Comparision on the Low-Temperature NH3-SCR Performance of γ-Fe2O3 Catalysts Prepared by Two Different Methods


Abstract
:1. Introduction
2. Results and Discussion
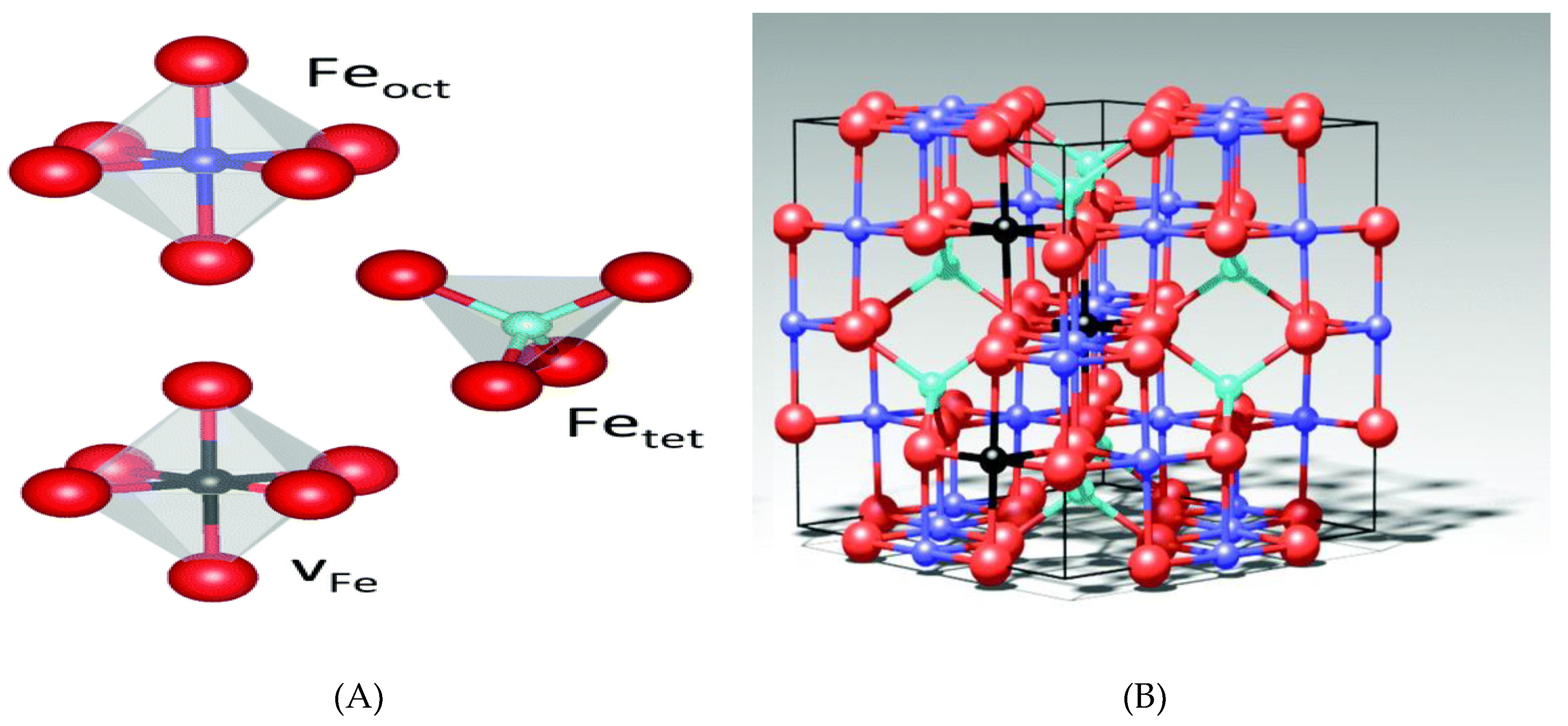
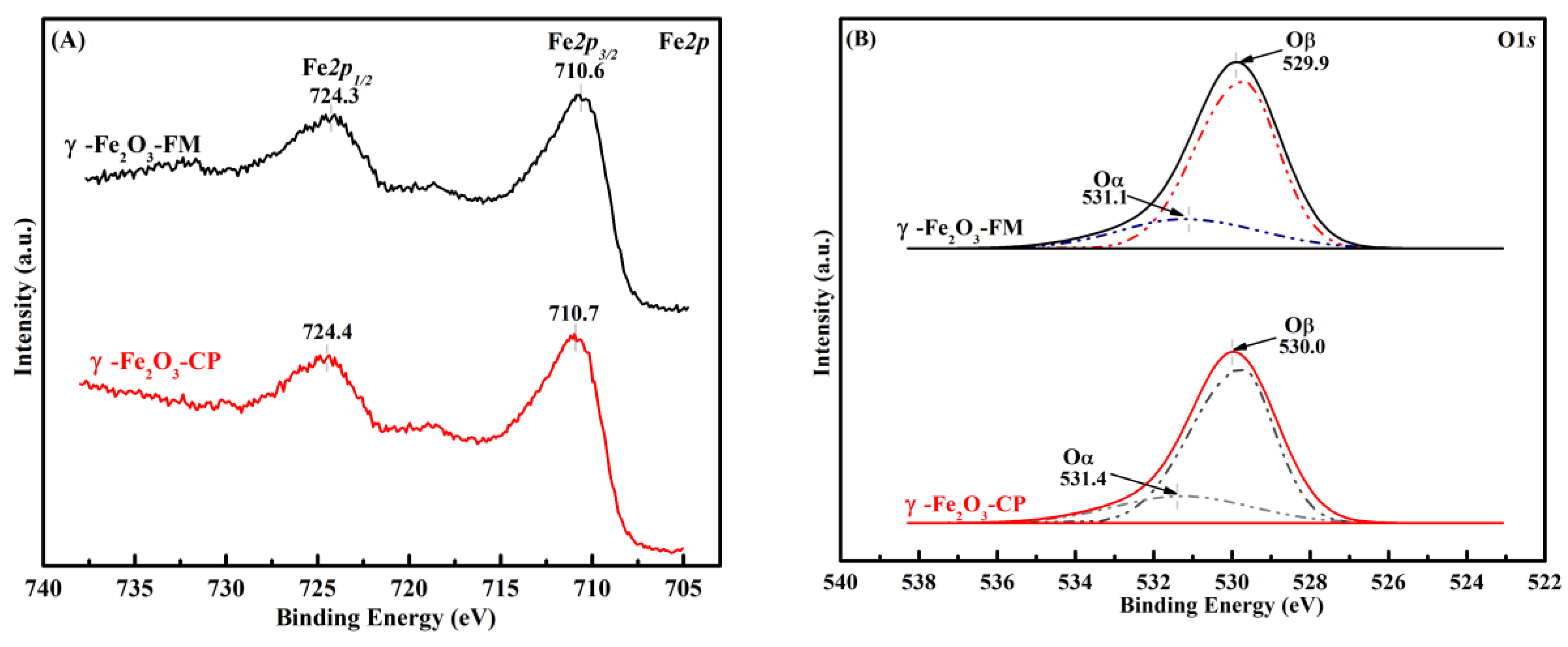
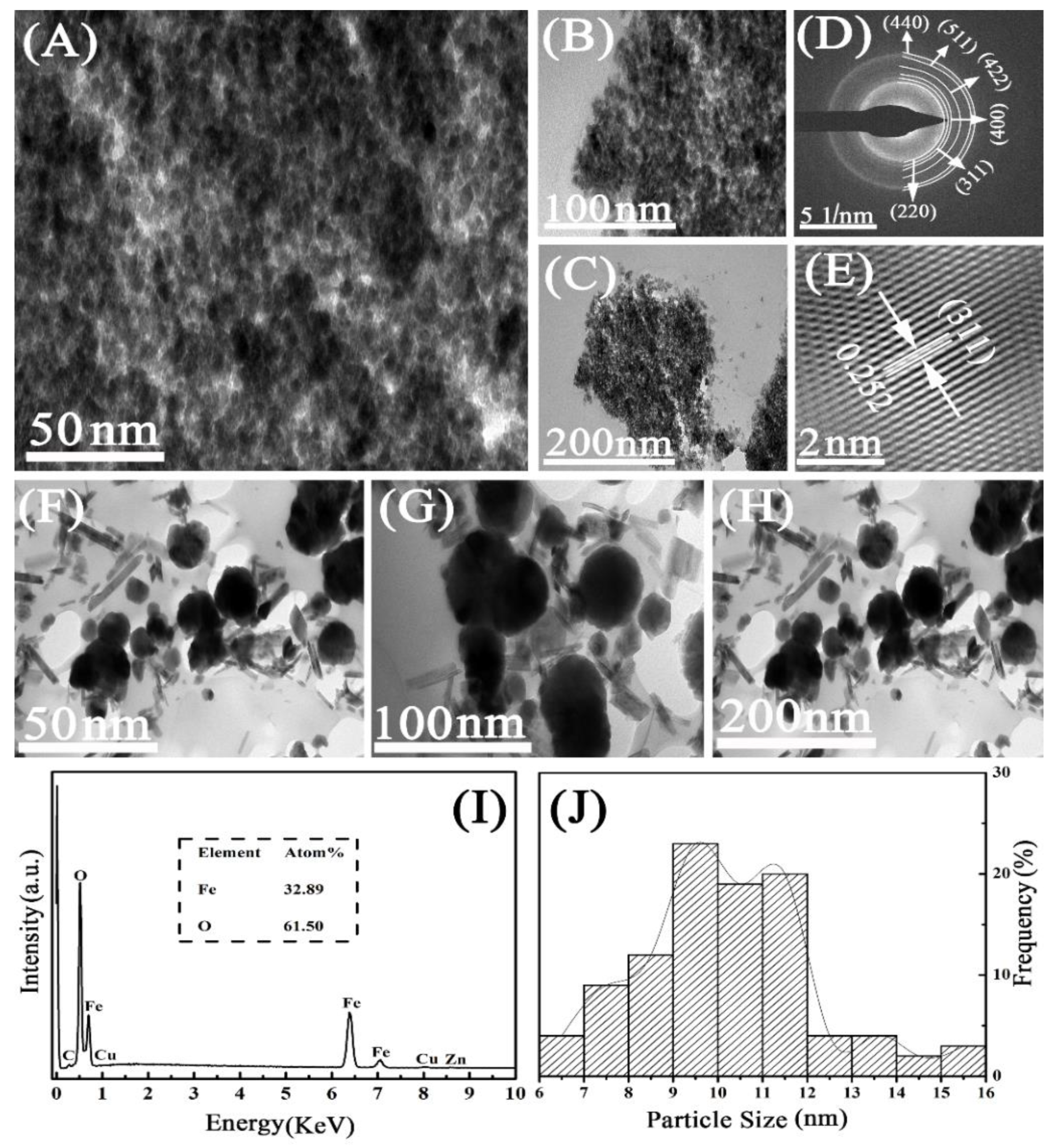
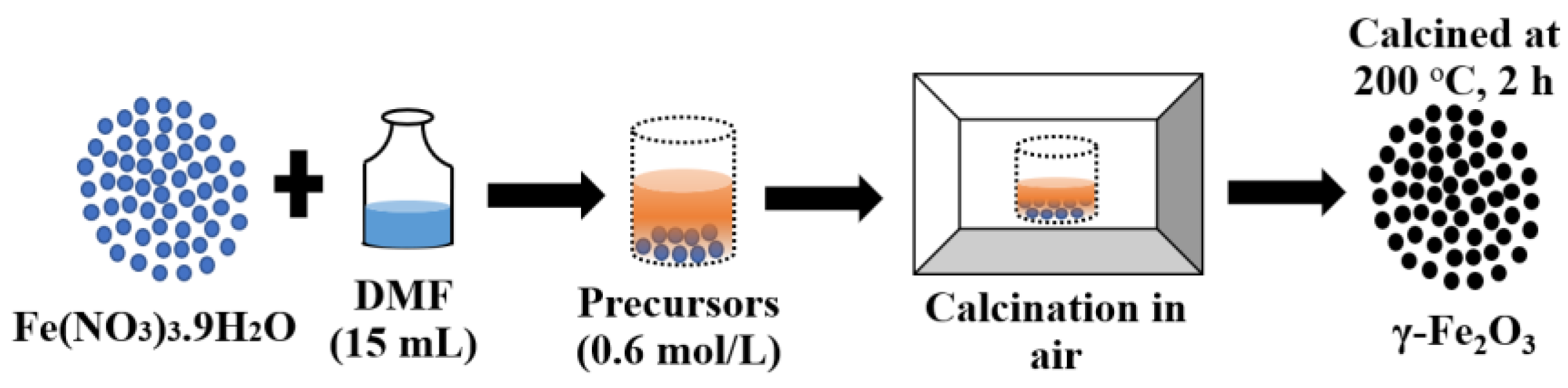
2.1. Characterization
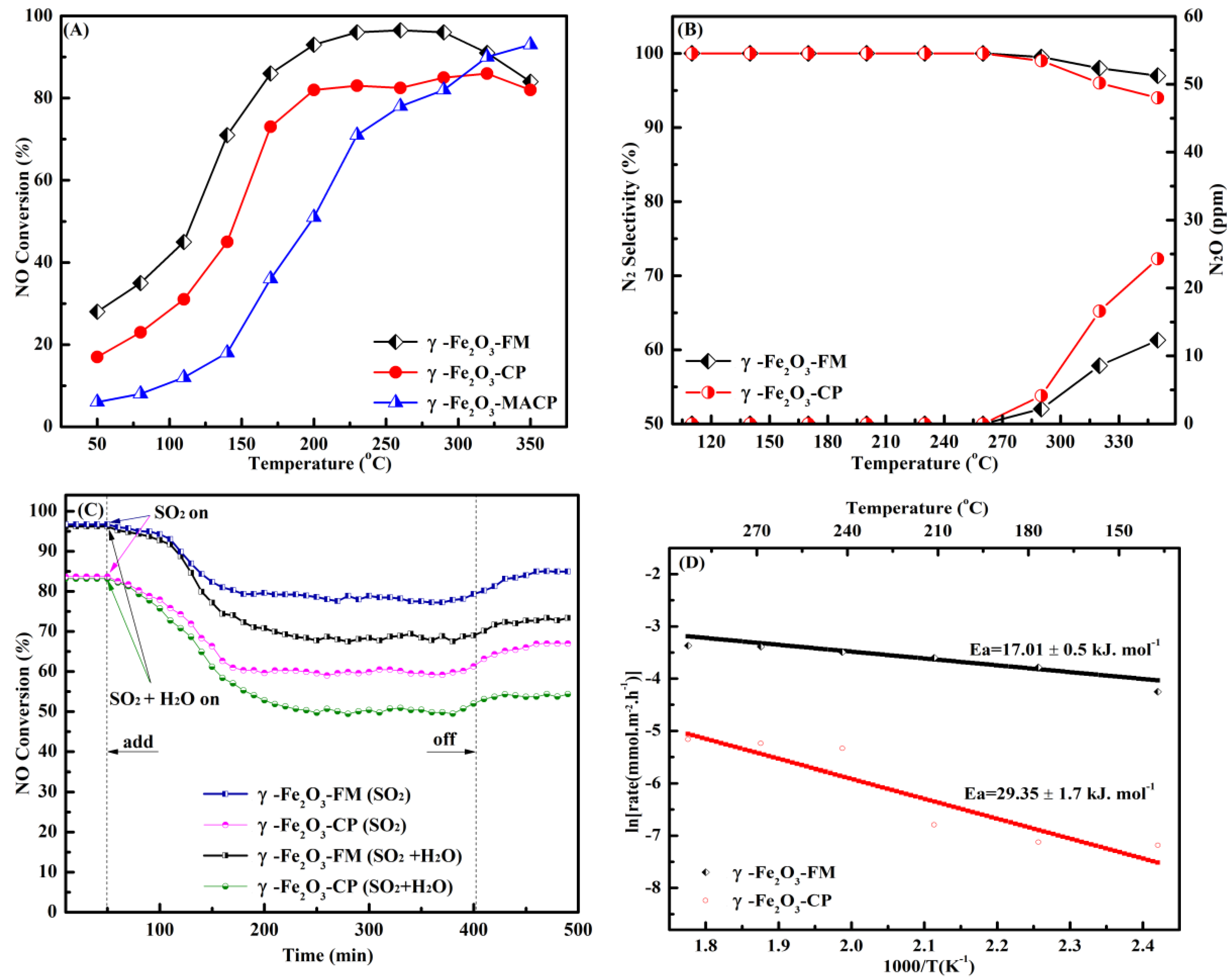
2.2. Low-Temperature NH3-SCR Performance
2.3. NO and NH3 Oxidation Activity
2.4. In Situ DRIFTS Study of γ-Fe2O3-FM
2.4.1. Ammonia Adsorbents over γ-Fe2O3-FM
2.4.2. Nitric Oxide Plus Oxygen Adsorption over γ-Fe2O3-FM
2.4.3. Ammonia Ad Species Reaction with Nitrogen Oxides
2.4.4. Nitrogen Oxides Ad Species Reaction with Ammonia
3. Materials and Methods
3.1. Materials and Reagents
3.2. Experimental Setup and Governing Equations
3.3. Characterization Used
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, F.; Asakura, K.; He, H.; Liu, Y.; Shan, W.; Shi, X. Influence of calcination temperature on iron titanate catalyst for the selective catalytic reduction of NOx with NH3. Catal. Today 2011, 164, 520–527. [Google Scholar] [CrossRef]
- Husnain, N.; Wang, E.; Li, K.; Anwar, M.T.; Mehmood, A.; Gul, M.; Li, D.; Jinda, M. Iron oxide-based catalysts for low-temperature selective catalytic reduction of NOx with NH3. Rev. Chem. Eng. 2019, 35, 239–264. [Google Scholar] [CrossRef]
- Husnain, N.; Wang, E.; Fareed, S. Low-temperature selective catalytic reduction of NO with NH3 over natural iron ore catalyst. Catalysts 2019, 9, 956. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Liu, T.; Zhao, N.; Yang, X.; Deng, L. Iron-doped Mn-Ce/TiO2 catalyst for low temperature selective catalytic reduction of NO with NH3. J. Environ. Sci. 2010, 22, 1447–1454. [Google Scholar] [CrossRef]
- Yang, S.J.; Wang, C.Z.; Ma, L.; Peng, Y.; Qu, Z.; Yan, N.Q. Substitution of WO3 in V2O5/WO3-TiO2 by Fe2O3 for selective catalytic reduction of NO with NH3. Catal. Sci. Technol. 2013, 3, 161–168. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Y.; Chen, B.; Zhu, T.; Ma, L. Novel Fe-Ce-Ti catalyst with remarkable performance for the selective catalytic reduction of NOx by NH3. Catal. Sci. Technol. 2016, 6, 6688–6696. [Google Scholar] [CrossRef]
- Wang, X.; Gui, K. Fe2O3 particles as superior catalysts for low temperature selective catalytic reduction of NO with NH3. J. Environ. Sci. 2013, 25, 2469–2475. [Google Scholar] [CrossRef]
- Liu, C.; Yang, S.; Ma, L.; Peng, Y.; Hamidreza, A.; Chang, H. Comparison on the performance of α-Fe2O3 and γ-Fe2O3 for selective catalytic reduction of nitrogen oxides with ammonia. Catal. Lett. 2013, 143, 697–704. [Google Scholar] [CrossRef]
- Lu, C.; Li, J.; Peng, Y.; Wang, D.; Gan, L.; Ma, Y. De-reducibility mechanism of titanium on maghemite catalysts for the SCR reaction: An in situ DRIFTS and quantitative kinetics study. Appl. Catal. B Environ. 2017, 221, 556–564. [Google Scholar]
- Xu, H.; Ni, K.; Li, X.; Fang, G.; Fan, G. Structural transformation of Pd-α-Fe2O3 and Pd-γ-Fe2O3 catalysts and application in the CO oxidation reaction. RSC Adv. 2017, 7, 51403–51410. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, M.; Sultan, S.; Ahmad, I.; Ullah, H.; Yaseen, M.; Amir, A. Adsorptive desulfurization of model oil using untreated, acid-activated and magnetite nanoparticle loaded bentonite as adsorbent. J. Saudi. Chem. Soc. 2017, 21, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Pehkonen, S.O. Aqueous free radial chemistry of mercury in the presence of iron oxides and ambient aerosol. Atmos. Environ. 1997, 31, 4125–4137. [Google Scholar] [CrossRef]
- Stevin, M.; Wagner, M.; Schmid, M.; Parkinson, G.S.; Diebodd, U. Surface point defects on bulk oxides: Atomically-resolved scanning probe microscopy. Chem. Soc. Rev. 2017, 46, 1772–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Liu, F.; Xie, L.; Shan, W.; He, H. NH3-SCR performance of fresh and hydrothermally aged Fe-ZSM-5 in standard and fast selective catalytic reduction reactions. Environ. Sci. Technol. 2013, 47, 3293–3298. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, F.; Li, H.; Yang, Q.; Wang, L.; Li, X. Low-Temperature selective catalytic reduction of NOx with NH3 over Fe–Mn mixed-oxide catalysts containing Fe3Mn3O8 phase. Ind. Eng. Chem. Res. 2011, 51, 202–212. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Q.; Li, X.; Peng, Y.; Li, B.; Si, W. Preparation of γ-Fe2O3 catalysts and their deNOx performance: Effects of precipitation conditions. Chem. Eng. Technol. 2018, 41, 1019–1026. [Google Scholar] [CrossRef]
- Vidal-Vidal, J.; Rivas, J.; López-Quintela, M.A. Synthesis of monodisperse maghemite nanoparticles by the microemulsion method. Colloids Surf. A Physicochem. Eng. Asp. 2006, 288, 44–51. [Google Scholar] [CrossRef]
- Zare, N.; Zabardasti, A.; Mohammadi, A.; Azarbani, F. Synthesis of spherical Fe3O4 nanoparticles from the thermal decomposition of iron (III) nano-structure complex: DFT studies and evaluation of the biological activity. Bioorg. Chem. 2018, 80, 334–346. [Google Scholar] [CrossRef]
- Aliahmad, M.; Nasiri, M.N. Synthesis of maghemite (γ-Fe2O3) nanoparticles by thermal-decomposition of magnetite (Fe3O4) nanoparticles. Mater. Sci. Pol. 2013, 31, 264–268. [Google Scholar] [CrossRef]
- Al-Gaashani, R.; Radiman, S.; Tabet, N.; Daud, A.R. Rapid synthesis and optical properties of hematite (α-Fe2O3) nanostructures using a simple thermal decomposition method. J. Alloys. Compd. 2013, 550, 395–401. [Google Scholar] [CrossRef]
- Ismail, A.A. Synthesis and characterization of Y2O3/Fe2O3/TiO2 nanoparticles by sol-gel method. Appl. Catal. B Environ. 2005, 58, 115–121. [Google Scholar] [CrossRef]
- Alagiri, M.; Hamid, S.B.A. Sol-gel synthesis of α-Fe2O3 nanoparticles and its photocatalytic application. J. Sol.-Gel. Sci. Technol. 2015, 74, 783–789. [Google Scholar] [CrossRef]
- Reda, S.M. Synthesis of ZnO and Fe2O3 nanoparticles by sol-gel method and their application in dye-sensitized solar cells. Mater. Sci. Semicond. Process. 2010, 13, 417–425. [Google Scholar] [CrossRef]
- Lassoued, A.; Dkhil, B.; Gadri, A.; Ammar, S. Control of the shape and size of iron oxide (α-Fe2O3) nanoparticles synthesized through the chemical precipitation method. Results. Phys. 2017, 7, 3007–3015. [Google Scholar] [CrossRef]
- Khodadadi-Moghaddam, M.; Salimi, F.; Sahebalzamani, H.; Jafari, N. Synthesis of Fe2O3 nanoparticles via various methods and coating with silica for drug immobilization. Synth. React. Inorg. Met. Nano-Metal. Chem. 2013, 43, 1224–1227. [Google Scholar] [CrossRef]
- Zhang, G.; Huang, X.; Tang, Z. Enhancing Water resistance of a Mn-based catalyst for low temperature selective catalytic reduction reaction by modifying super hydrophobic layers. ACS Appl. Mater. Interfaces 2019, 11, 36598–36606. [Google Scholar] [CrossRef]
- Duan, Z.; Chi, K.; Liu, J.; Shi, J.; Zhao, Z.; Wei, Y. The catalytic performances and reaction mechanism of nanoparticle Cd/Ce-Ti oxide catalysts for NH3-SCR reaction. RSC Adv. 2017, 7, 50127–50134. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wang, J.; Wang, J.; Shen, M. The influence of Si/Al ratios on adsorption and desorption characterizations of Pd/Beta served as cold-start catalysts. Materials 2019, 12, 1045. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wan, Y.; Li, Y.; Zhan, S.; Guan, Q.; Tian, Y. Low-temperature selective catalytic reduction of NO with NH3 over Mn2O3-doped Fe2O3 hexagonal microsheets. ACS Appl. Mater. Interfaces 2016, 8, 5224–5233. [Google Scholar] [CrossRef]
- Shan, H.; Liu, C.; Liu, L.; Zhang, J.; Li, H.; Liu, Z. Excellent toluene sensing properties of SnO2-Fe2O3 interconnected nanotubes. ACS Appl. Mater. Interfaces 2013, 5, 6376–6380. [Google Scholar] [CrossRef]
- Ning, R.; Chen, L.; Li, E.; Liu, X.; Zhu, T. Applicability of V2O5-WO3/TiO2 catalysts for the SCR denitrification of alumina calcining flue gas. Catalysts 2019, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Jin, R.; Liu, Y.; Wang, H. Ceria modified MnOx/TiO2 as a superior catalyst for NO reduction with NH3 at low-temperature. Catal. Commun. 2008, 9, 2217–2220. [Google Scholar] [CrossRef]
- Yu, J.; Si, Z.; Chen, L.; Wu, X.; Weng, D. Selective catalytic reduction of NOx by ammonia over phosphate-containing Ce0.75Zr0.25O2 solids. Appl. Catal. B Environ. 2015, 163, 223–232. [Google Scholar] [CrossRef]
- Xu, L.; Yang, Q.; Hu, L.; Wang, D.; Peng, Y.; Shao, Z.; Lu, C.; Li, J. Insights over titanium modified FeMgOx catalysts for selective catalytic reduction of NOx with NH3: Influence of precursors and crystalline structures. Catalysts 2019, 9, 560. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, H.; Ma, Z.; Zhang, X. Selective catalytic reduction of NO with NH3 over CuOx-carbonaceous materials. Catal. Commun. 2012, 17, 8–12. [Google Scholar] [CrossRef]
- Xie, G.; Liu, Z.; Zhu, Z.; Liu, Q.; Ge, J.; Huang, Z. Simultaneous removal of SO2 and NOx from flue gas using a CuO/Al2O3 catalyst sorbent: II. Promotion of SCR activity by SO2 at high temperatures. J. Catal. 2004, 224, 42–49. [Google Scholar] [CrossRef]
- Roy, S.; Viswanath, B.; Hegde, M.S.; Madras, G. Low-temperature selective catalytic reduction of NO with NH3 over Ti0.9M0.1O2−δ (M = Cr, Cr, Mn, Fe, Co, Cu). J. Phys. Chem. C 2008, 112, 6002–6012. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, B.; Liu, Y.; Zhao, W.; Guan, B. Experimental study on a low-temperature SCR catalyst based on MnOx/TiO2 prepared by sol-gel method. J. Hazard. Mater. 2007, 145, 488–494. [Google Scholar] [CrossRef]
- Madia, G.; Koebel, M.; Elsener, M.; Wokaun, A. The effect of an oxidation precatalyst on the NOx reduction by ammonia SCR. Ind. Eng. Chem. Res. 2002, 41, 3512–3517. [Google Scholar] [CrossRef]
- Koebel, M.; Madia, G.; Elsener, M. Selective catalytic reduction of NO and NO2 at low temperatures. Catal. Today 2002, 73, 239–247. [Google Scholar] [CrossRef]
- Madia, G.; Koebel, M.; Elsener, M.; Wokaun, A. Side reactions in the selective catalytic reduction of NOx with various NO2 fractions. Ind. Eng. Chem. Res. 2002, 41, 4008–4015. [Google Scholar] [CrossRef]
- Liu, F.; He, H.; Ding, Y.; Zhang, C. Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3. Appl. Catal. B Environ. 2009, 93, 194–204. [Google Scholar] [CrossRef]
- Cai, S.; Shi, L.; Huang, L.; Zhang, D.; Hu, H.; Li, H. Mechanistic aspects of deNOx processing over TiO2 supported Co–Mn oxide catalysts: Structure–activity relationships and In Situ DRIFTs analysis. ACS Catal. 2015, 5, 6069–6077. [Google Scholar]
- Cai, S.; Zhang, D.; Shi, L.; Li, H.; Huang, L.; Hu, H. In Situ DRIFTs investigation of the low-temperature reaction mechanism over Mn-doped Co3O4 for the selective catalytic reduction of NOx with NH3. J. Phys. Chem. C. 2015, 119, 22924–22933. [Google Scholar]
- Kijlstra, W.S.; Brands, D.S.; Poels, E.K.; Bliek, A. Mechanism of the selective catalytic reduction of NO by NH3 over MnOx/Al2O3. J. Catal. 1997, 218, 208–218. [Google Scholar] [CrossRef]
- Gu, T.; Liu, Y.; Weng, X.; Wang, H.; Wu, Z. The enhanced performance of ceria with surface sulfation for selective catalytic reduction of NO by NH3. Catal. Commun. 2010, 12, 310–313. [Google Scholar] [CrossRef]
- Cataluna, R.; Arcoya, A.; Martínez-Arias, A.; Conesa, J.C.; Seoane, X.L.; Soria, J. NO reaction at surface oxygen vacancies generated in cerium oxide. J. Chem. Soc. Farad. Trans. 2004, 91, 1679–1687. [Google Scholar]
- Underwood, G.M.; Miller, T.M.; Grassian, V.H. Transmission FT-IR and knudsen cell study of the heterogeneous reactivity of gaseous nitrogen dioxide on mineral oxide particles. J. Phys. Chem. A 1999, 103, 6184–6190. [Google Scholar] [CrossRef]
- Cao, D.; Li, H.; Pan, L.; Li, J.; Wang, X.; Jing, P. High saturation magnetization of γ-Fe2O3 nano-particles by a facile one-step synthesis approach. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Santos, P.; Marzan, L.M. N, N-Diamethylformamide as a reaction medium for metal nanoparticle synthesis. Adv. Funct. Mater. 2009, 19, 679–688. [Google Scholar] [CrossRef]
- Muzart, J. N, N-Diamethylformamide: Much more than a solvent. Tetrahedron 2009, 65, 8313–8323. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | SBET (m2·g−1) a | Pore Volume (cm3·g−1) b | Pore Diameter (nm) c | Particle Size (nm) |
|---|---|---|---|---|
| γ-Fe2O3-FM | 194.91 | 0.271 | 5.419 | 10.30 |
| γ-Fe2O3-CP | 56.4 | 0.195 | 12.72 | 84.8 |
| γ-Fe2O3-MACP [16] | 29.4 | 0.13 | 17.1 | 92.6 |
| Sample | Percentage by Atomicity (at%) | Percentage by Weight (wt%) | O/Fe | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fe | O | C | Cu | Fe | O | C | Cu | Atomicity Ratio | Weight Ratio | |
| γ-Fe2O3-FM | 32.89 | 61.50 | 1.38 | 1.21 | 61.15 | 32.76 | 0.55 | 2.57 | 1.86 | 0.53 |
| γ-Fe2O3-CP | 39.79 | 57.68 | 1.39 | 0.84 | 66.69 | 27.56 | 0.52 | 2.56 | 1.45 | 0.42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Husnain, N.; Wang, E.; Fareed, S.; Tuoqeer Anwar, M. Comparision on the Low-Temperature NH3-SCR Performance of γ-Fe2O3 Catalysts Prepared by Two Different Methods. Catalysts 2019, 9, 1018. https://doi.org/10.3390/catal9121018
Husnain N, Wang E, Fareed S, Tuoqeer Anwar M. Comparision on the Low-Temperature NH3-SCR Performance of γ-Fe2O3 Catalysts Prepared by Two Different Methods. Catalysts. 2019; 9(12):1018. https://doi.org/10.3390/catal9121018
Chicago/Turabian StyleHusnain, Naveed, Enlu Wang, Shagufta Fareed, and Muhammad Tuoqeer Anwar. 2019. "Comparision on the Low-Temperature NH3-SCR Performance of γ-Fe2O3 Catalysts Prepared by Two Different Methods" Catalysts 9, no. 12: 1018. https://doi.org/10.3390/catal9121018
APA StyleHusnain, N., Wang, E., Fareed, S., & Tuoqeer Anwar, M. (2019). Comparision on the Low-Temperature NH3-SCR Performance of γ-Fe2O3 Catalysts Prepared by Two Different Methods. Catalysts, 9(12), 1018. https://doi.org/10.3390/catal9121018