In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts

Abstract
:
1. Introduction
2. Results and Discussion
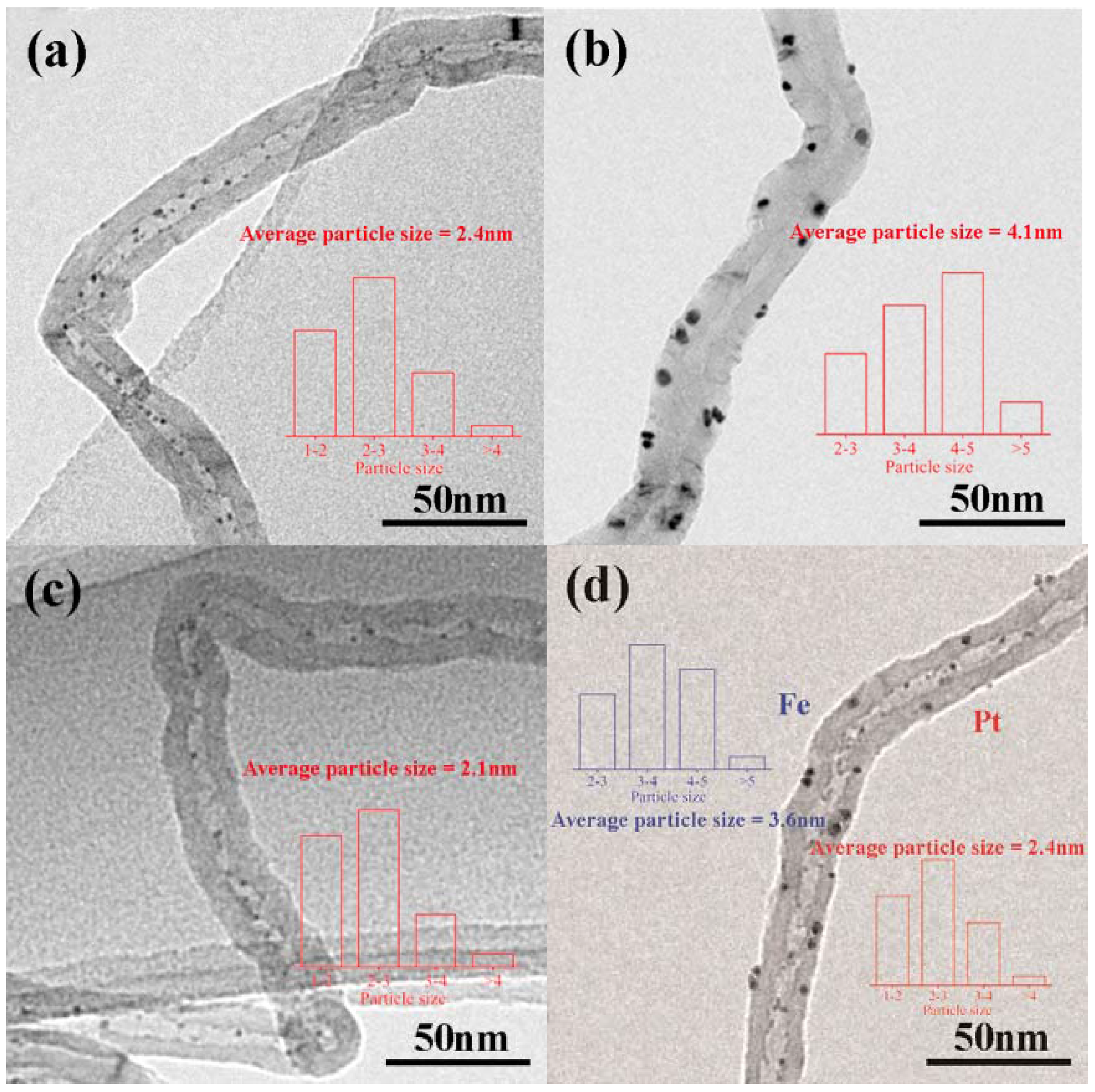
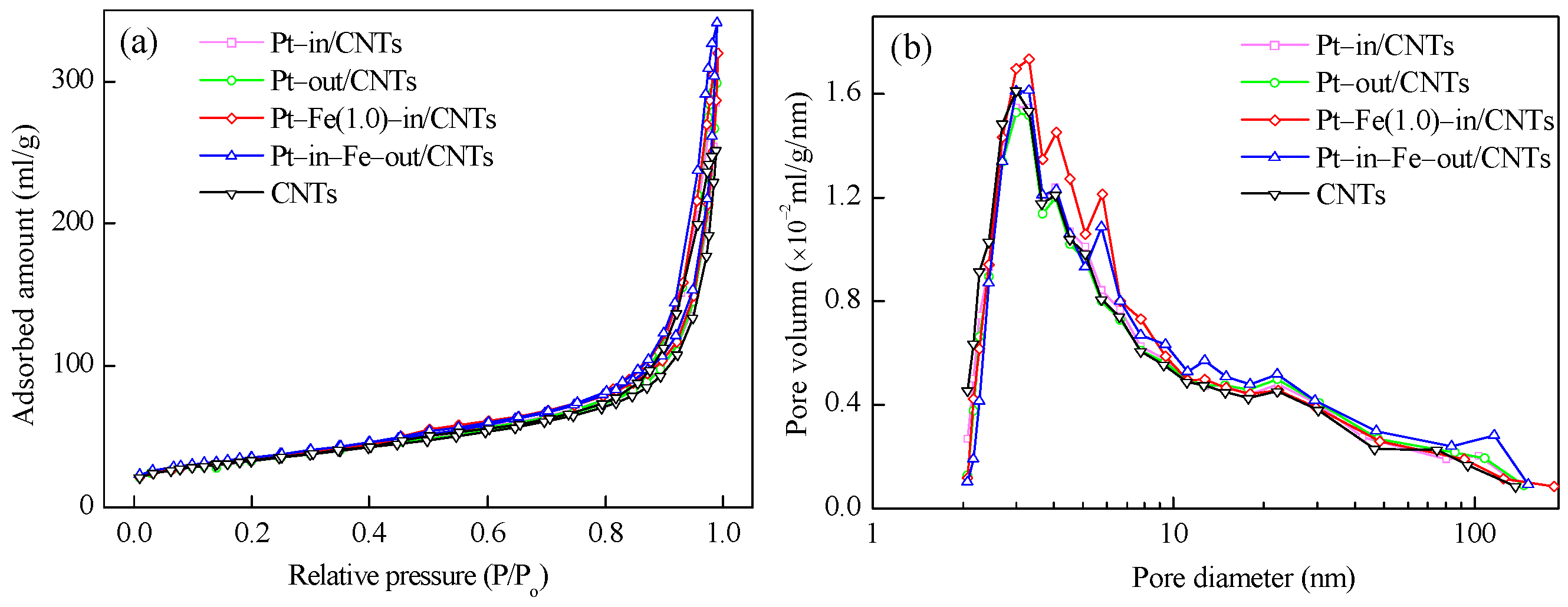
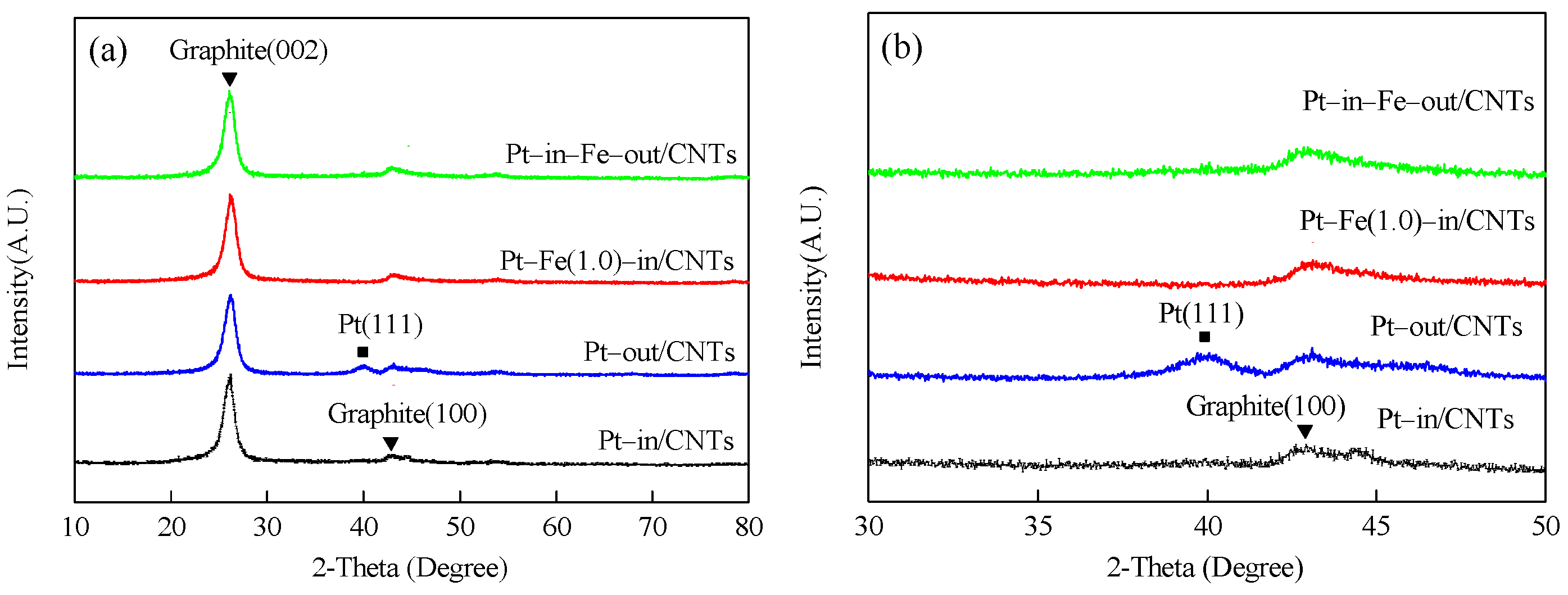
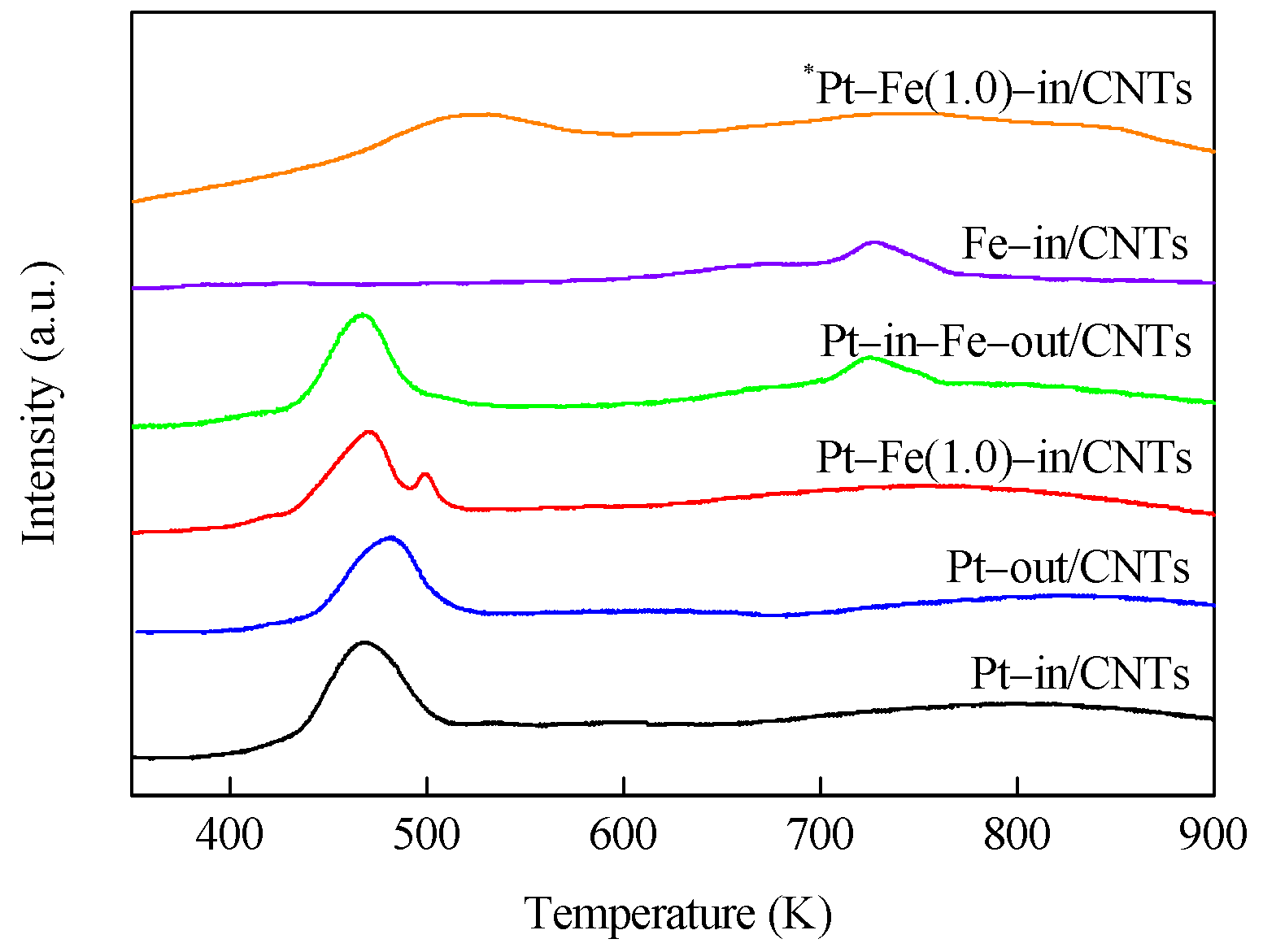
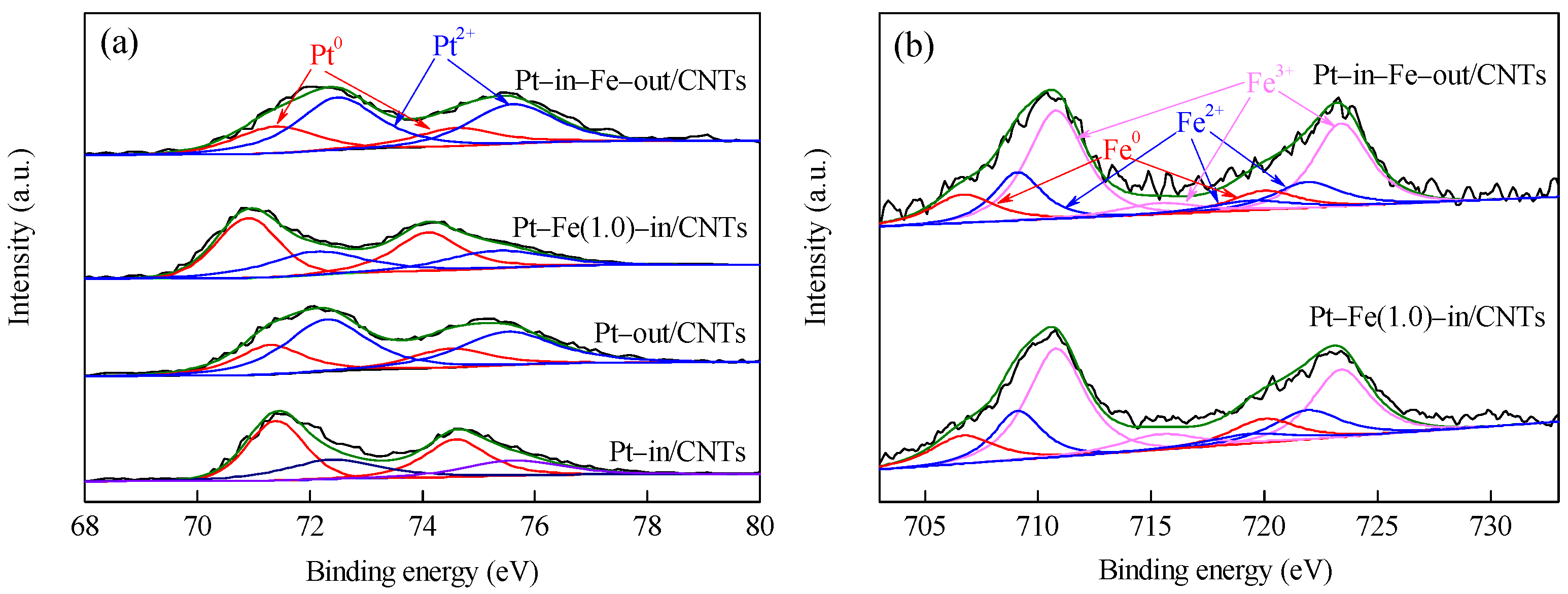
2.1. Catalyst Characterization
2.2. Catalytic Activity of Catalysts
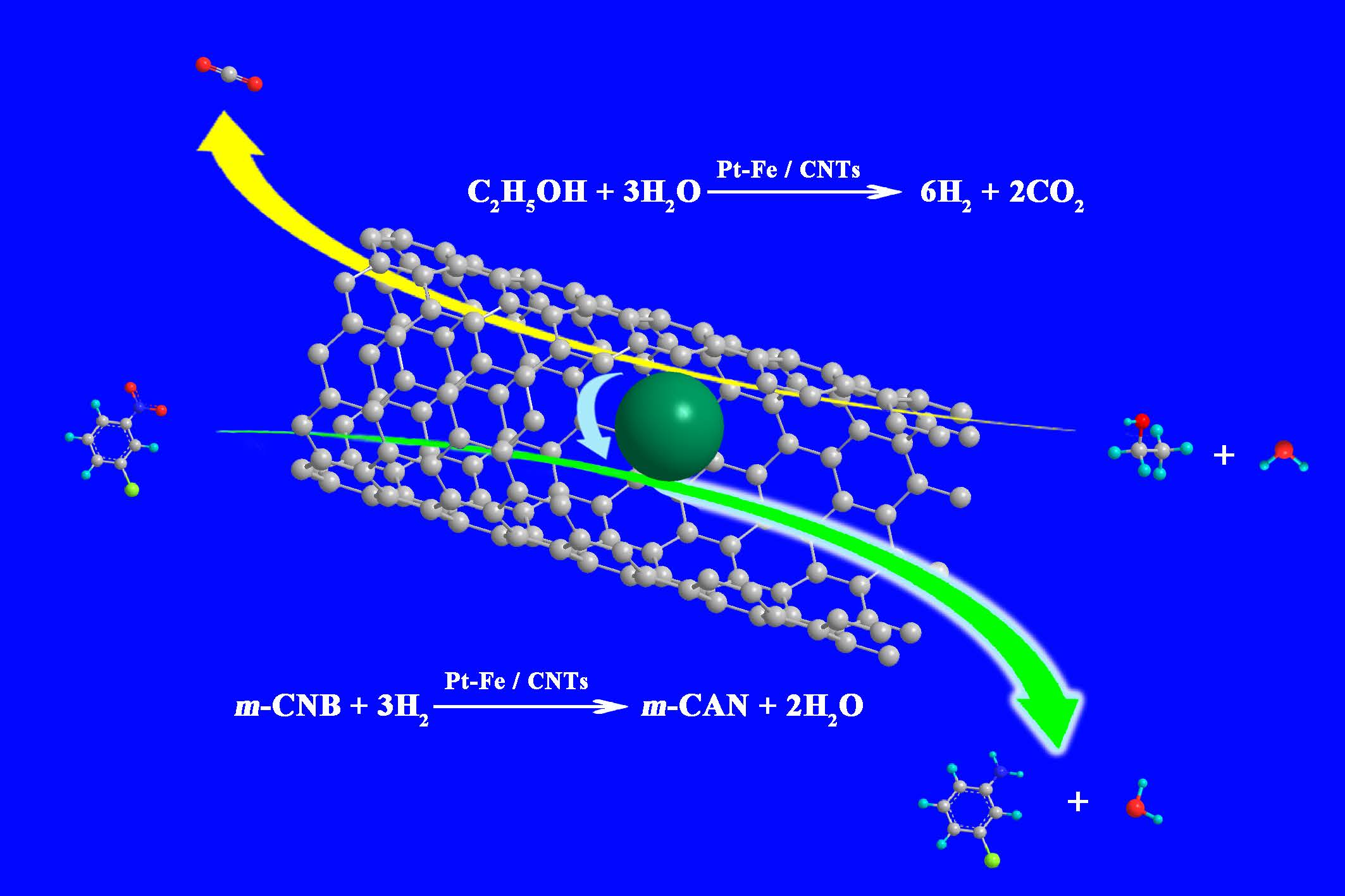
2.3. In-Situ Hydrogenation of m-CNB
3. Materials and Methods
3.1. Chemicals
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Catalytic Test
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiong, J.; Chen, J.; Zhang, J. Liquid-phase hydrogenation of O-chloronitrobenzene over supported nickel catalysts. Catal. Commun. 2007, 8, 345–350. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Long, J.; Liu, Q.; Li, Y.; Wang, C.; Zhang, Q.; Lv, W.; Zhang, X.; Qiu, S.; Wang, T.; et al. In situ hydrogenation of model compounds and raw bio-oil over Raney Ni catalyst. Energy Convers. Manag. 2015, 89, 188–196. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Wang, C.; Wang, C.; Ma, L.; Wang, T.; Zhang, X.; Zhang, Q. In-situ hydrogenation of model compounds and raw bio-oil over Ni/CMK-3 catalyst. Fuel Process. Technol. 2017, 161, 226–231. [Google Scholar] [CrossRef]
- Xu, Y.; Qiu, S.; Long, J.; Wang, C.; Chang, J.; Tan, J.; Liu, Q.; Ma, L.; Wang, T.; Zhang, Q. In situ hydrogenation of furfural with additives over a RANEY®Ni catalyst. RSC. Adv. 2015, 5, 91190–91195. [Google Scholar] [CrossRef]
- Fisk, C.A.; Morgan, T.; Ji, Y.; Crocker, M.; Crofcheck, C.; Lewis, S.A. Bio-oil upgrading over platinum catalysts using in situ generated hydrogen. Appl. Catal. A 2009, 358, 150–156. [Google Scholar] [CrossRef]
- Nozawa, T.; Mizukoshi, Y.; Yoshida, A.; Naito, S. Aqueous phase reforming of ethanol and acetic acid over TiO2 supported Ru catalysts. Appl. Catal. B 2014, 146, 221–226. [Google Scholar] [CrossRef]
- Nozawa, T.; Yoshida, A.; Hikichi, S.; Naito, S. Effects of Re addition upon aqueous phase reforming of ethanol over TiO2 supported Rh and Ir catalysts. Int. J. Hydrogen Energy 2015, 40, 4129–4140. [Google Scholar] [CrossRef]
- Xiong, H.; DeLaRiva, A.; Wang, Y.; Datye, A.K. Low-temperature aqueous-phase reforming of ethanol on bimetallic PdZn catalysts. Catal. Sci. Technol. 2014, 5, 254–263. [Google Scholar] [CrossRef]
- Sankar, M.; Dimitratos, N.; Miedziak, P.J.; Wells, P.P.; Kiely, C.J.; Hutchings, G.J. Designing bimetallic catalysts for a green and sustainable future. Chem. Soc. Rev. 2012, 41, 8099–8139. [Google Scholar] [CrossRef] [PubMed]
- Santo, V.D.; Gallo, A.; Naldoni, A.; Guidotti, M.; Psaro, R. Bimetallic heterogeneous catalysts for hydrogen production. Catal. Today 2012, 197, 190–205. [Google Scholar] [CrossRef]
- Kim, M.C.; Kim, T.W.; Kim, H.J.; Kim, C.U.; Bae, J.W. Aqueous phase reforming of polyols for hydrogen production using supported PtFe bimetallic catalysts. Renew. Energy 2016, 95, 396–403. [Google Scholar] [CrossRef]
- Han, X.; Zhou, R.; Lai, G.; Yue, B.; Zheng, X. Effect of transition metal (Cr, Mn, Fe, Co, Ni and Cu) on the hydrogenation properties of chloronitrobenzene over Pt/TiO2 catalysts. J. Mol. Catal. A 2004, 209, 83–87. [Google Scholar] [CrossRef]
- Cano, O.A.; González, C.A.R.; Paz, J.F.H.; Madrid, P.A.; Casillas, P.E.G.; Hernández, A.C.M.; Pérez, C.A.M. Catalytic activity of palladium nanocubes/multiwalled carbon nanotubes structures for methyl dye removal. Catal. Today 2017, 282, 168–173. [Google Scholar] [CrossRef]
- Yang, L.; Lin, G.D.; Zhang, H.B. Highly efficient Pd-ZnO catalyst doubly promoted by CNTs and Sc2O3 for methanol steam reforming. Appl. Catal. A 2013, 455, 137–144. [Google Scholar] [CrossRef]
- Stassi, J.P.; Zgolicz, P.D.; de Miguel, S.R.; Scelza, O.A. Formation of different promoted metallic phases in PtFe and PtSn catalysts supported on carbonaceous materials used for selective hydrogenation. J. Catal. 2013, 306, 11–29. [Google Scholar] [CrossRef]
- Deng, D.; Yu, L.; Chen, X.; Wang, G.; Jin, L.; Pan, X.; Deng, J.; Sun, G.; Bao, X. Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction. Angew. Chem. Int. Ed. 2013, 52, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Yang, G.; Endo, Y.; Tsubaki, N. The solvent effects during preparation of Fischer-Tropsch synthesis catalysts: Improvement of reducibility, dispersion of supported cobalt and stability of catalyst. Catal. Today 2009, 142, 85–89. [Google Scholar] [CrossRef]
- Sirijaruphan, A.; Goodwin, J.G.; Rice, R.W. Effect of Fe promotion on the surface reaction parameters of Pt/γ-Al2O3 for the selective oxidation of CO. J. Catal. 2004, 224, 304–313. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, Y.W. Hydrogenation of p-chloronitrobenzene on Mo, La, Fe, and W-modified NiCoB nanoalloy catalysts. J. Noncryst. Solids 2010, 356, 839–847. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- Chen, A.A.; Xu, X.S.; Hua, Y.X.; Gu, H.Z.; Yan, X.H. Fe3O4 modified alumina supported ruthenium catalyst for novel in-situ liquid phase catalytic hydrogenation. Acta Phys. Chim. Sin. 2013, 29, 799–805. [Google Scholar]
- Chen, W.; Fan, Z.; Pan, X.; Bao, X. Effect of confinement in carbon nanotubes on the activity of Fischer-Tropsch iron catalyst. J. Am. Chem. Soc. 2008, 130, 9414–9419. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, C.; Oubenali, M.; Galletti, A.M.R.; Serp, P.; Vannucci, G. Novel microwave synthesis of ruthenium nanoparticles supported on carbon nanotubes active in the selective hydrogenation of p-chloronitrobenzene to p-chloroaniline. Appl. Catal. A 2012, 421, 99–107. [Google Scholar] [CrossRef]
- Yan, X.; Sun, J.; Wang, Y.; Yang, J. A Fe-promoted Ni-P amorphous alloy catalyst (Ni-Fe-P) for liquid phase hydrogenation of m- and p-chloronitrobenzene. J. Mol. Catal. A 2006, 252, 17–22. [Google Scholar] [CrossRef]
- Xu, X.; Li, X.; Gu, H.; Huang, Z.; Yan, X. A highly active and chemoselective assembled Pt/C(Fe) catalyst for hydrogenation of o-chloronitrobenzene. Appl. Catal. A 2012, 429, 17–23. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Ji, H.; Wei, Y.; Wu, N.; Zuo, B.; Wang, Q. Magnetic nanocomposite catalysts with high activity and selectivity for selective hydrogenation of ortho-chloronitrobenzene. J. Catal. 2005, 229, 114–118. [Google Scholar] [CrossRef]
- Bossola, F.; Pereira-Hernández, X.I.; Evangelisti, C.; Wang, Y.; Santo, V.D. Investigation of the promoting effect of Mn on a Pt/C catalyst for the steam and aqueous phase reforming of glycerol. J. Catal. 2017, 349, 75–83. [Google Scholar] [CrossRef]
- Kourtelesis, M.; Panagiotopoulou, P.; Verykios, X. Influence of structural parameters on the reaction of low temperature ethanol steam reforming over Pt/Al2O3 catalsyts. Catal. Today 2015, 258, 247–255. [Google Scholar] [CrossRef]
- Bilal, M.; Jackson, S.D. Ethanol steam reforming over Pt/Al2O3 catalysts: The effect of impurities on selectivity and catalyst deactivation. Appl. Catal. A 2017, 529, 98–107. [Google Scholar] [CrossRef]
- He, C.; Zheng, J.; Wang, K.; Lin, H.; Wang, J.Y.; Yang, Y. Sorption enhanced aqueous phase reforming of glycerol for hydrogen production over Pt–Ni supported on multi-walled carbon nanotubes. Appl. Catal. B 2015, 162, 401–411. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. A first-principles study of methanol decomposition on Pt(111). J. Am. Chem. Soc. 2002, 124, 7193–7201. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, J.; Li, X.; Yan, X. Liquid-phase hydrogenation of nitrobenzene to aniline using solvent hydrogen from ethanol reforming. Chin. J. Catal. 2006, 27, 559–561. [Google Scholar]
- Larimi, A.S.; Kazemeini, M.; Khorasheh, F. Aqueous phase reforming of glycerol using highly active and stable Pt0.05CexZr0.95–xO2 ternary solid solution catalysts. Appl. Catal. A 2016, 523, 230–240. [Google Scholar] [CrossRef]
- Ciftci, A.; Michel Ligthart, D.A.J.; Oben Sen, A.; van Hoof, A.J.F.; Friedrich, H.; Hensen, E.J.M. Pt-Re synergy in aqueous-phase reforming of glycerol and the water-gas shift reaction. J. Catal. 2014, 311, 88–101. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, Y.; Wang, T.; Ma, L.; Zhang, Q.; Zhang, X.; Zhang, X. In-situ hydrogenation of lignin depolymerization model compounds to cyclohexanol. J. Fuel Chem. Technol. 2013, 41, 443–448. [Google Scholar] [CrossRef]
- Soares, A.V.H.; Atia, H.; Armbruster, U.; Passos, F.B.; Martin, A. Platinum, palladium and nickel supported on Fe3O4 as catalysts for glycerol aqueous-phase hydrogenolysis and reforming. Appl. Catal. A 2017, 548, 179–190. [Google Scholar] [CrossRef]
- He, Z.; Yang, M.; Wang, X.; Zhao, Z.; Duan, A. Effect of the transition metal oxide supports on hydrogen production from bio-ethanol reforming. Catal. Today 2012, 194, 2–8. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Tao, Z.; Zhang, C.; Xiang, H.; Li, Y. Study on an iron-manganese Fischer-Tropschsynthesis catalyst prepared from ferrous sulfate. Fuel Process. Technol. 2009, 90, 1247–1251. [Google Scholar] [CrossRef]
- Wan, H.J.; Wu, B.S.; Li, T.Z.; Tao, Z.C.; Xia, A.; Xiang, H.W.; Li, Y.W. Study of an iron-based Fischer-Tropsch synthesis catalyst incorporated with SiO2. J. Fuel Chem. Technol. 2007, 35, 589–594. [Google Scholar] [CrossRef]
- Smit, E.D.; Beale, A.M.; Nikitenko, S.; Weckhuysen, B.M. Local and long range order in promoted iron-based Fischer-Tropsch catalysts: A combined in situ X-ray absorption spectroscopy/wide angle X-ray scattering study. J. Catal. 2009, 262, 244–256. [Google Scholar] [CrossRef]
- Deluga, G.; Salge, J.R.; Schmidt, L.D.; Verykios, X.E. Renewable hydrogen from ethanol by autothermal reforming. Science 2004, 303, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Gu, H.; Yan, X. A novel transfer hydrogenation with high hydrogen utilization for the hydrogenation of halogenated nitrobenzene without hydrodehalogenation. Catal. Lett. 2009, 132, 16–21. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y. A novel liquid system of catalytic hydrogenation. Sci. China Ser. B 2007, 50, 746–753. [Google Scholar] [CrossRef]
- Liu, L.; Qiao, B.; Chen, Z.; Zhang, J.; Deng, Y. Novel chemoselective hydrogenation of aromatic nitro compounds over ferric hydroxide supported nanocluster gold in the presence of CO and H2O. Chem. Commun. 2009, 6, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Fatsikostas, A.N.; Kondarides, D.I.; Verykios, X.E. Production of hydrogen for fuel cells by reformation of biomass-derived ethanol. Catal. Today 2002, 75, 145–155. [Google Scholar] [CrossRef]
- Yin, A.Y.; Guo, X.Y.; Dai, W.L.; Fan, K.N. The synthesis of propylene glycol and ethylene glycol from glycerol using Raney Ni as a versatile catalyst. Green Chem. 2009, 11, 1514–1516. [Google Scholar] [CrossRef]
- Kong, D.; Jiang, T.; Zhang, Y.; Cao, F. Catalytic performance of activated carbon supported Pt–Ni biometallic catalyst for glycerol in situ hydrogenolysis. Chem. J. Chin. Univ. 2016, 36, 1140–1147. [Google Scholar]
- Tessonnier, J.P.; Ersen, O.; Weinberg, G.; Pham-Huu, C.; Su, D.S.; Schlögl, R. Selective deposition of metal nanoparticles inside or outside multiwalled carbon nanotubes. ACS Nano 2009, 3, 2081–2089. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m²/g) | Vpore (cm³/g) | Dpore (nm) | dPt1 (nm) | dFe2 (nm) | Pt 3 (wt %) | Pt0/Pt2+ 4 | Fe0/Fe2+/Fe3+ 5 |
|---|---|---|---|---|---|---|---|---|
| CNTs 6 | 113.5 | 0.39 | 13.8 | — | — | — | — | — |
| Pt–in/CNTs | 111.5 | 0.45 | 16.3 | 2.4 | — | 1.93 | 52.8/47.2 | — |
| Pt–out/CNTs | 111.3 | 0.44 | 16.3 | 4.1 | — | 1.92 | 31.5/68.5 | — |
| Pt–Fe(1.0)–in/CNTs | 121.7 | 0.51 | 15.8 | 2.1 7 | — | 1.91 | 60.3/39.7 | 19.1/24.4/56.5 |
| Pt–in–Fe–out/CNTs | 118.6 | 0.48 | 16.2 | 2.4 | 3.6 | 1.91 | 31.7/68.3 | 16.2/23.6/60.2 |
| Catalysts | Conversion (%) | Selectivity (%) | TOF 2 | ||
|---|---|---|---|---|---|
| m-CAN | AN | Others | |||
| CNTs | <1 | — | — | — | — |
| Pt–in/CNTs | 91.5 | 94.6 | 5.0 | 0.4 | 18.6 |
| Pt–out/CNTs | 60.4 | 96.3 | 3.1 | 0.3 | 12.3 |
| Catalysts | Conversion (%) | Selectivity (%) | TOF 2 | ||
|---|---|---|---|---|---|
| m-CAN | AN | Others | |||
| Pt–in/CNTs | 12.3 | 97.7 | 2.3 | 0 | 10.0 |
| Fe–in/CNTs | <1 | — | — | — | — |
| Pt–Fe(0.5)–in/CNTs | 18.4 | 97.3 | 2.5 | 0.2 | 15.0 |
| Pt–Fe(0.75)–in/CNTs | 67.5 | 97.1 | 2.6 | 0.3 | 54.9 |
| Pt–Fe(1.0)–in/CNTs | 94.3 | 95.6 | 4.0 | 0.4 | 76.7 |
| Pt–Fe(1.25)–in/CNTs | 83.2 | 94.8 | 5.1 | 0.1 | 67.6 |
| Pt–Fe(1.5)–in/CNTs | 29.6 | 93.4 | 6.4 | 0.2 | 24.1 |
| Pt–in–Fe–out/CNTs | 20.5 | 93.8 | 6.2 | 0 | 16.7 |
| H2O/C2H5OH 2 | T (K) | P (MPa) | Conversion (%) | Selectivity (%) | TOF 3 | ||
|---|---|---|---|---|---|---|---|
| m-CAN | AN | Others | |||||
| 1/9 | 473 | 3 | 71.5 | 96.4 | 3.5 | 0.1 | 58.1 |
| 2/8 | 473 | 3 | 85.4 | 96.1 | 3.7 | 0.2 | 69.4 |
| 3/7 | 473 | 3 | 94.3 | 95.6 | 4.0 | 0.4 | 76.7 |
| 4/6 | 473 | 3 | 89.7 | 95.9 | 3.7 | 0.4 | 72.9 |
| 3/7 | 453 | 3 | 91.2 | 96.8 | 2.9 | 0.3 | 74.1 |
| 3/7 | 433 | 3 | 70.8 | 97.4 | 2.4 | 0.2 | 57.6 |
| 3/7 | 473 | 4 | 91.6 | 95.4 | 4.1 | 0.5 | 74.5 |
| 3/7 | 473 | 2 | 92.3 | 96.2 | 3.6 | 0.2 | 75.0 |
| 3/7 | 473 | 1 | 92.1 | 96.3 | 3.4 | 0.3 | 74.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Liang, J.; Zhu, W.; Song, H.; Wang, K.; Li, C. In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts. Catalysts 2018, 8, 62. https://doi.org/10.3390/catal8020062
Li F, Liang J, Zhu W, Song H, Wang K, Li C. In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts. Catalysts. 2018; 8(2):62. https://doi.org/10.3390/catal8020062
Chicago/Turabian StyleLi, Feng, Jinrong Liang, Wenxi Zhu, Hua Song, Keliang Wang, and Cuiqin Li. 2018. "In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts" Catalysts 8, no. 2: 62. https://doi.org/10.3390/catal8020062
APA StyleLi, F., Liang, J., Zhu, W., Song, H., Wang, K., & Li, C. (2018). In-Situ Liquid Hydrogenation of m-Chloronitrobenzene over Fe-Modified Pt/Carbon Nanotubes Catalysts. Catalysts, 8(2), 62. https://doi.org/10.3390/catal8020062