as an alternative to biooxidation of prochiral sulfides for efficient production of enantiopure sulfoxides. Bioresour. Technol. 2011, 102, 1537–1542. [Google Scholar] [CrossRef]
- He, Y.C.; Ma, C.L.; Yang, Z.X.; Zhou, M.; Xing, Z.; Ma, J.T.; Yu, H.L. Highly enantioselective oxidation of phenyl methyl sulfide and its derivatives into optically pure (S)-sulfoxides with Rhodococcus sp. CCZU10-1 in an n-octane-water biphasic system. Appl. Microbiol. Biotechnol. 2013, 97, 10329–10337. [Google Scholar] [CrossRef]
- Chen, Y.; Zhuoa, J.; Zhenga, D.; Tiana, S.; Lib, Z. Stereoselective oxidation of sulfides to optically active sulfoxides withresting cells of Pseudomonas monteilii CCTCC M2013683. J. Mol. Catal. B Enzym. 2014, 106, 100–104. [Google Scholar] [CrossRef]
- Kylosovaa, T.I.; Elkina, A.A.; Grishkoc, V.V.; Ivshina, I.B. Biotransformation of prochiral sulfides into (R)-sulfoxides using immobilized Gordonia terrae IEGM 136 cells. J. Mol. Catal. B Enzym. 2016, 123, 8–13. [Google Scholar] [CrossRef]
- Lim, M.L.; DeWayne Brooks, M.; Boothe, M.A.; Krzmarzick, M.J. Novel bacterial diversity is enriched with chloroperoxidase-reacted organic matter under anaerobic conditions. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Juarez-Moreno, K.; de León, J.N.D.; Zepeda, T.A.; Vazquez-Duhalt, R.; Fuentes, S. Oxidative transformation of dibenzothiophene by chloroperoxidase enzyme immobilized on (1D)-γ-Al2O3 nanorods. J. Mol. Catal. B Enzym. 2015, 115, 90–95. [Google Scholar] [CrossRef]
- Pereira, P.C.; Arends, I.W.C.E.; Sheldon, R.A. Optimizing the chloroperoxidase-glucose oxidase system: The effect of glucose oxidase on activity and enantioselectivity. Process Biochem. 2015, 50, 746–751. [Google Scholar] [CrossRef]
- O’Mahony, G.E.; Kelly, P.; Lawrence, S.E.; Maguire, A.R. Synthesis of Enantioenriched Sulfoxides. ARKIVOC 2011, 2011, 1–110. [Google Scholar]
- Kadri, T.; Rouissi, T.; Brar, s.K.; Cledon, M.; Sarma, S.; Verma, M. Biodegradation of polycyclic aromatic hydrocarbons (PAHs) by fungal enzymes: A review. J. Environ. Sci. 2017, 51, 52–74. [Google Scholar] [CrossRef]
- Gao, F.; Wang, L.; Liu, Y.; Wang, S.; Jiang, Y.; Hu, M.; Li, S.; Zhai, Q. Enzymatic synthesis of (R)-modafinil by chloroperoxidase-catalyzed enantioselective sulfoxidation of 2-(diphenylmethylthio)acetamide. Biochem. Eng. J. 2015, 93, 243–249. [Google Scholar] [CrossRef]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Matsui, T.; Dekishima, Y.; Ueda, M. Biotechnological production of chiral organic sulfoxides: Current state and perspectives. Appl. Microbiol. Biotechnol. 2014, 98, 7699–7706. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Haughey, S.A.; Kennedy, M.A.; McMurray, B.T.; Sheldrake, G.N.; Allen, C.C.R.; Dalton, H.; Sproule, K. Toluene and naphthalene dioxygenase-catalysed sulfoxidation of alkyl aryl sulfides. J. Chem. Soc. Perkin Trans. 1998, 1, 1929–1934. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; McMurray, B.; Haughey, S.A.; Allen, C.C.R.; Hamilton, J.T.G.; McRoberts, W.C.; More O’Ferrall, R.A.; Nikodinovic-Runic, J.; Coulombele, L.A.; et al. Bacterial dioxygenase- and monooxygenase-catalysed sulfoxidation of benzo[b]thiophenes. Org. Biomol. Chem. 2012, 10, 782–790. [Google Scholar] [CrossRef]
- Porter, J.L.; Sabatini, S.; Manning, J.; Tavanti, M.; Galman, J.L.; Turner, N.J.; Flitsch, S.L. Cloning, expression and characterisation of P450-Hal1 (CYP116B62) from Halomonas sp. NCIMB 172: A self-sufficient P450 with high expression and diverse substrate scope. Enzym. Microb. Technol. 2018, 113, 1–8. [Google Scholar] [CrossRef]
- Ciaramella, A.; Minerdi, D.; Gilardi, G. Catalytically self-sufficient cytochromes P450 for green production of fine chemicals. Rend. Fis. Acc. Lincei 2017, 28 (Suppl. 1), S169–S181. [Google Scholar] [CrossRef]
- Li, R.-J.; Xu, J.-H.; Chen, Q.; Zhao, J.; Li, A.-T.; Yu, H.-L. Enhancing the catalytic performance of a CYP116B monooxygenase by transdomain combination mutagenesis. ChemCatChem 2018, 10, 2962–2968. [Google Scholar] [CrossRef]
- O’Reilly, E.; Corbett, M.; Hussain, S.; Kelly, P.P.; Richardson, D.; Flitsch, S.L.; Turner, N.J. Substrate promiscuity of cytochrome P450 RhF. Catal. Sci. Technol. 2013, 3, 1490–1492. [Google Scholar] [CrossRef]
- Uehara, S.; Kawano, M.; Murayama, N.; Uno, Y.; Utoh, M.; Inoue, T.; Sasaki, E.; Yamazaki, H. Oxidation of R- and S-omeprazole stereoselectively mediated by liver microsomal cytochrome P450 2C19 enzymes from cynomolgus monkeys and common marmosets. Biochem. Pharmacol. 2016, 120, 56–62. [Google Scholar] [CrossRef]
- Gao, P.; Li, A.; Lee, H.H.; Wang, D.I.C.; Li, Z. Enhancing enantioselectivity and productivity of P450-catalyzed asymmetric sulfoxidation with an aqueous/ionic liquid biphasic system. ACS Catal. 2014, 4, 3763–3771. [Google Scholar] [CrossRef]
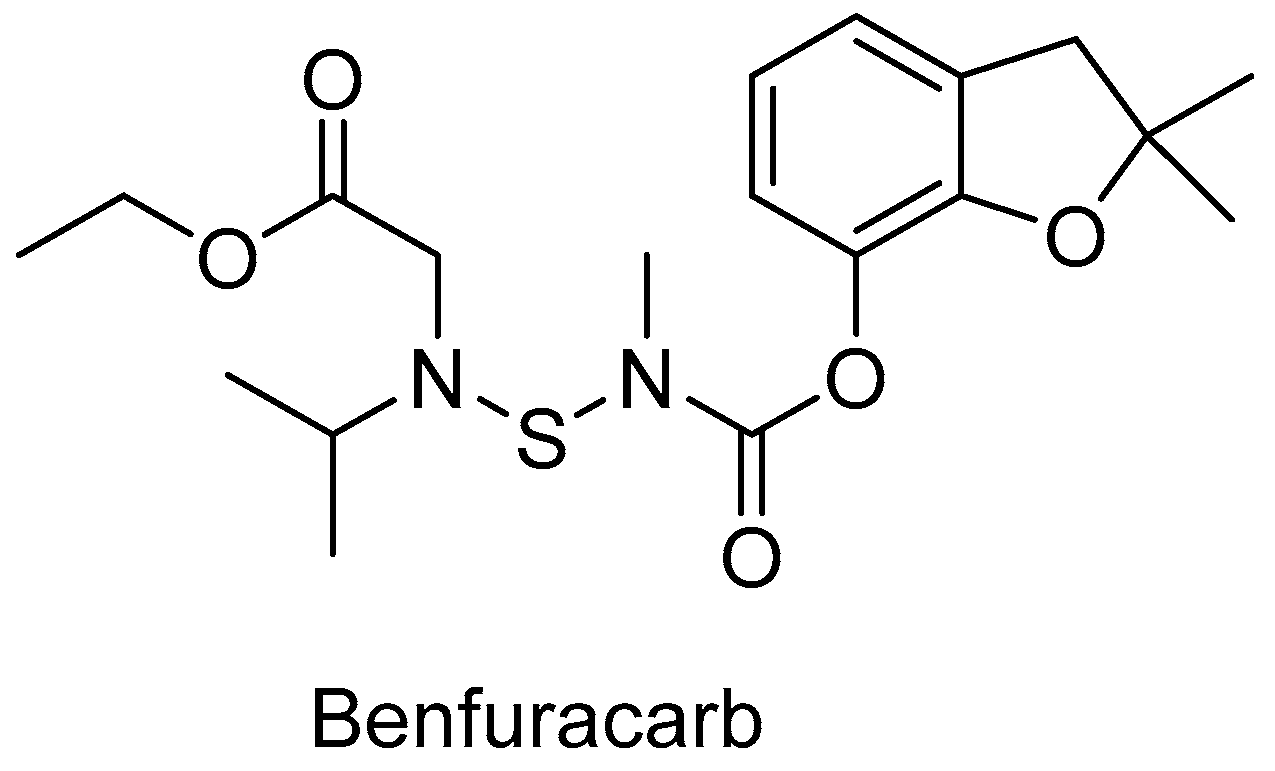
- Abass, K.; Reponen, P.; Mattila, S.; Rautio, A.; Pelkonen, O. Human variation and CYP enzyme contribution in benfuracarb metabolism in human in vitro hepatic models. Toxicol. Lett. 2014, 224, 300–309. [Google Scholar] [CrossRef]
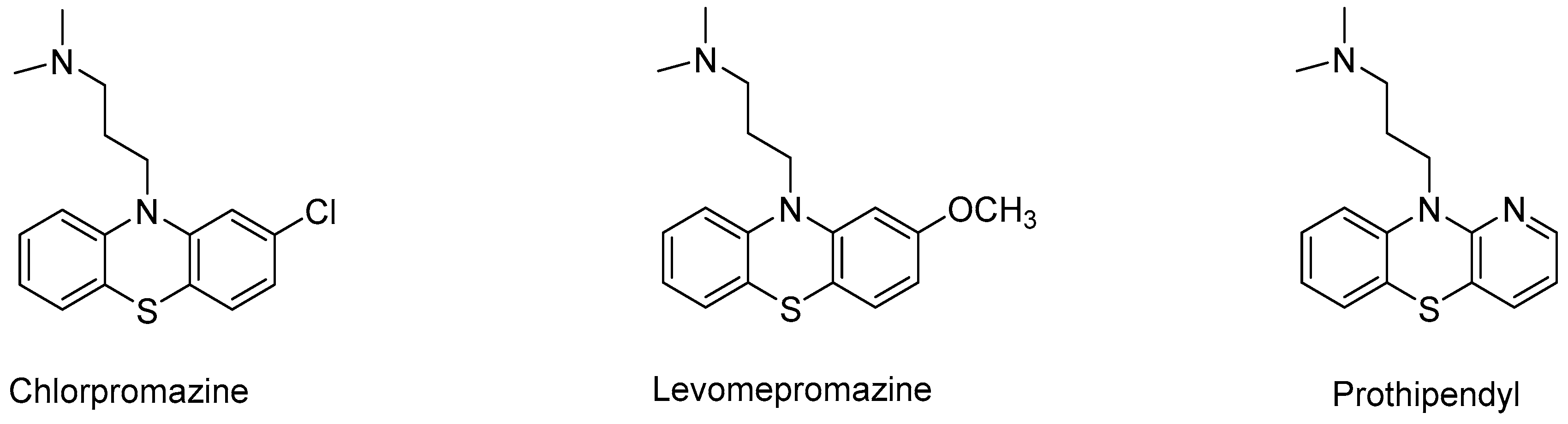
- Wójcikowski, J.; Daniel, W.A. Influence of antidepressant drugs on chlorpromazine metabolism in human liver—An in vitro study. Pharmacol. Rep. 2010, 62, 1062–1069. [Google Scholar] [CrossRef]
- Wójcikowski, J.; Basińska, A.; Daniel, W.A. The cytochrome P450-catalyzed metabolism of levomepromazine: A phenothiazine neuroleptic with a wide spectrum of clinical application. Biochem. Pharmacol. 2014, 90, 188–195. [Google Scholar] [CrossRef]
- Krämer, M.; Broecker, S.; Madea, B.; Hess, C. Confirmation of metabolites of the neuroleptic drug prothipendyl using human liver microsomes, specific CYP enzymes and authentic forensic samples—Benefit for routine drug testing. J. Pharm. Biomed. Anal. 2017, 145, 517–524. [Google Scholar] [CrossRef]
- Worsch, A.; Eggimann, F.K.; Girhard, M.; von Bühler, C.J.; Tieves, F.; Czaja, R.; Vogel, A.; Grumaz, C.; Sohn, K.; Lütz, S.; et al. A novel cytochrome P450 monooxygenase from Streptomyces platensis resembles activities of human drug metabolizing P450s. Biotechnol. Bioeng. 2018, 115, 2156–2166. [Google Scholar] [CrossRef]
- Rettie, A.E.; Lawton, M.P.; Sadeque, A.J.M.; Meier, G.P.; Philpot, R.M. Prochiral sulfoxidation as a probe for multiple forms of the microsomal flavin-containing monooxygenase: Studies with rabbit FMO1, FMO2, FMO3, and FMO5 expressed in Escherichia coli. Arch. Biochem. Biophys. 1994, 311, 369–377. [Google Scholar] [CrossRef]
- Catucci, G.; Gao, C.; Sadeghi, S.J.; Gilardi, G. Chemical applications of Class B flavoprotein monooxygenases. Rend. Fis. Acc. Lincei 2017, 28 (Suppl. 1), S195–S206. [Google Scholar] [CrossRef]
- Hamman, M.A.; Haehner-Daniels, B.D.; Wrighton, S.A.; Rettie, A.E.; Hall, S.D. Stereoselective sulfoxidation of sulindac sulfide by flavin-containing monooxygenases. Biochem. Pharmacol. 2000, 60, 7–17. [Google Scholar] [CrossRef]
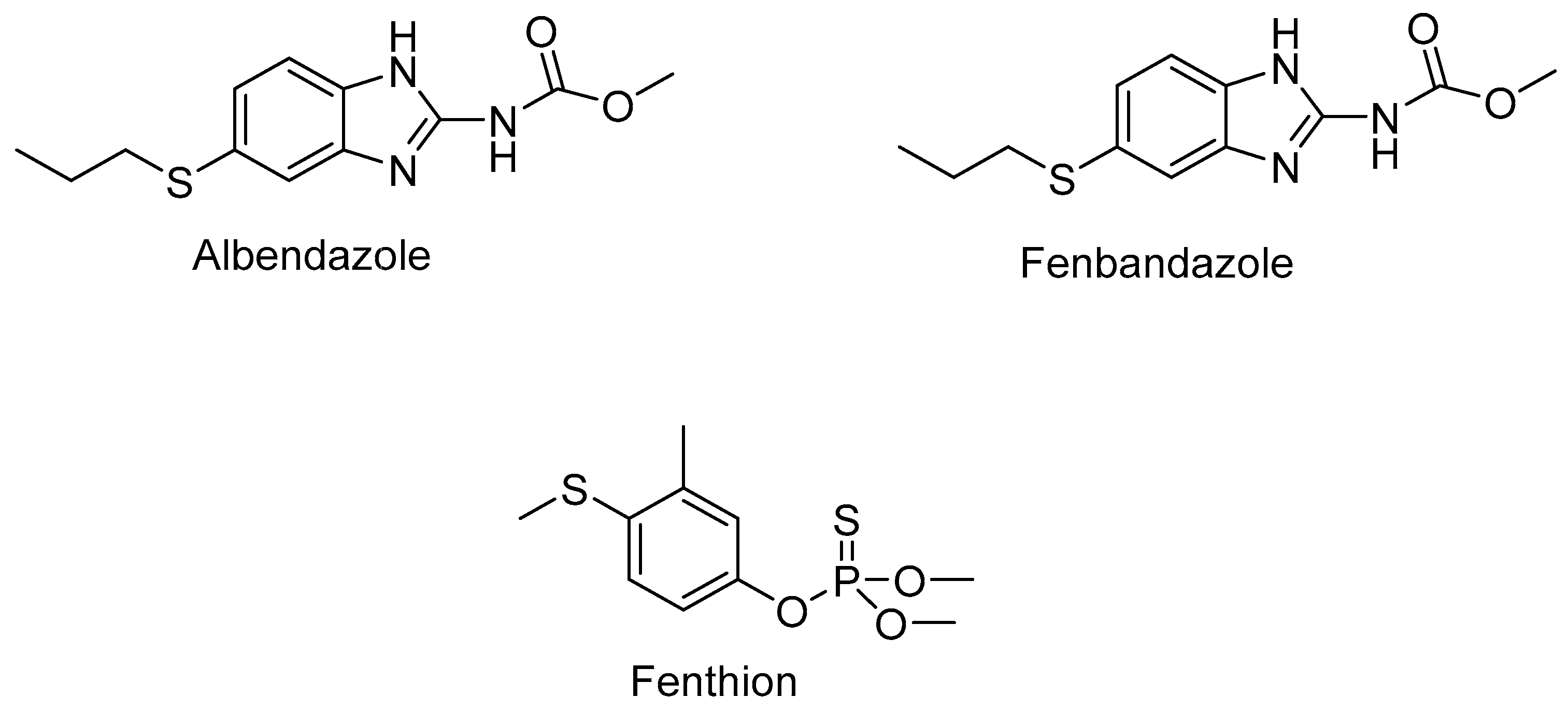
- Virkel, G.; Lifschitz, J.; Sallovitz, J.; Pis, A.; Lanusse, C. Comparative hepatic and extrahepatic enantioselective sulfoxidation of albendazole and fenbendazole in sheep and cattle. Drug Metab. Dispos. 2004, 32, 536–544. [Google Scholar] [CrossRef]
- Furnes, B.; Schlenk, D. Evaluation of xenobiotic N- and S-oxidation by variant flavin-containing monooxygenase 1 (FMO1) enzymes. Toxicol. Sci. 2004, 78, 196–203. [Google Scholar] [CrossRef]
- Fiorentini, F.; Geier, M.; Binda, C.; Winkler, M.; Faber, K.; Hall, M.; Mattevi, A. Biocatalytic characterization of human FMO5: Unearthing Baeyer-Villiger reactions in humans ACS Chem. Biol. 2016, 11, 1039–1048. [Google Scholar] [CrossRef]
- Gul, T.; Krzek, M.; Permentier, H.P.; Fraaije, M.W.; Bischoff, R. Microbial flavoprotein monooxygenases as mimics of mammalian flavin-containing monooxygenases for the enantioselective preparation of drug metabolites. Drug Metab. Dispos. 2016, 44, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Mthethwa, K.S.; Kassier, K.; Engel, J.; Kara, S.; Smit, M.S.; Opperman, D.J. Fungal BVMOs as alternatives to cyclohexanone monooxygenase. Enzym. Microb. Technol. 2017, 106, 11–17. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, F.; Xu, N.; Wu, Y.-Q.; Zheng, Y.-C.; Zhao, Q.; Lin, G.; Yu, H.-L.; Xu, J.-H. The discovery of two native Baeyer-Villiger monooxygenases for asymmetric synthesis of bulky chiral sulfoxides. Appl. Environ. Microbiol. 2018, 84, e00638-18. [Google Scholar] [CrossRef]
- Bisagni, S.; Summers, B.; Kara, S.; Hatti-Kaul, R.; Grogan, G.; Mamo, G.; Hollmann, F. Exploring the substrate specificity and enantioselectivity of a Baeyer–Villiger monooxygenase from Dietzia sp. D5: Oxidation of sulfides and aldehydes. Top. Catal. 2014, 57, 366–375. [Google Scholar] [CrossRef]
- Kohl, A.; Srinivasamurthy, V.; Böttcher, D.; Kabisch, J.; Bornscheuer, U.T. Co-expression of an alcohol dehydrogenase and a cyclohexanone monooxygenase for cascade reactions facilitates the regeneration of the NADPH cofactor. Enzym. Microb. Technol. 2018, 108, 53–58. [Google Scholar] [CrossRef]
- Torres Pazmiño, D.E.; Dudek, H.M.; Fraaije, M.W. Baeyer-Villiger monooxygenases: Recent advances and future challenges. Curr. Opin. Chem. Biol. 2010, 14, 138–144. [Google Scholar] [CrossRef]
- Sheng, D.W.; Ballou, D.P.; Massey, V. Mechanistic studies of cyclohexanone monooxygenase: Chemical properties of intermediates involved in catalysis. Biochemistry 2001, 40, 11156–11167. [Google Scholar] [CrossRef]
- Donoghue, N.A.; Trudgill, P.W. The metabolism of cyclohexanol by Acinetobacter NCIB 9871. Eur. J. Biochem. 1975, 60, 1–7. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggero, N.; Pasta, P.; Ottolina, G. Enantioselective oxidation of sulfides to sulfoxides catalysed by bacterial cyclohexanone monooxygenases. Chem. Commun. 1996, 20, 2303–2307. [Google Scholar] [CrossRef]
- Reetz, M.T.; Daligault, F.; Brunner, B.; Hinrichs, H.; Deege, A. Directed Evolution of Cyclohexanone Monooxygenases: Enantioselective Biocatalysts for the Oxidation of Prochiral Thioethers. Angew. Chem. 2004, 116, 4170–4173. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Rodriguez, C.; Rioz-Martinez, A.; Gotor, V. Improvement of the biocatalytic properties of ane phenylacetone monooxygenase mutant in hydrophilic organic solvents. Enzym. Microb. Technol. 2012, 50, 43–49. [Google Scholar] [CrossRef]
- Rodriguez, C.; de Gonzalo, G.; Gotor, V. Optimization of oxidative bioconversions catalyzed by phenylacetone monooxygenase from Thermobifida fusca. J. Mol. Catal. B Enzym. 2012, 74, 138–143. [Google Scholar] [CrossRef]
- Bisagni, S.; Abolhalaj, M.; de Brevern, A.G.; Rebehmed, J.; Hatti-Kaul, R.; Mamo, G. Enhancing the activity of a Dietzia sp. D5 Baeyer-Villiger monooxygenase towards cyclohexanone by saturation mutagenesis. ChemistrySelect 2017, 2, 7169–7177. [Google Scholar] [CrossRef]
- Zhang, Z.-G.; Lonsdale, R.; Sanchis, J.; Reetz, M.T. Extreme Synergistic Mutational Effects in the Directed Evolution of a Baeyer-Villiger Monooxygenase as Catalyst for Asymmetric Sulfoxidation. J. Am. Chem. Soc. 2014, 136, 17262–17272. [Google Scholar] [CrossRef]
- Rioz-Martínez, A.; de Gonzalo, G.; Torres Pazmiño, D.E.; Fraaije, M.W.; Gotor, V. Enzymatic synthesis of novel chiral sulfoxides employing Baeyer-Villiger monooxygenases. Eur. J. Org. Chem. 2010, 6409–6416. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Torres Pazmino, D.E.; Ottolina, G.; Fraaije, M.W.; Carrea, G. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB as an oxidative biocatalyst in the synthesis of optically active sulfoxides. Tetrahedron Asymm. 2006, 17, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Tischler, D. Microbial Styrene Degradation; Springer International Publishing: Basel, Switzerland, 2015. [Google Scholar]
- Tischler, D.; Schwabe, R.; Siegel, L.; Joffroy, K.; Kaschabek, S.R.; Scholtissek, A.; Heine, T. VpStyA1/VpStyA2B of Variovorax paradoxus EPS: An aryl alkyl sulfoxidase rather than a styrene epoxidizing monooxygenase. Molecules 2018, 23, 809. [Google Scholar] [CrossRef]
- Heine, T.; van Berkel, W.J.H.; Gassner, G.; van Pée, K.-H.; Tischler, D. Two-component FAD-dependent monooxygenases: Current knowledge and biotechnological opportunities. Biology 2018, 7, 42. [Google Scholar] [CrossRef]
- Pu, W.; Cui, C.; Guo, C.; Wu, Z.-L. Characterization of two styrene monooxygenases from marine microbes. Enzym. Microb. Technol. 2018, 112, 29–34. [Google Scholar] [CrossRef]
- Riedel, A.; Heine, T.; Westphal, A.H.; Conrad, C.; Rathsack, P.; van Berkel, W.J.H.; Tischler, D. Catalytic and hydrodynamic properties of styrene monooxygenases from Rhodococcus opacus 1CP are modulated by cofactor binding. AMB Express 2015, 5, 30. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
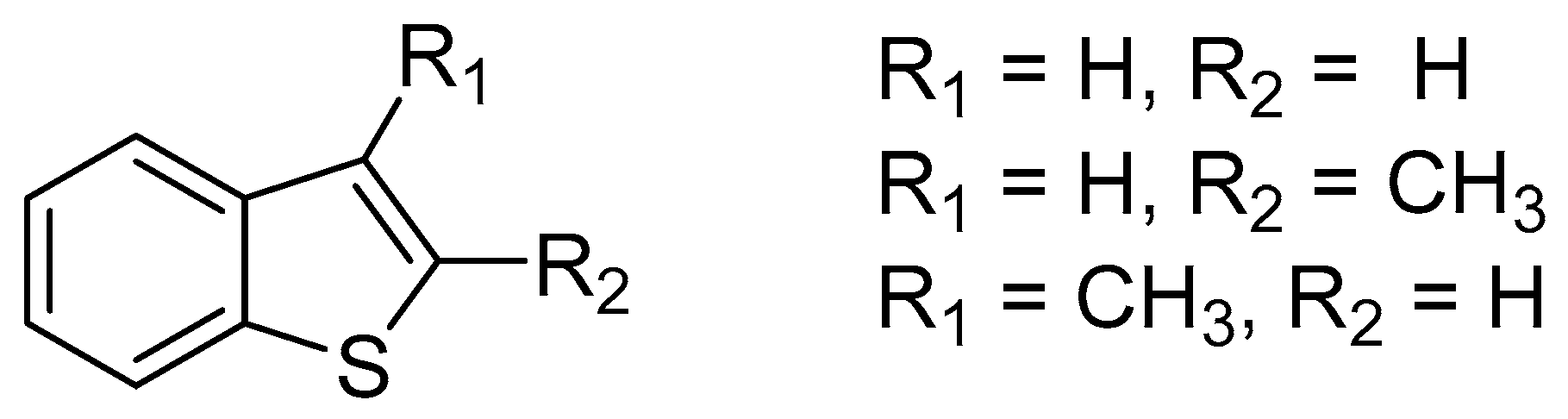
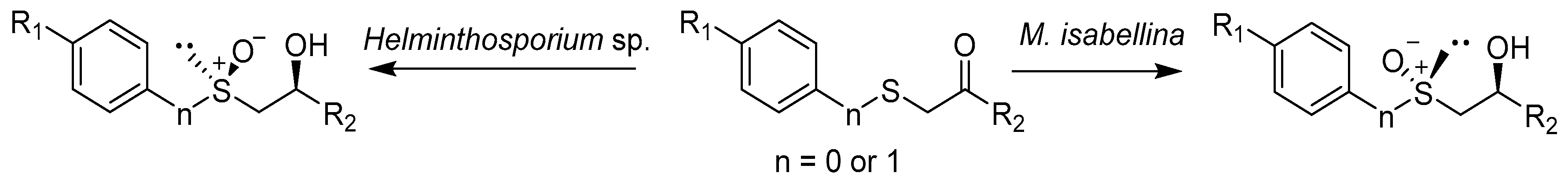
| Substrate | Biocatalyst | |||
|---|---|---|---|---|
| Helminthosporium sp. | M. isabellina | |||
| Yield | Stereoselectivity | Yield | Stereoselectivity | |
| R1 = H, R2 = CH3 | 90% | de 89% | 23% | de 46% |
| R1 = OCH3, R2 = CH3 | 70% | de 85% | 69% | de 52% |
| R1 = Br, R2 = CH3 | 30% | de 86% | 64% | de 50% |
| R1 = OCH3, R2 = Ph | 6% | de <5% | 48% | de 40% |
| R1 = H, R2 = CH3 | 66% | de 92% | 48% | de 40% |
| R1 = OCH3, R2 = CH3 | 72% | de 95% | 42% | de 85% |
| R1 = Br, R2 = CH3 | 72% | de 90% | 37% | de 40% |
| R1 = OCH3, R2 = Ph | 16% | de 26% | 69% | de 54% |
| R1 = Br, R2 =Ph | No products | 25% | de 28% | |
| Substrate | Yield (%) | Sulfoxide Configuration | ee (%) |
|---|---|---|---|
| C6H5SCH3 | 67 | rac | 2 |
| p-CH3C6H4SCH3 | 72 | R | 62 |
| m-CH3C6H4SCH3 | 60 | R | 10 |
| o-CH3C6H4SCH3 | 95 | rac | 4 |
| p-FC6H4SCH3 | 52 | R | 63 |
| p-ClC6H4SCH3 | 70 | R | 72 |
| p-BrC6H4SCH3 | 65 | R | 76 |
| p-NO2C6H4SCH3 | 83 | R | >98 |
| p-CNC6H4SCH3 | 43 | R | 85 |
| Substrate | Yield (%) | Sulfoxide Configuration | ee (%) |
|---|---|---|---|
| C6H5CH2SCH3 | 73 | R | 26 |
| C6H5CH2SC3H7 | 58 | R | 66 |
| C6H5C2H4SCH3 | 73 | R | 14 |
| C6H5C3H6SCH3 | 81 | rac | 3 |
| C6H5CH2SC6H5 | 65 | R | >98 |
| C6H5CH2SC3H6 C6H5 | 54 | R | 58 |
| C6H5CH2SC2H4 C6H5 | 50 | R | 78 |
| p-CH3OC6H4CH2SCH3 | 51 | R | 27 |
| m-CH3OC6H4CH2SCH3 | 54 | rac | 4 |
| o-CH3OC6H4CH2SCH3 | 82 | R | 44 |
| p-NO2C6H4CH2SCH3 | 58 | R | 76 |
| m-NO2C6H4CH2SCH3 | 15 | R | 52 |
| o-NO2C6H4CH2SCH3 | 41 | R | 70 |
| p-FC6H4CH2SCH3 | 61 | R | 62 |
| p-ClC6H4CH2SCH3 | 61 | R | 65 |
| p-BrC6H4CH2SCH3 | 60 | R | 76 |
| p-CNC6H4CH2SCH3 | 47 | R | 72 |
| p-CH3 C6H4CH2S C6H5 | 62 | R | 82 |
| p-CH3CONHC6H4CH2SCH3 | 63 | R | 39 |
| C6H5SCH2CN | 50 | S | 13 |
| p-CH3OC6H4SCH2CN | 91 | R | 88 |
| p-BrC6H4SCH2CN | 92 | R | 94 |
| Substrate | Yield (%) | Sulfoxide Configuration | de (%) |
|---|---|---|---|
| Methyl ester of N-MOC-S-methyl-L-Cys | 12 | R | 34 |
| Propyl ester N-MOC-S-methyl-L-Cys | 22 | R | 52 |
| Methyl ester of N-MOC-L-Met | 54 | R | 83 |
| Propyl ester of N-MOC-L-Met | 56 | R | 90 |
| Pentyl ester of N-MOC-L-Met | 59 | R | 93 |
| Heptyl ester of N-MOC-L-Met | 35 | R | >95 |
| Methyl ester of N-MOC-D-Met | 53 | R | >95 |
| Ethyl ester of N-t-Boc-D-Met | 69 | R | >95 |
| Pentyl ester of N-t-Boc-D-Met | 17 | R | >95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mączka, W.; Wińska, K.; Grabarczyk, M. Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts 2018, 8, 624. https://doi.org/10.3390/catal8120624
Mączka W, Wińska K, Grabarczyk M. Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts. 2018; 8(12):624. https://doi.org/10.3390/catal8120624
Chicago/Turabian StyleMączka, Wanda, Katarzyna Wińska, and Małgorzata Grabarczyk. 2018. "Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow" Catalysts 8, no. 12: 624. https://doi.org/10.3390/catal8120624
APA StyleMączka, W., Wińska, K., & Grabarczyk, M. (2018). Biotechnological Methods of Sulfoxidation: Yesterday, Today, Tomorrow. Catalysts, 8(12), 624. https://doi.org/10.3390/catal8120624