Recent Organic Transformations with Silver Carbonate as a Key External Base and Oxidant

 and
and 
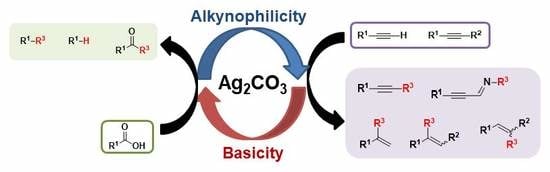
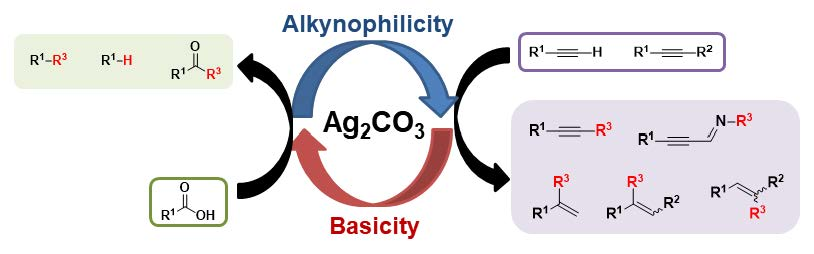
Abstract
:
{kind=link}
{kind=link}
{kind=link}
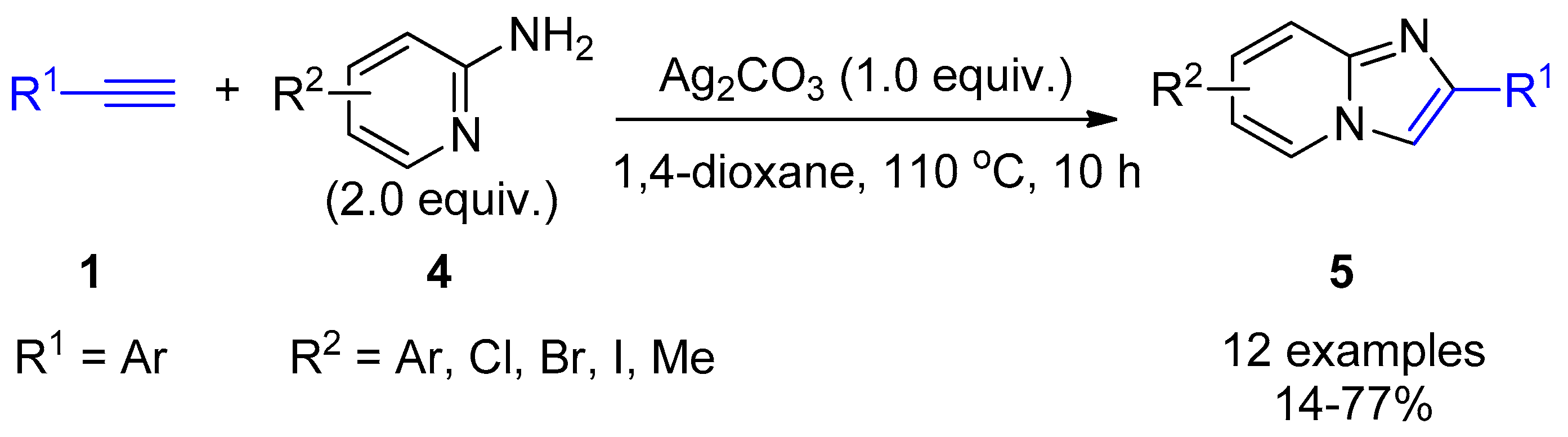{kind=link}
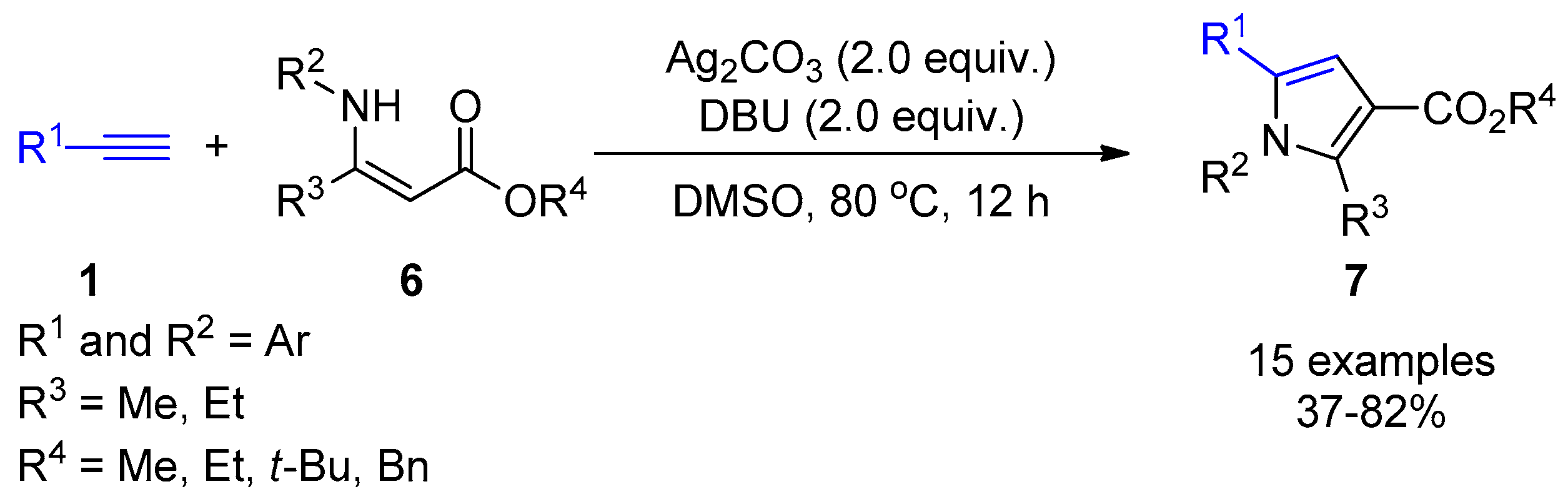{kind=link}
{kind=link}
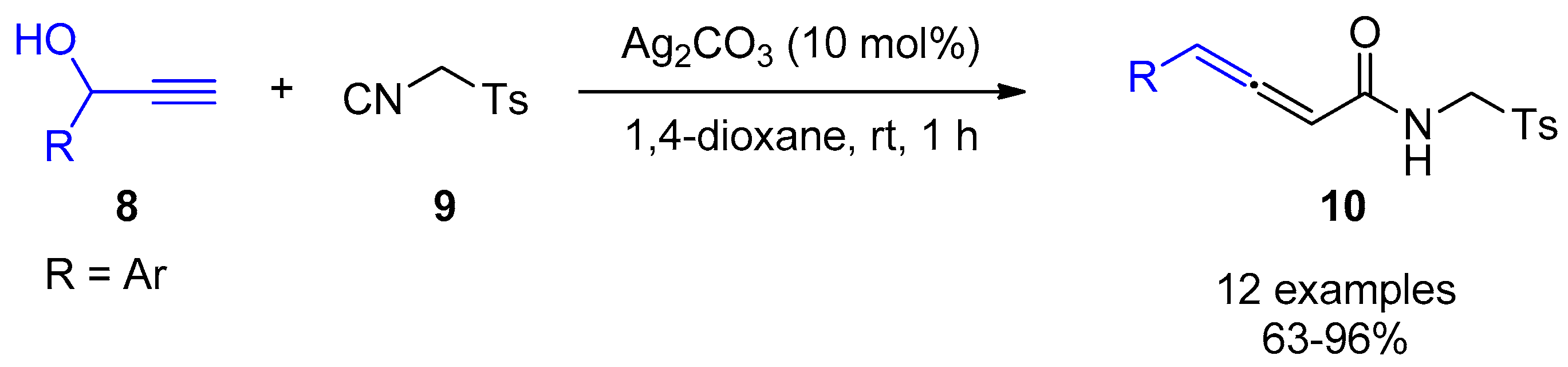{kind=link}
{kind=link}
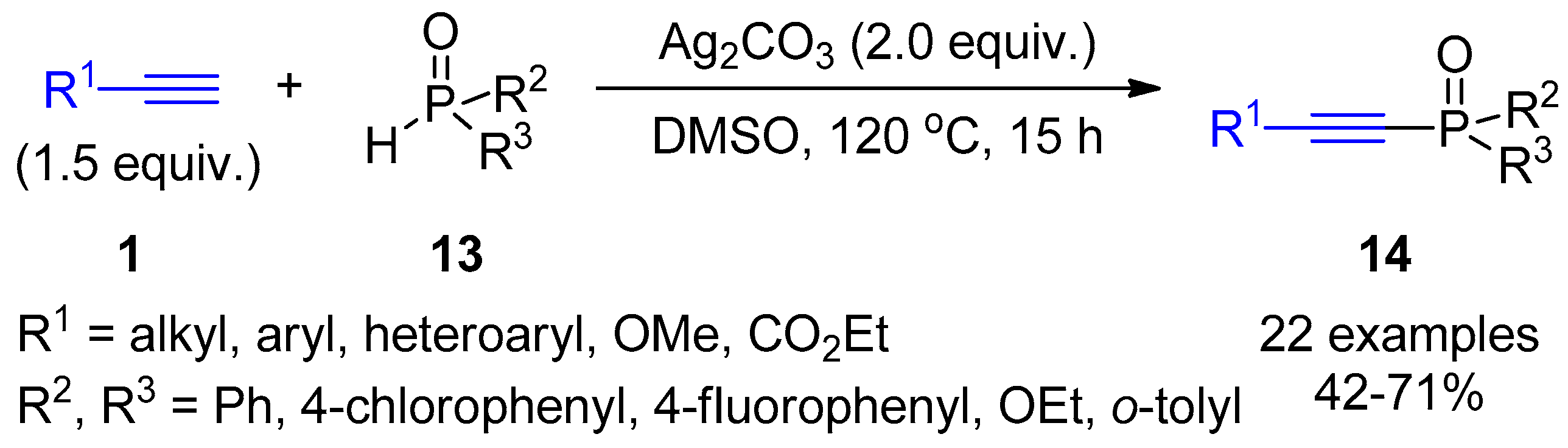{kind=link}
{kind=link}
{kind=link}
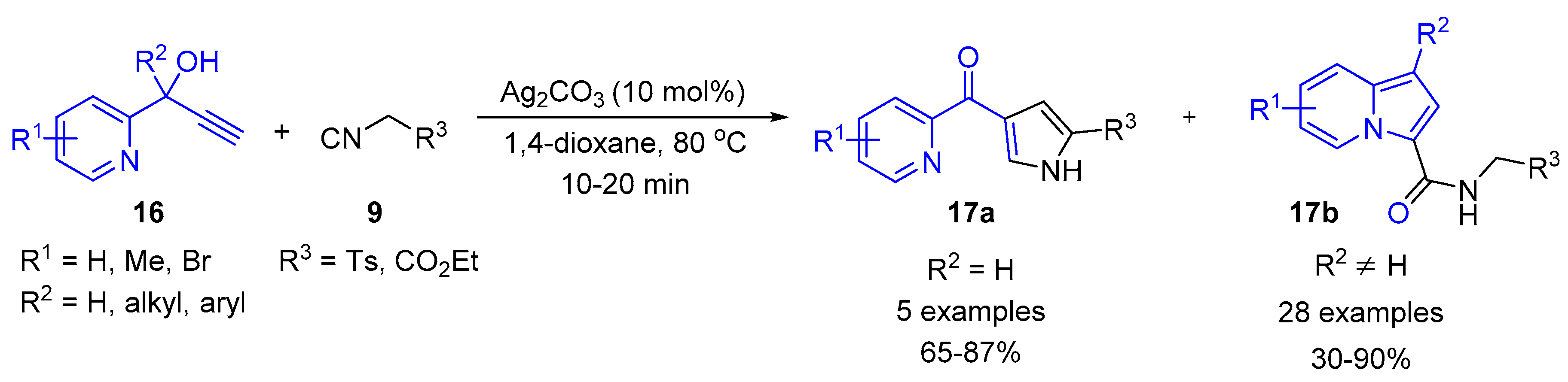{kind=link}
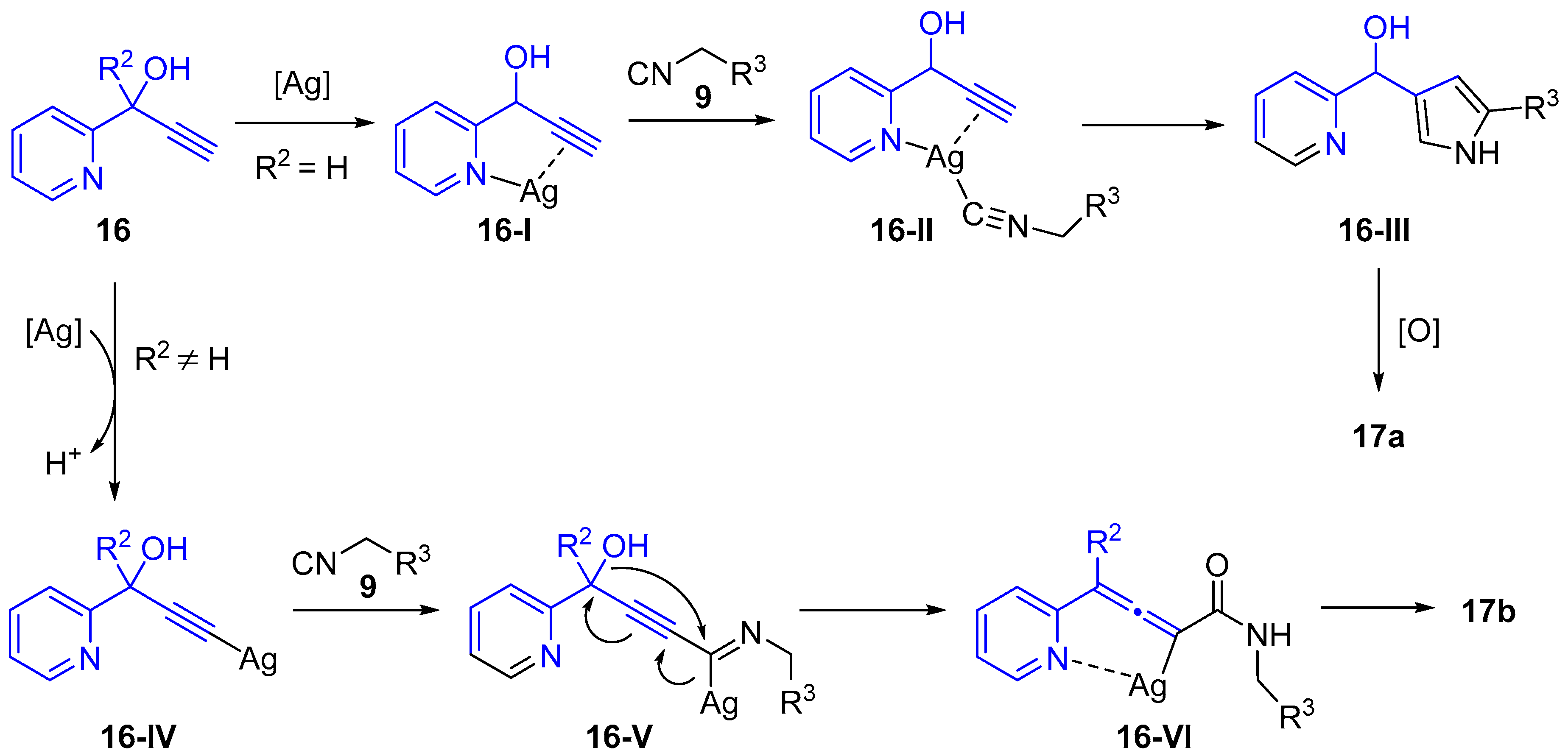{kind=link}
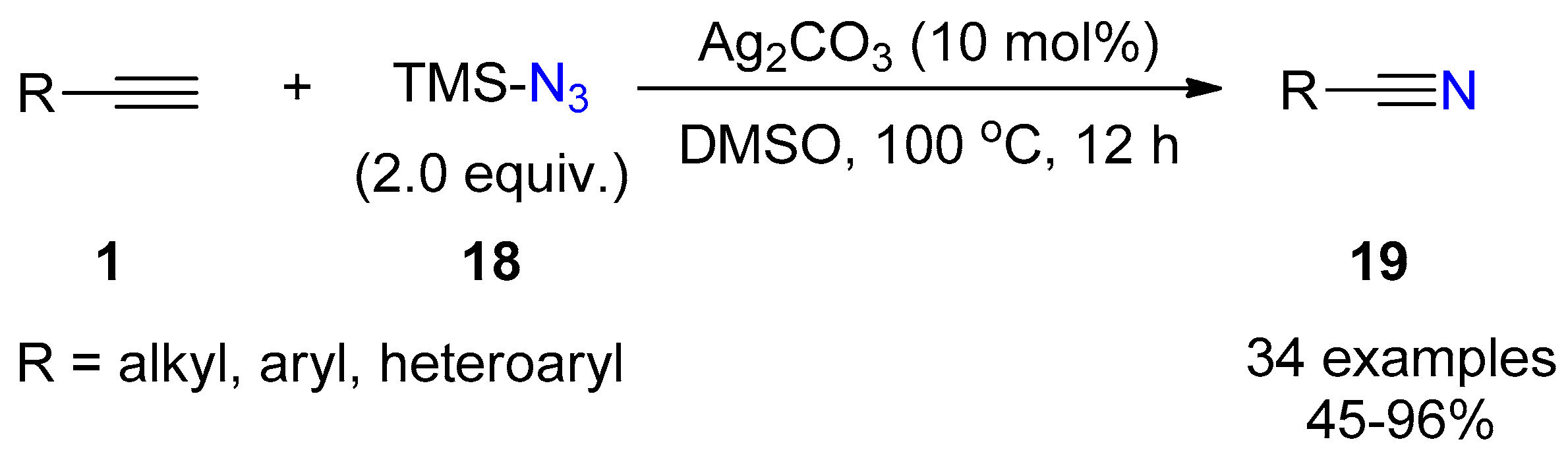{kind=link}
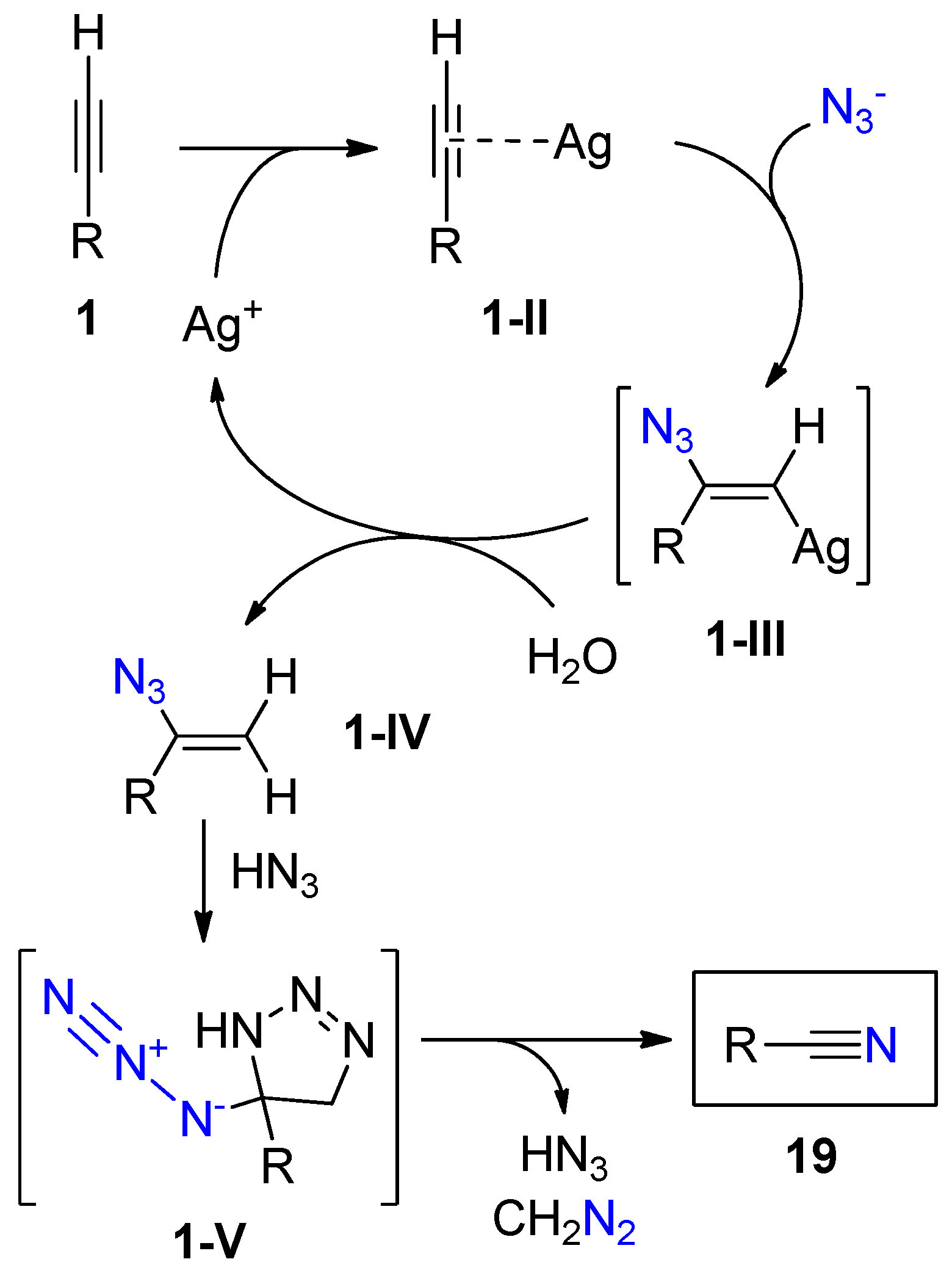{kind=link}
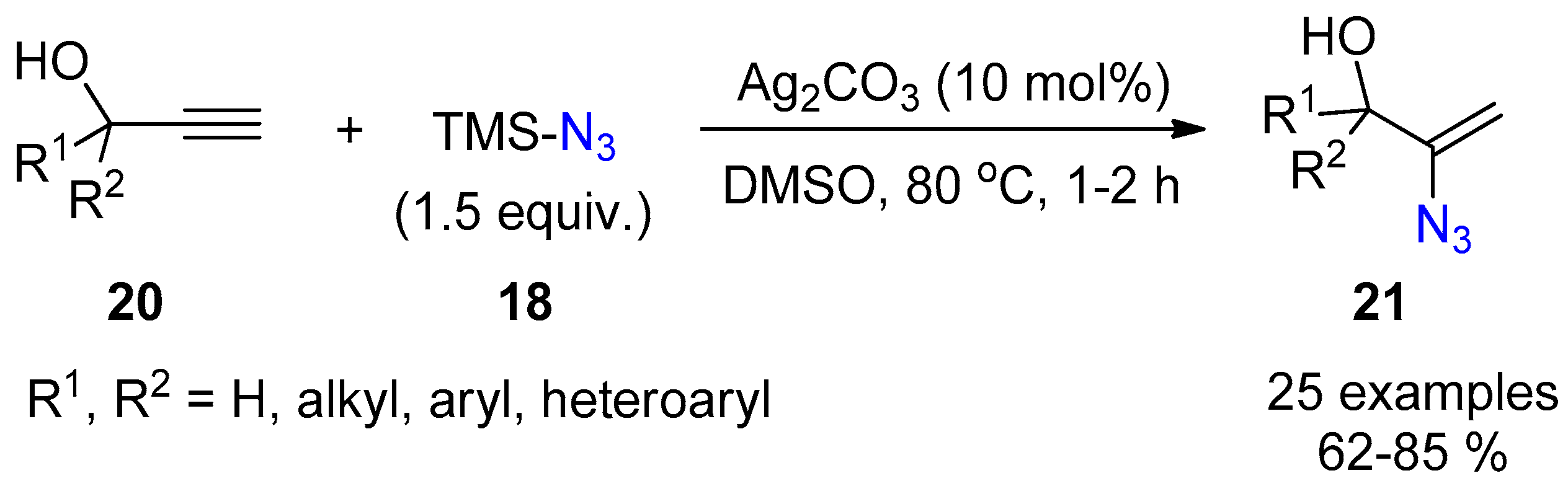{kind=link}
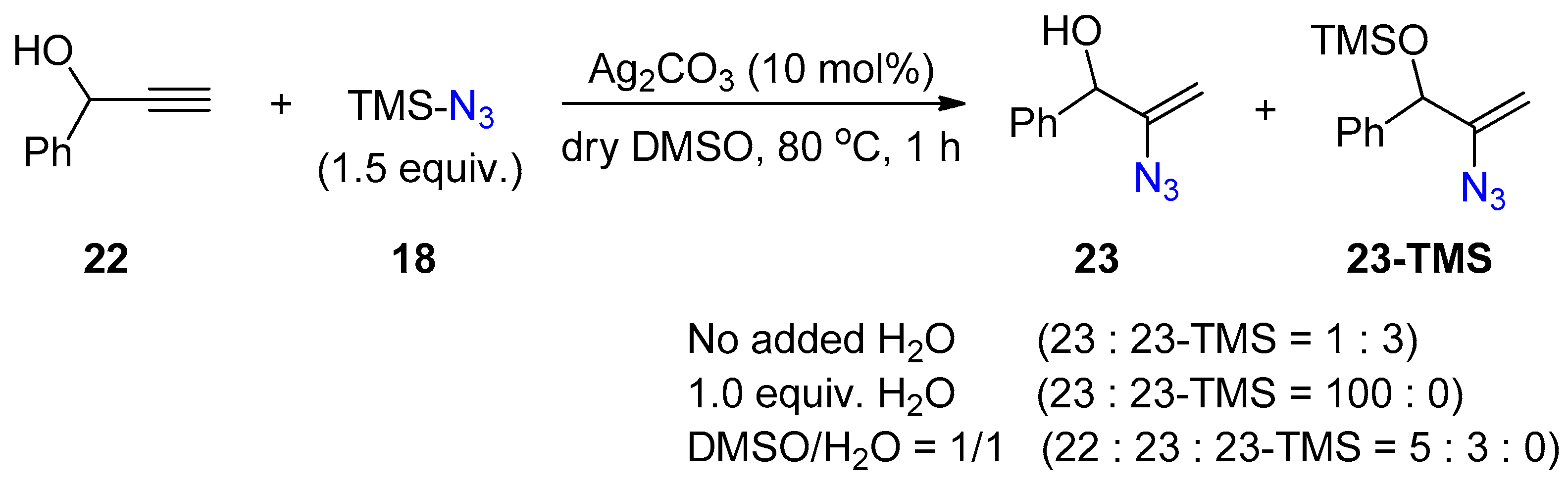{kind=link}
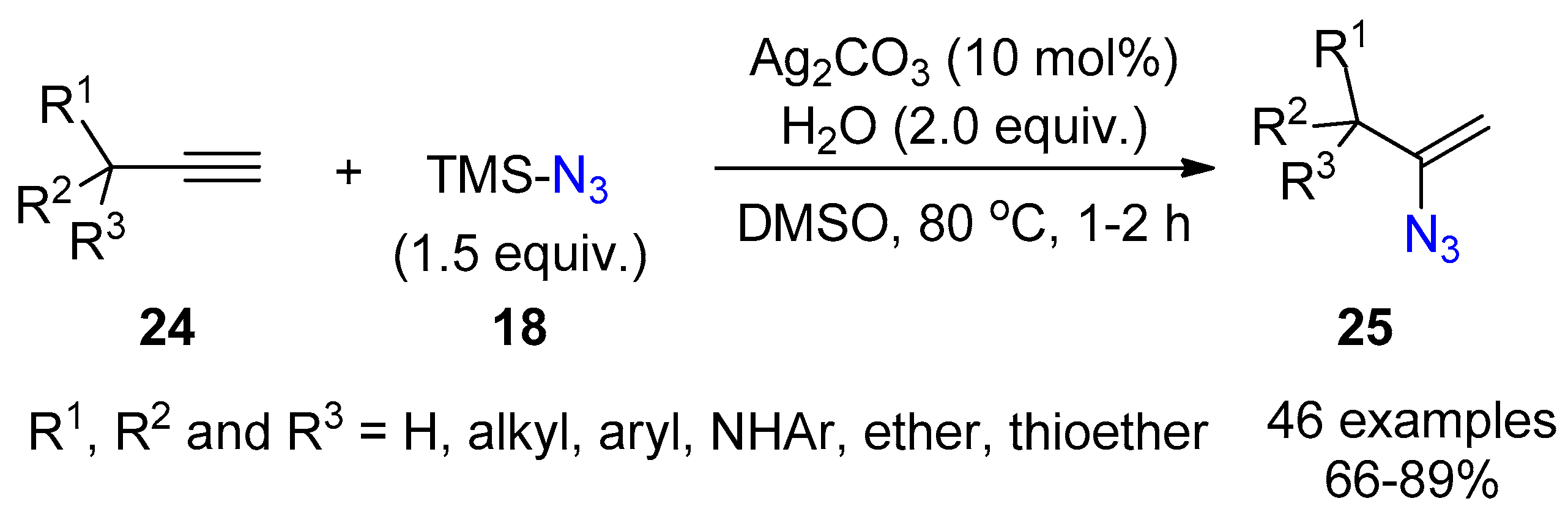{kind=link}
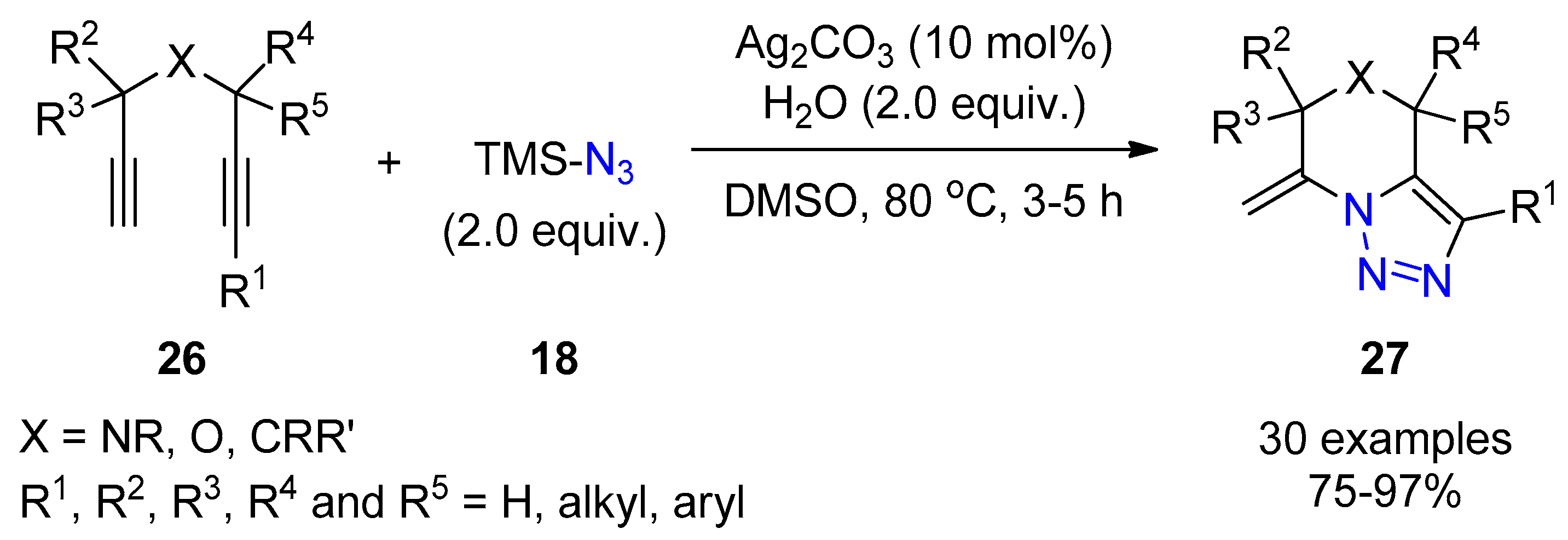{kind=link}
{kind=link}
{kind=link}
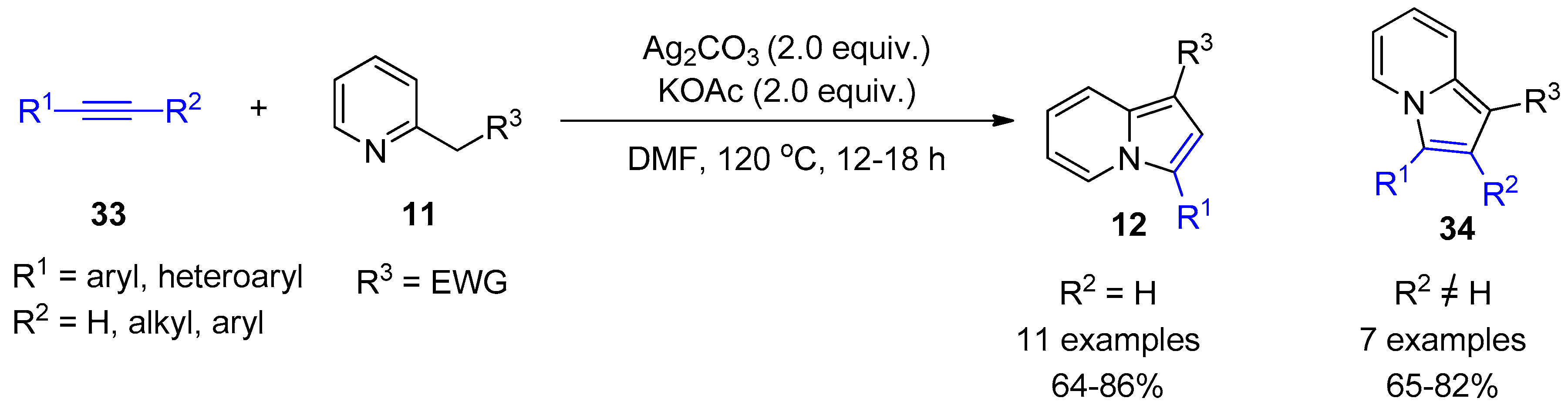{kind=link}
{kind=link}
{kind=link}
{kind=link}
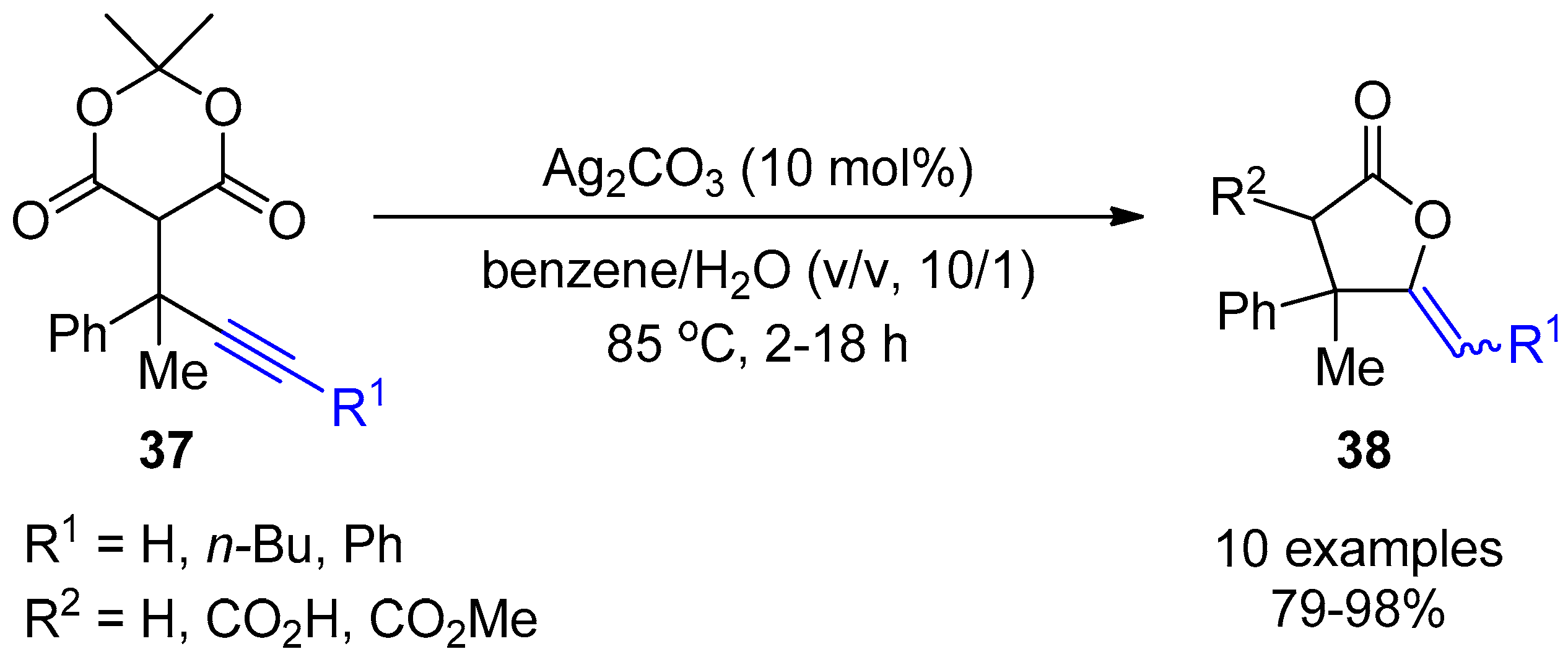{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
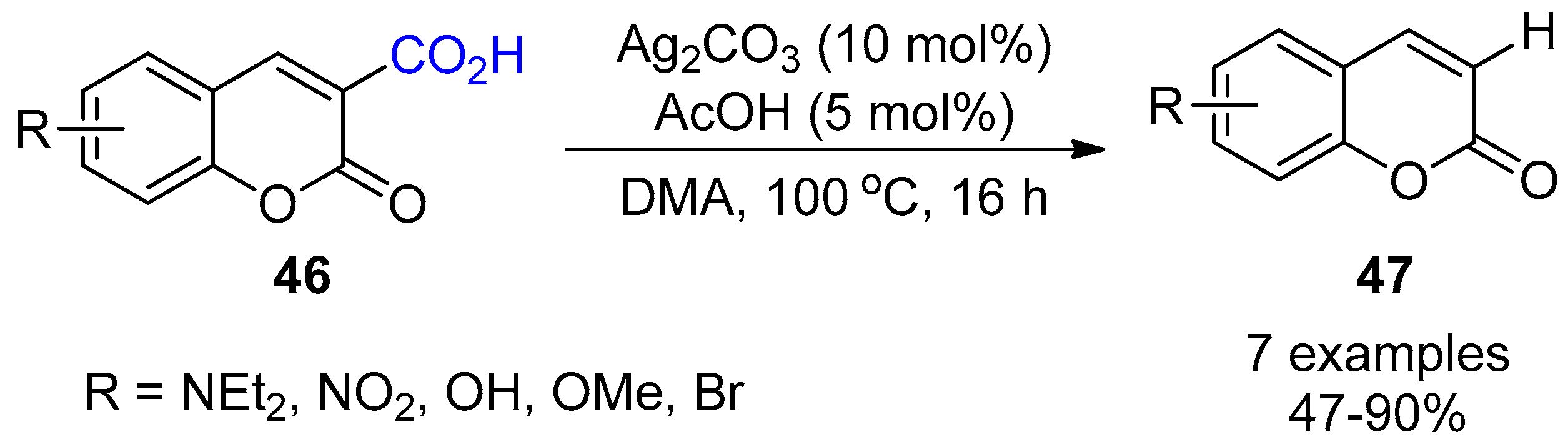{kind=link}
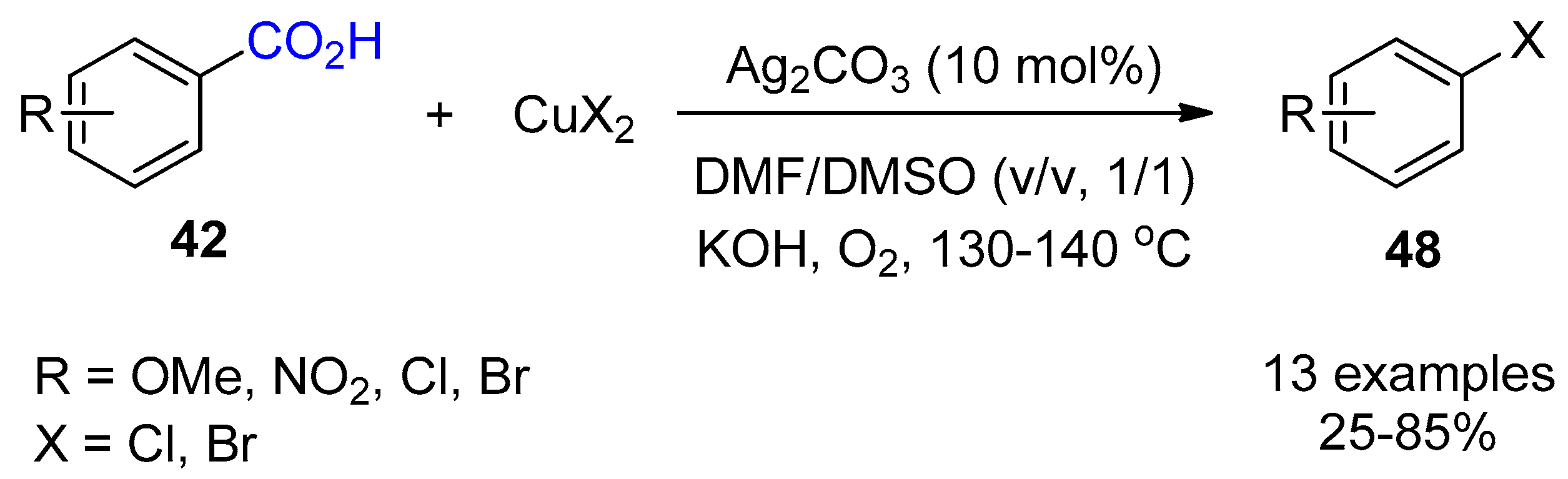{kind=link}
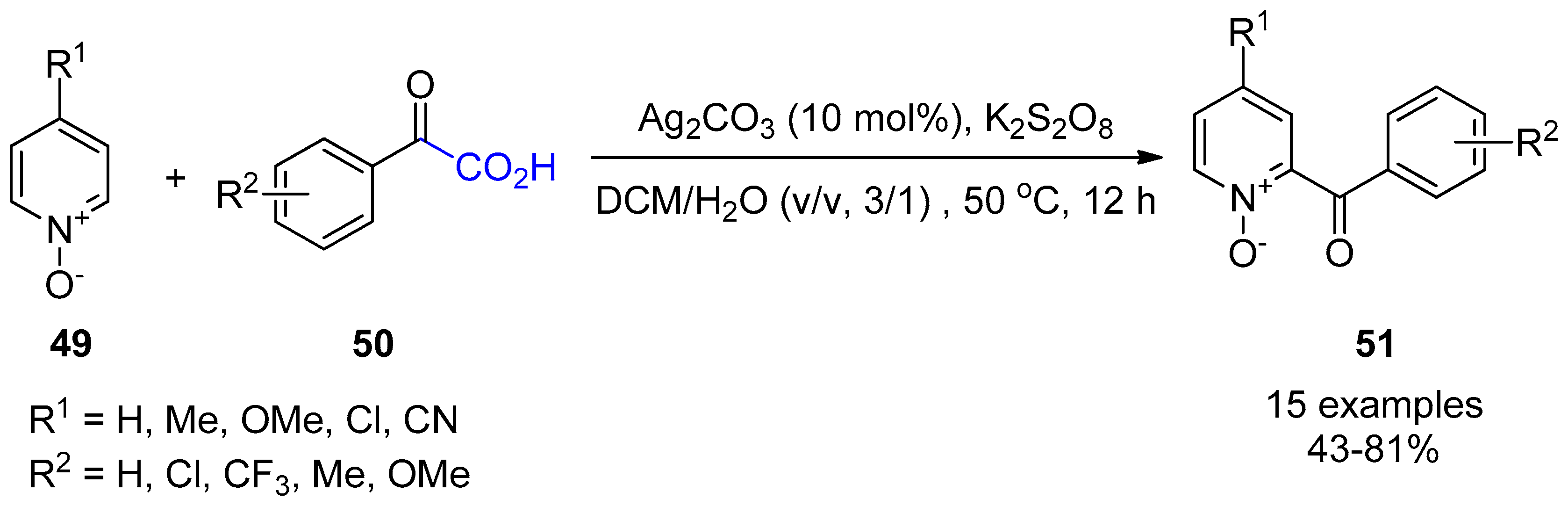{kind=link}
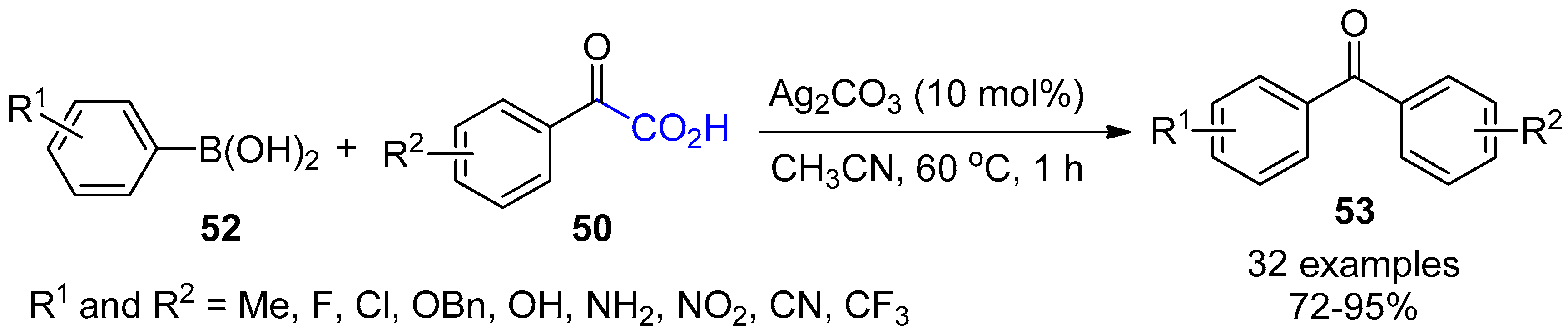{kind=link}
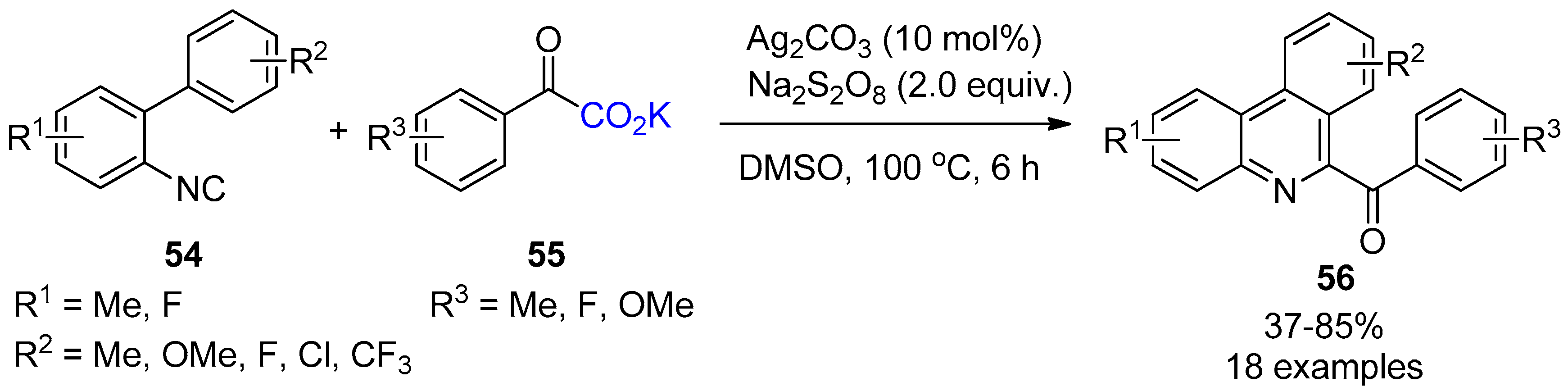{kind=link}
{kind=link}
{kind=link}
{kind=link}
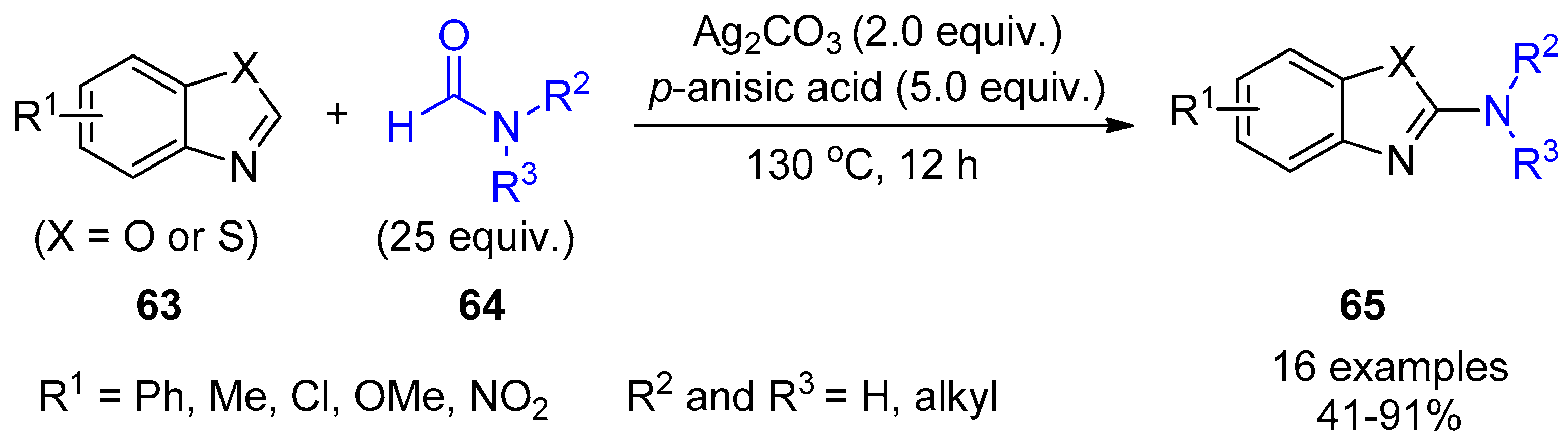{kind=link}
{kind=link}
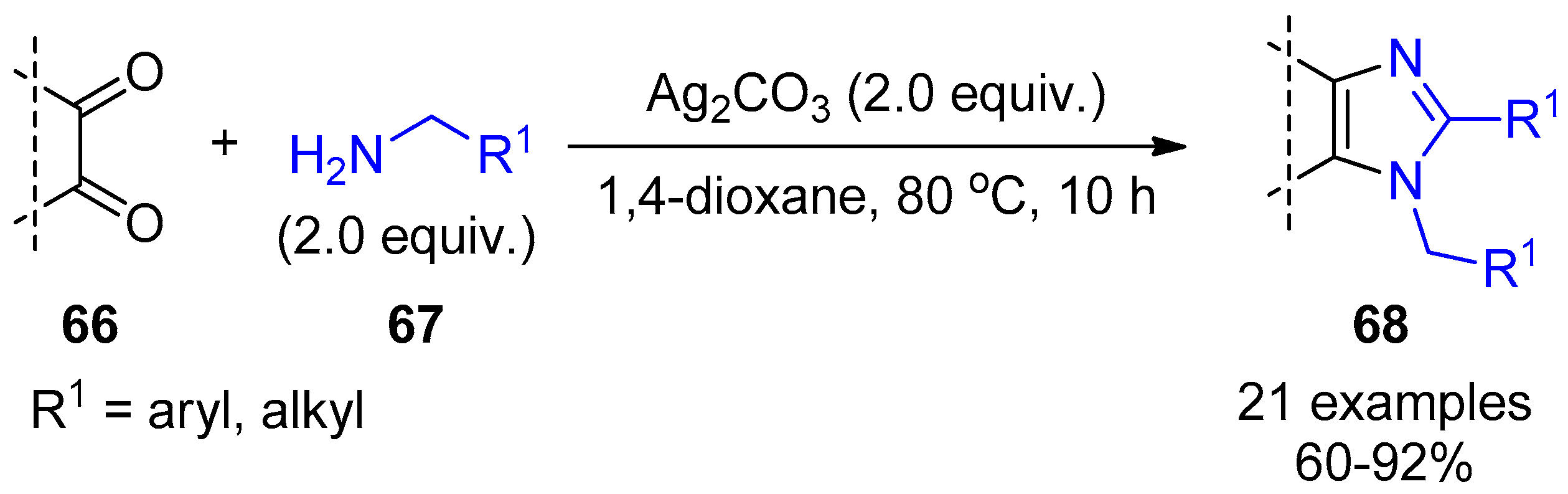{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
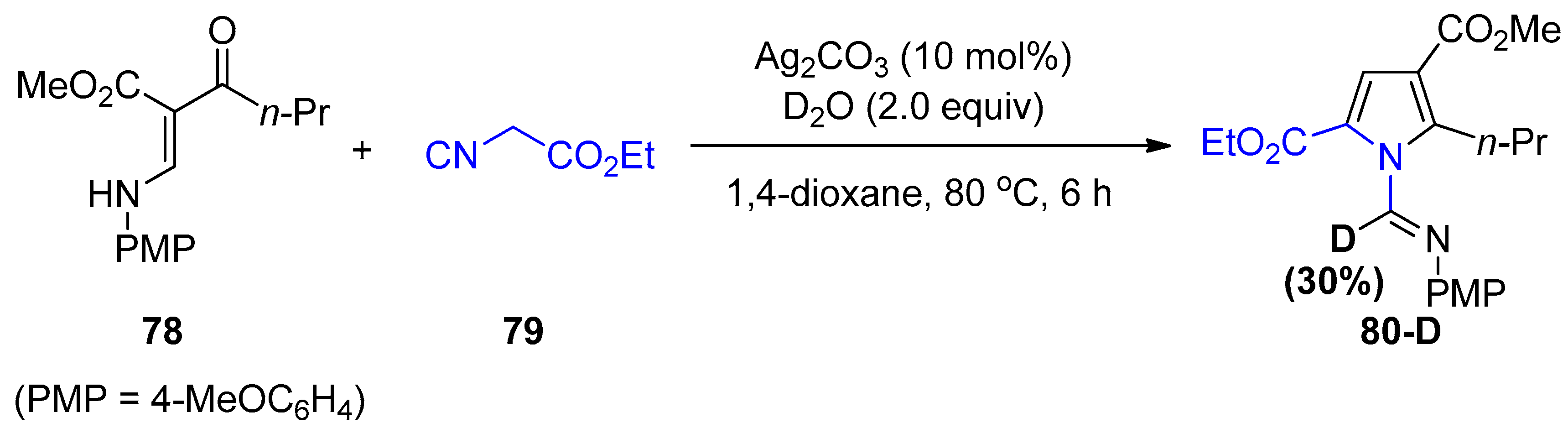{kind=link}
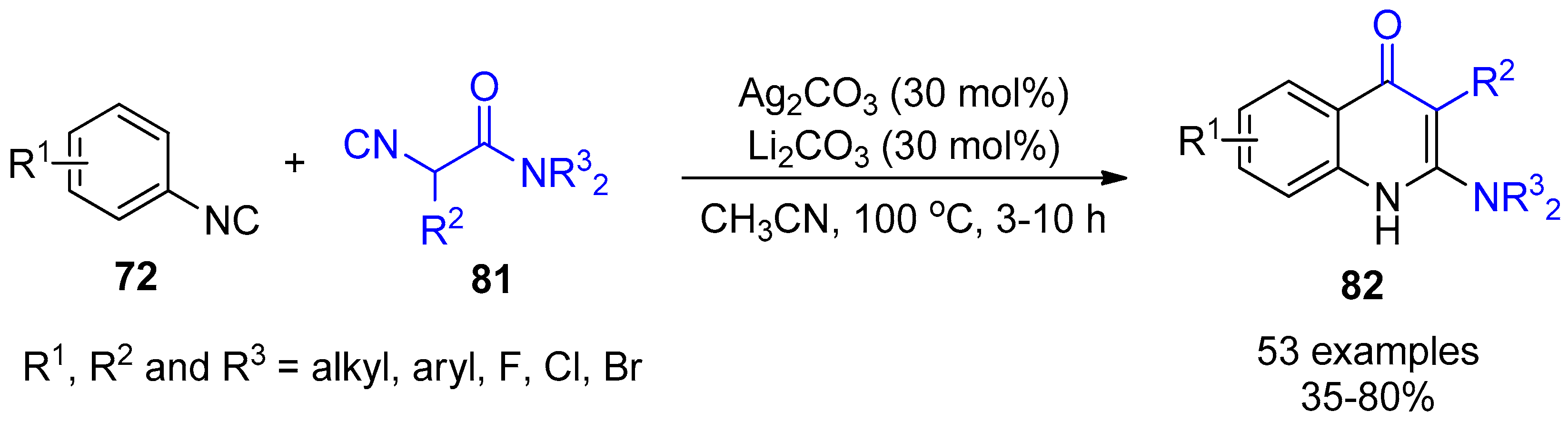{kind=link}
{kind=link}
{kind=link}
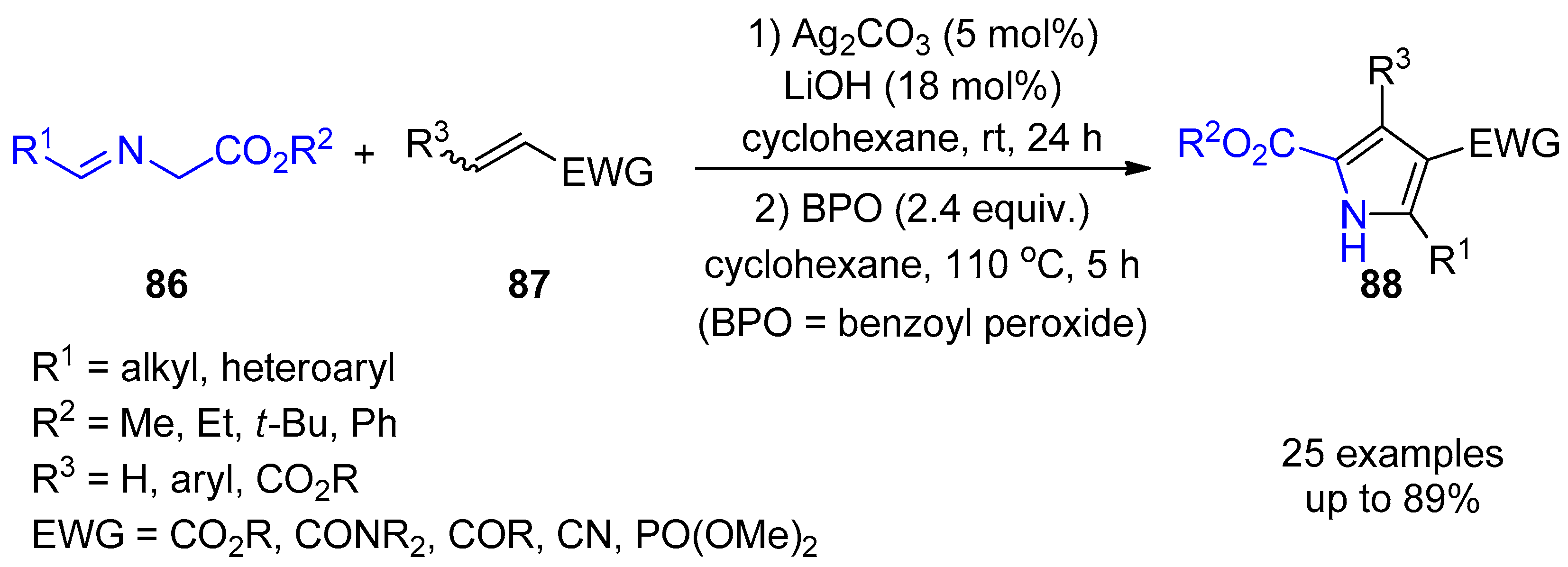{kind=link}
1. Introduction
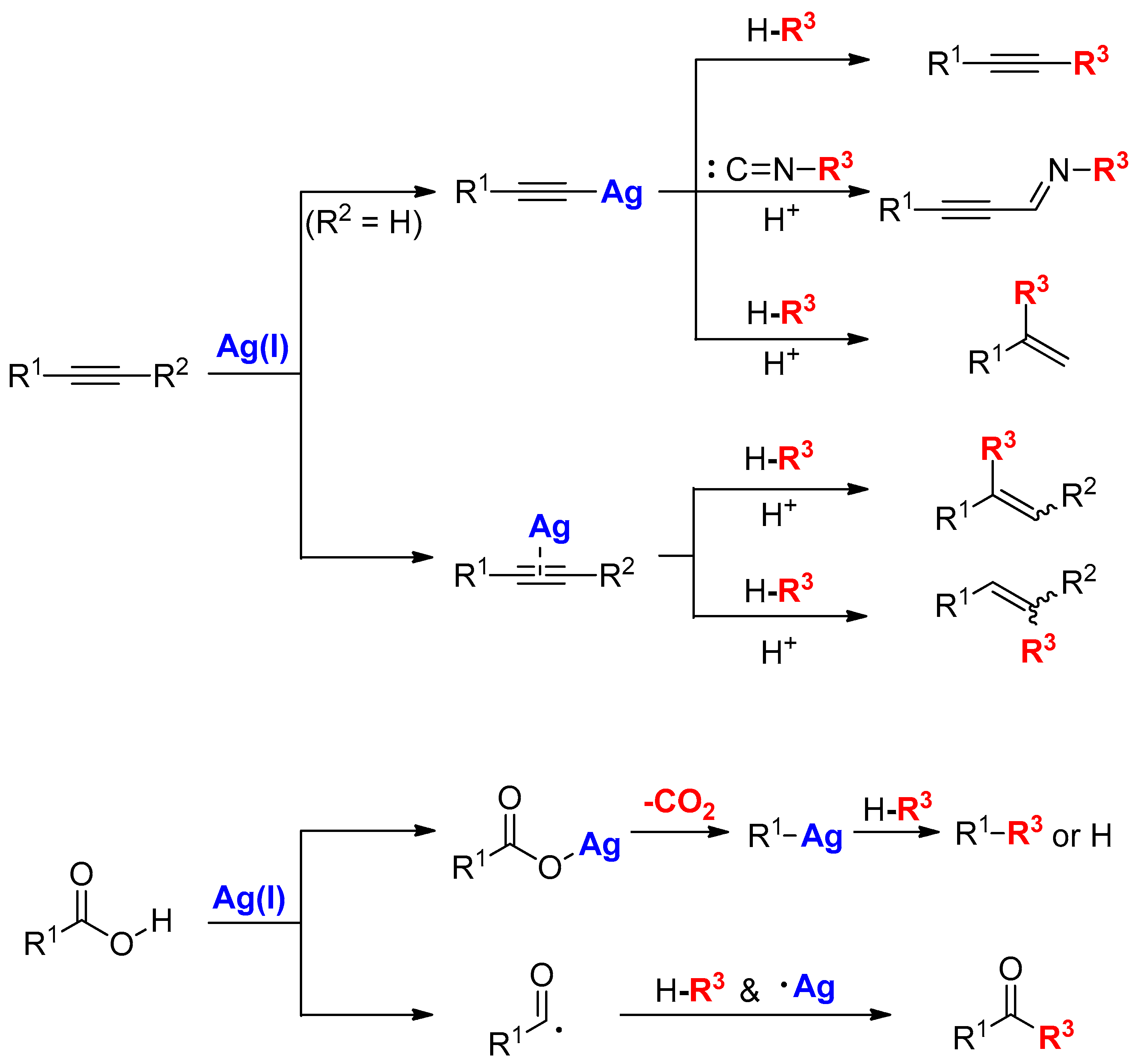
2. Activation of Alkynes Using Silver Carbonate
2.1. Terminal Alkynes: Cross-Coupling Reactions
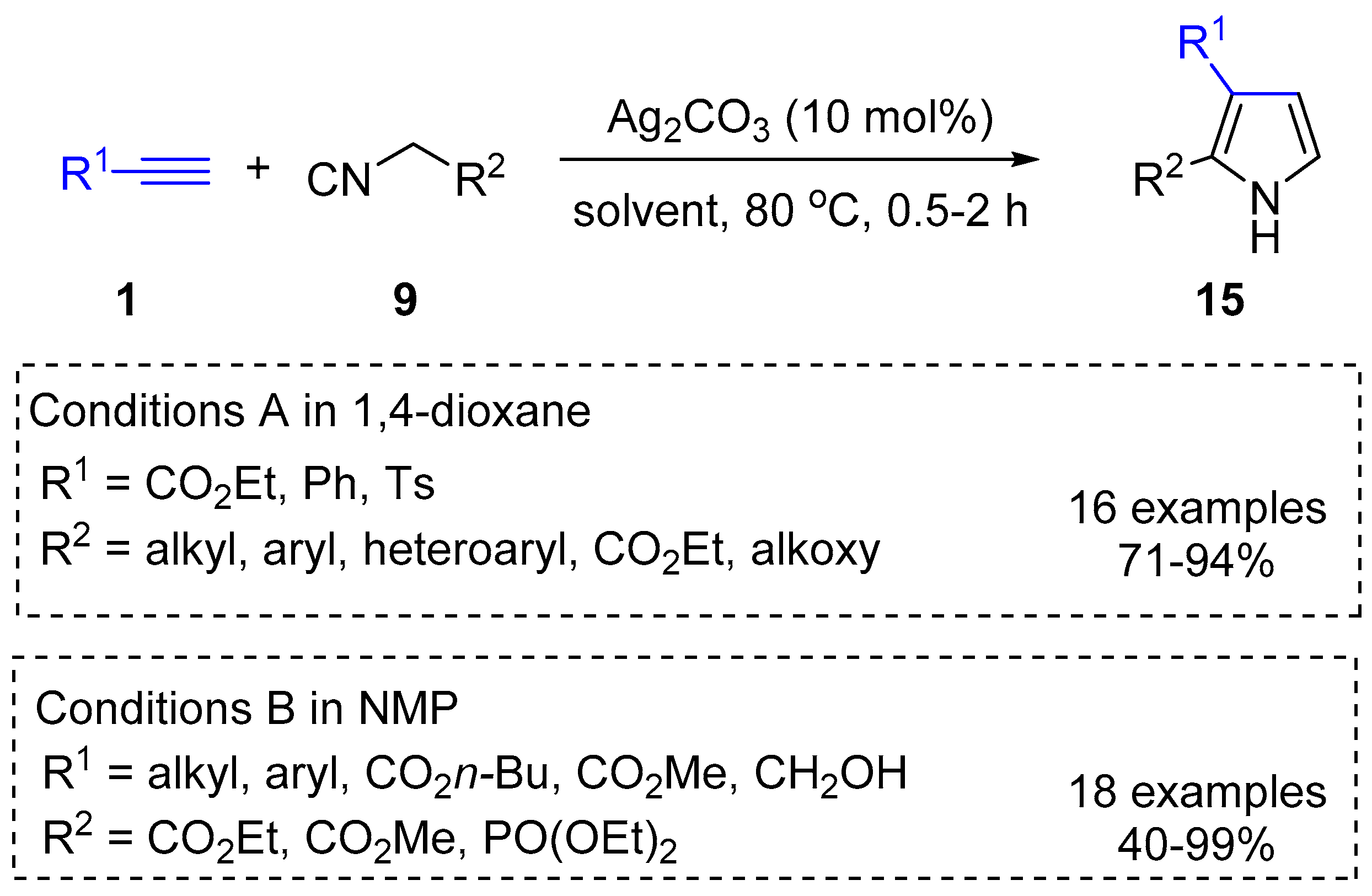
2.2. Terminal Alkynes: Cycloaddition Reactions
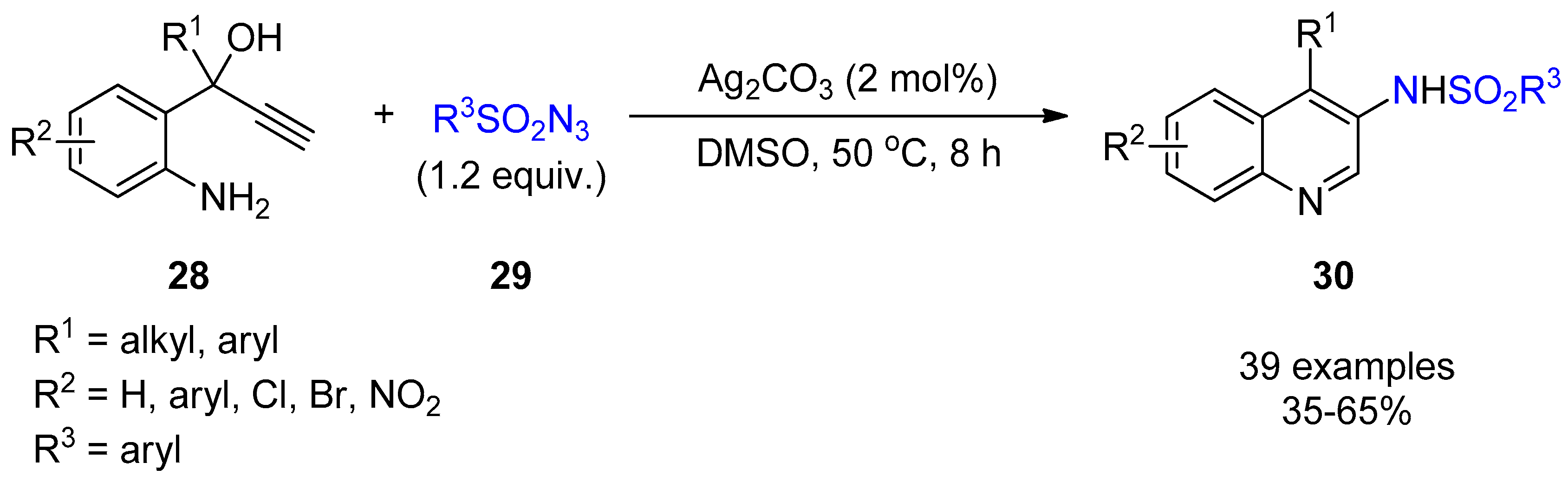
2.3. Terminal Alkynes: Reactions with Azide
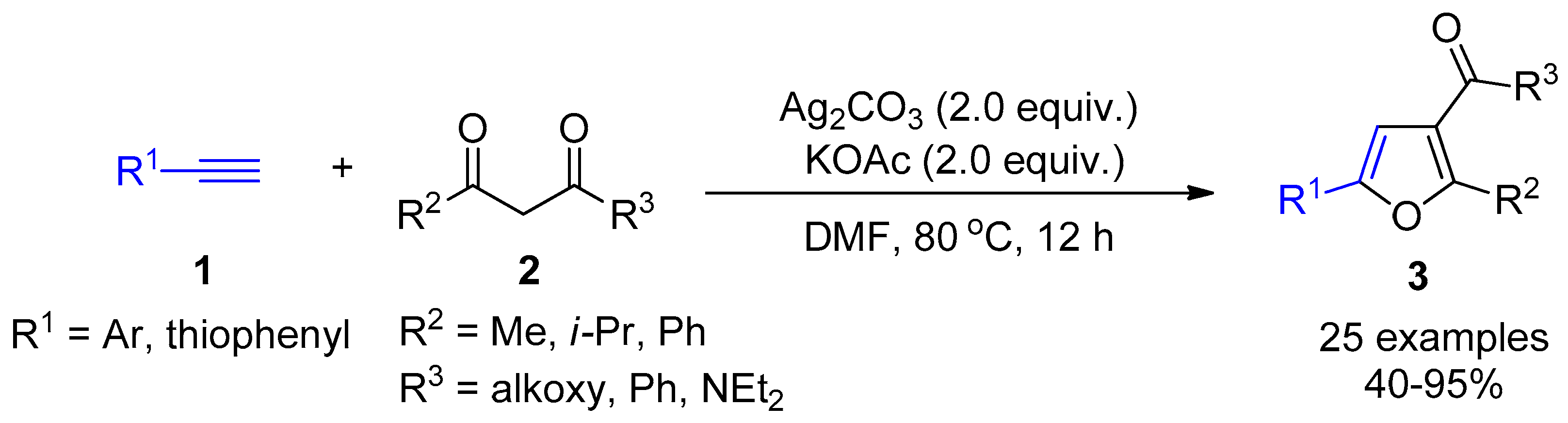
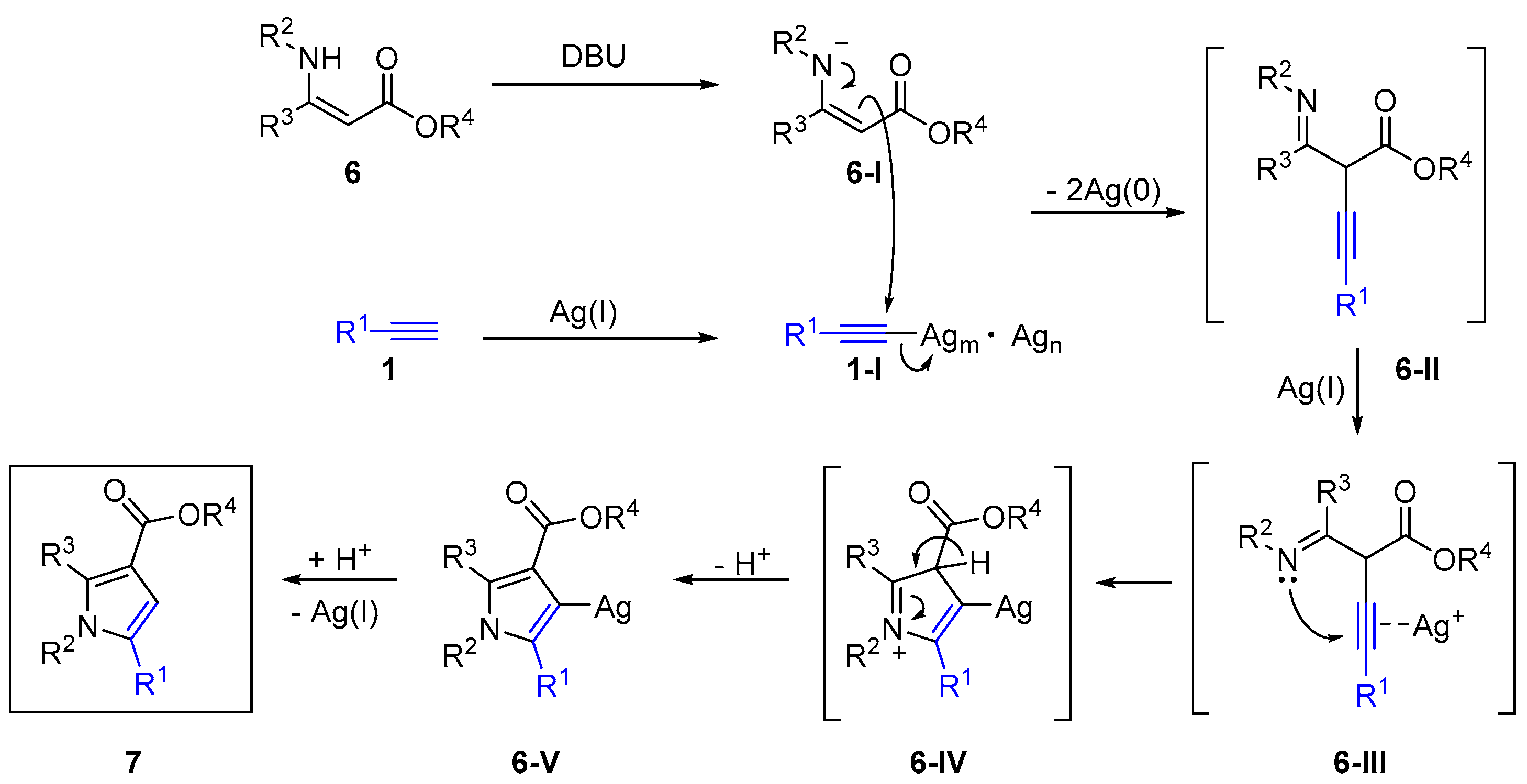
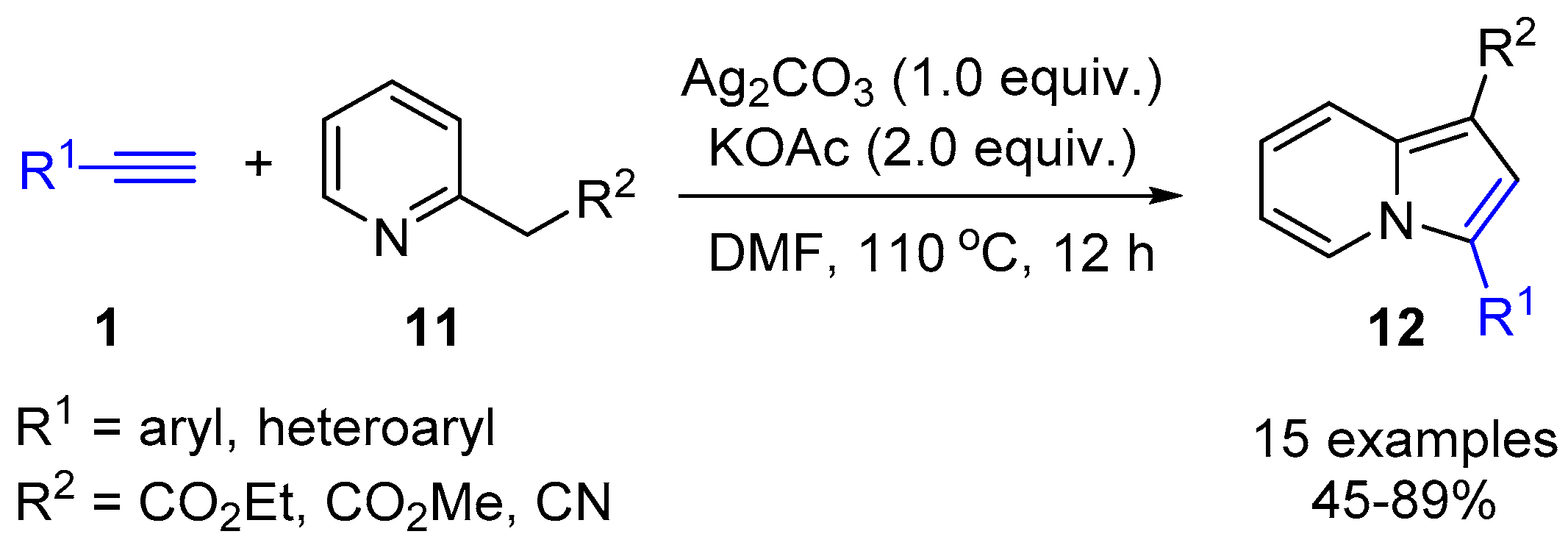
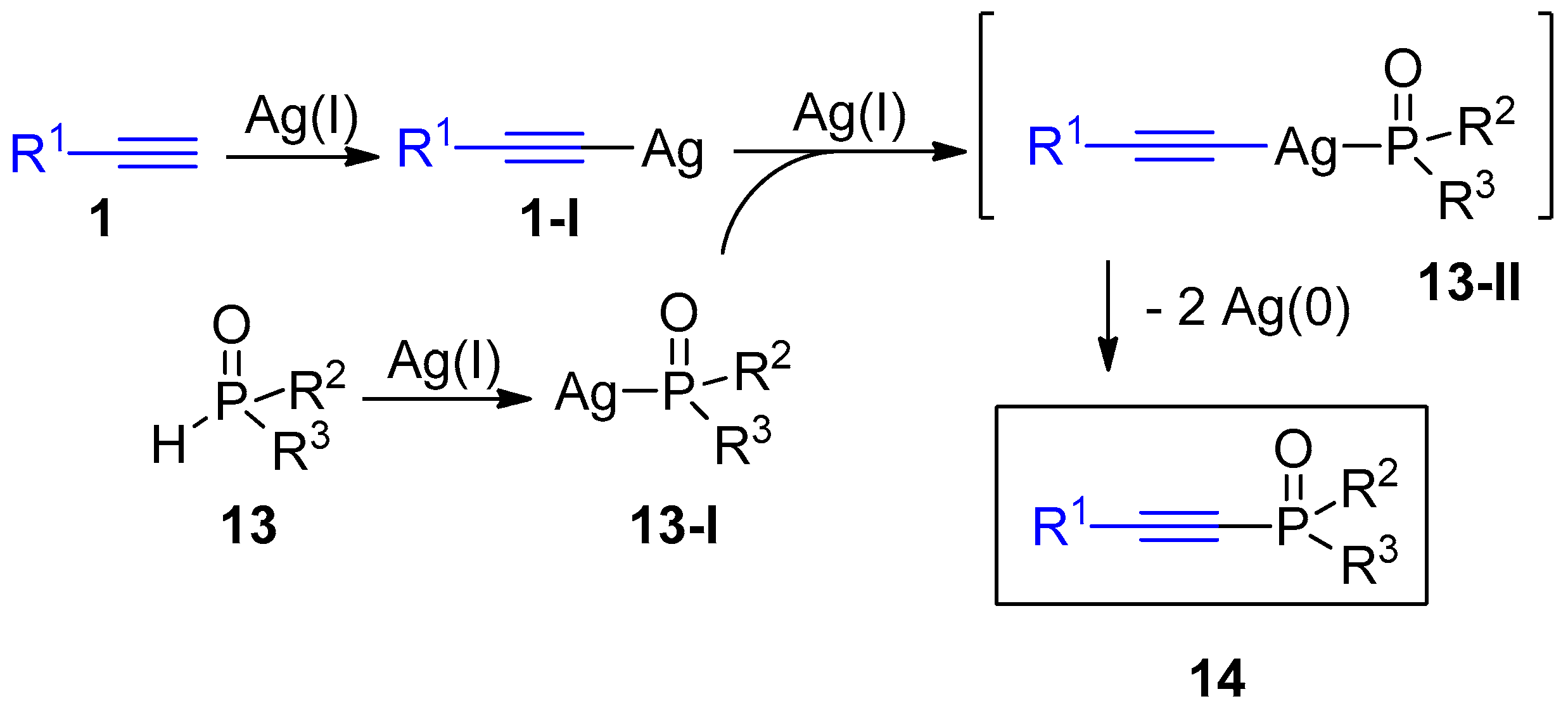
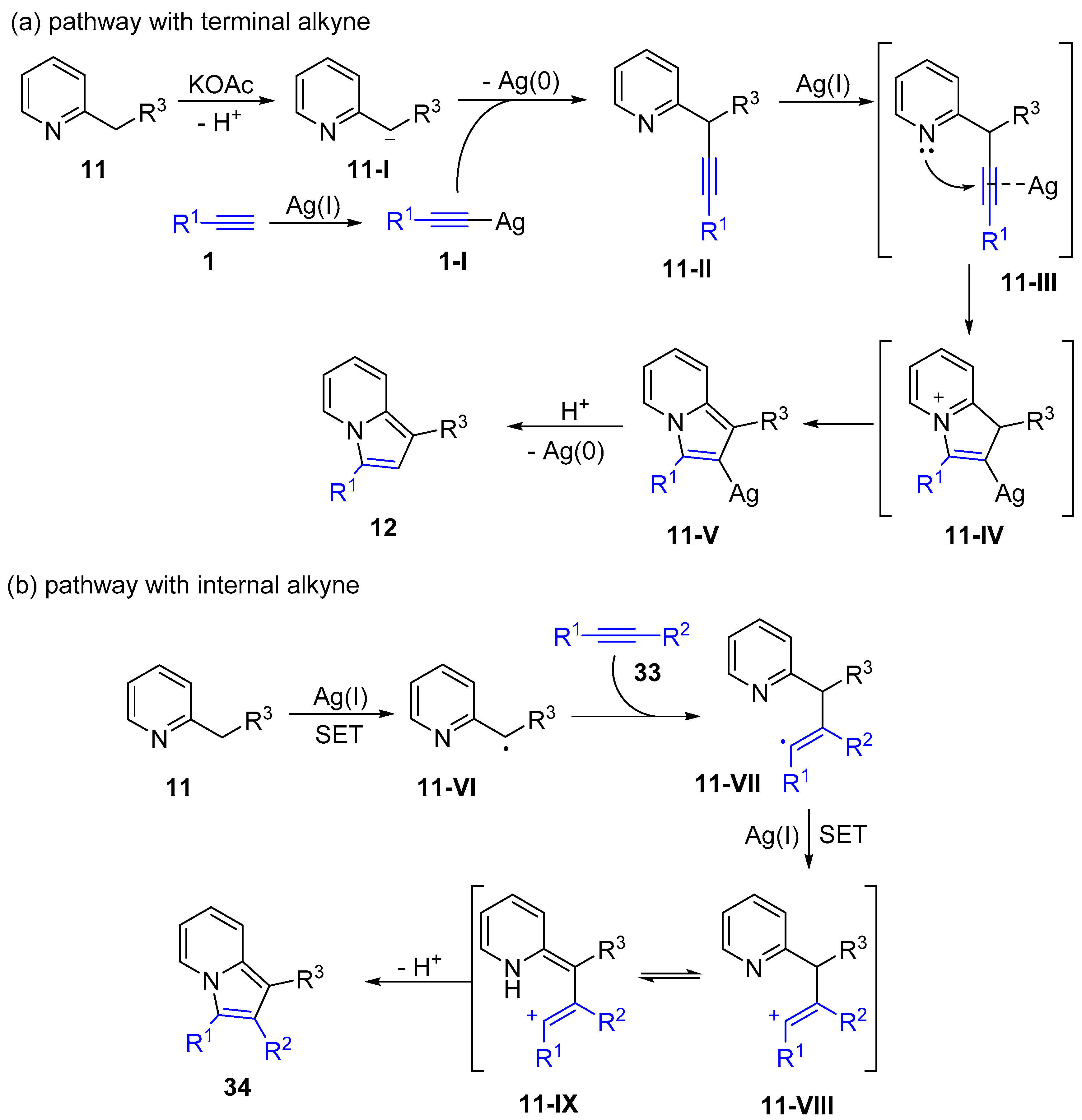
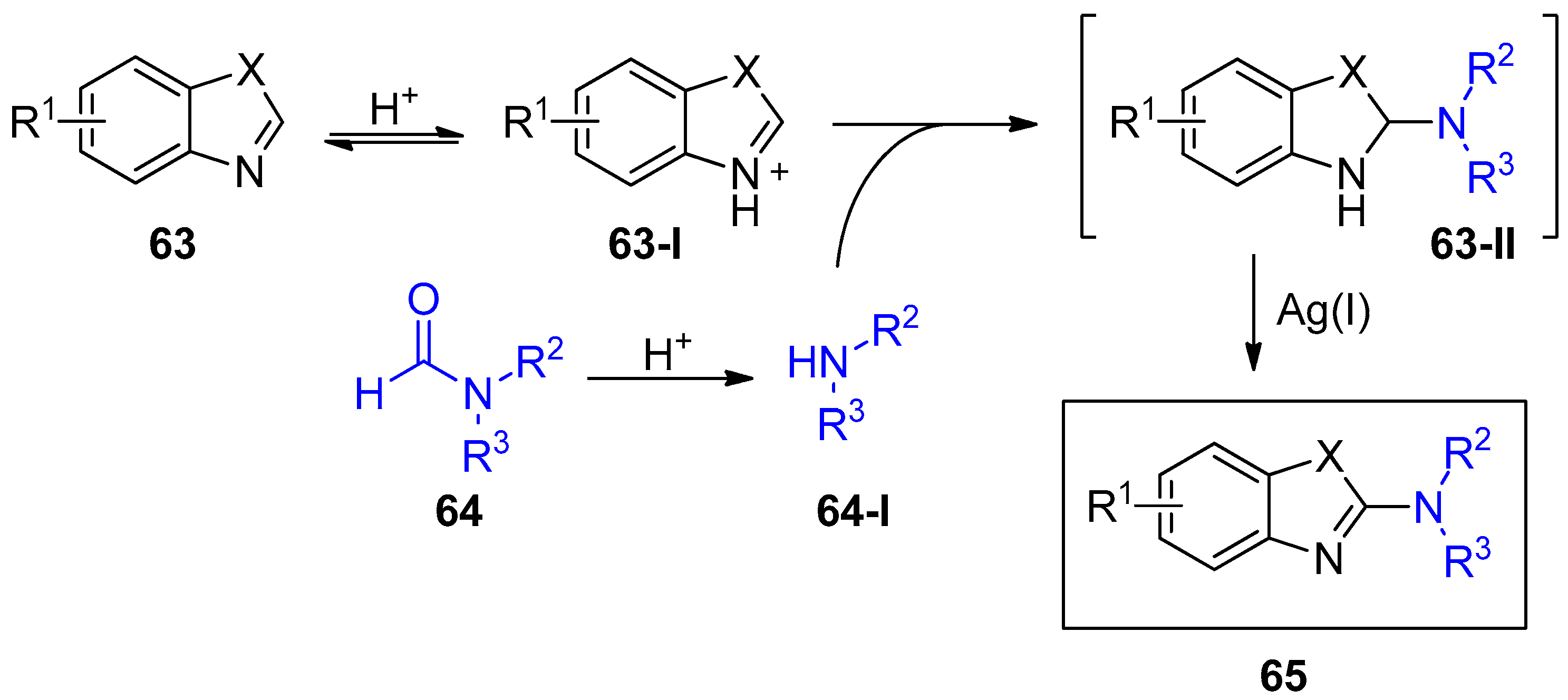
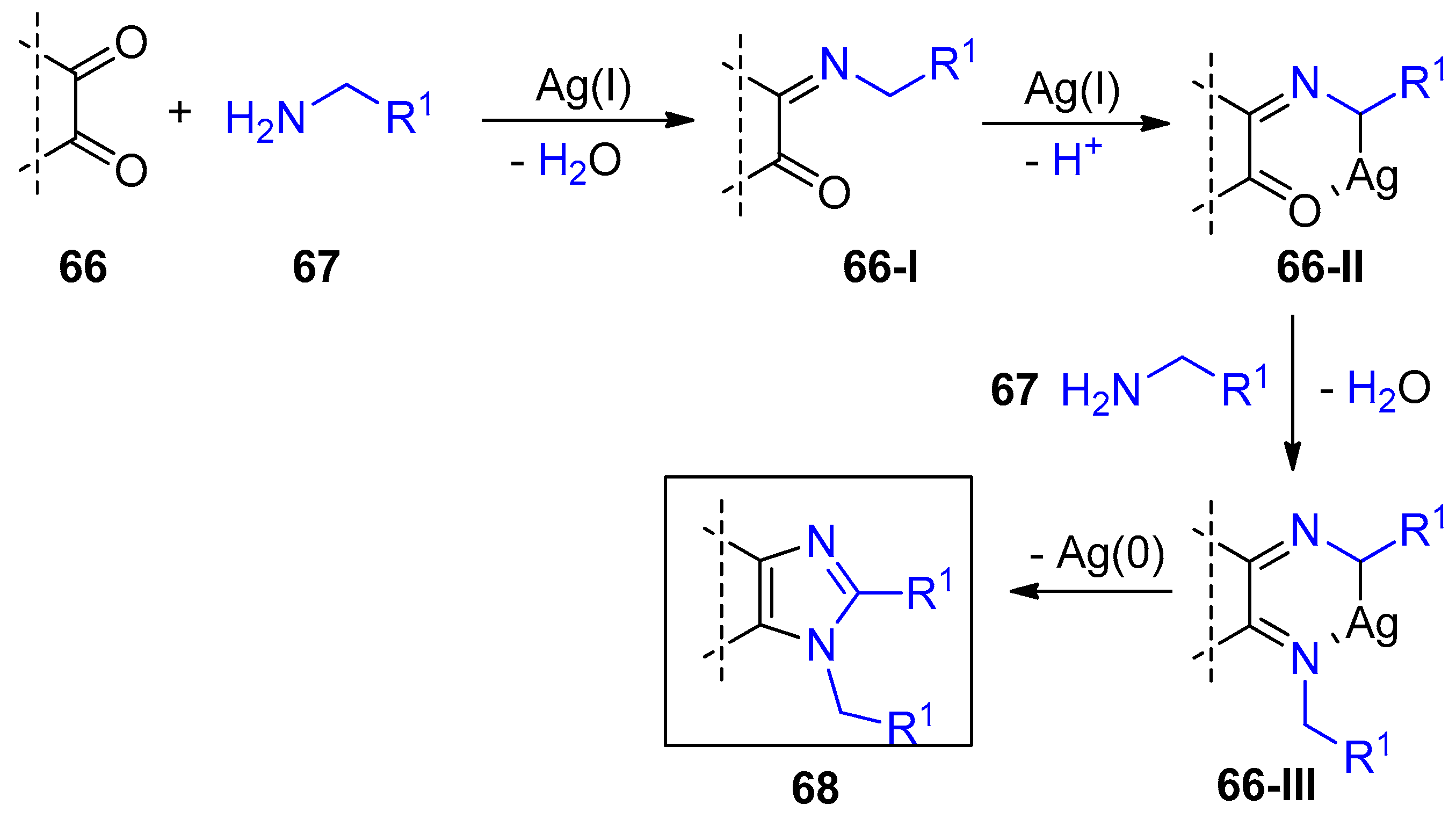
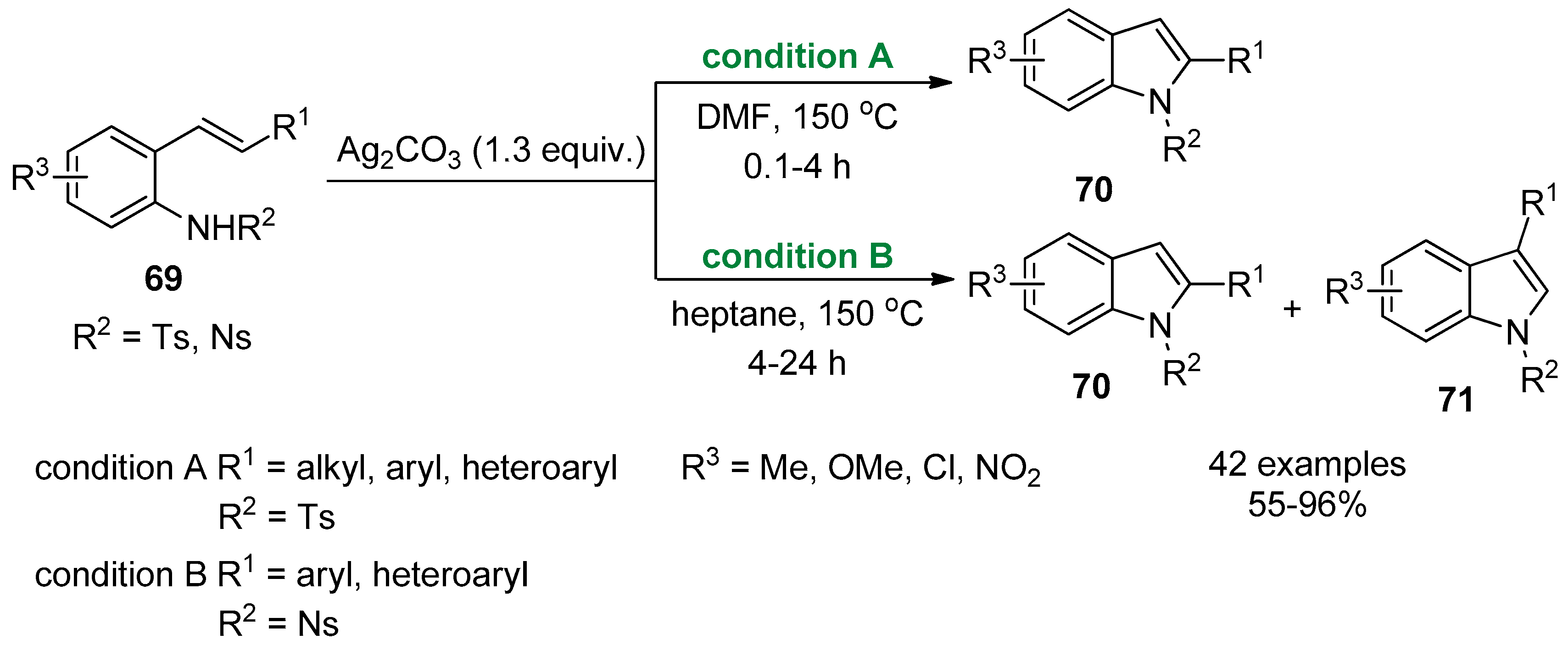
2.4. Internal Alkynes: Synthesis of Heterocyclic Compounds
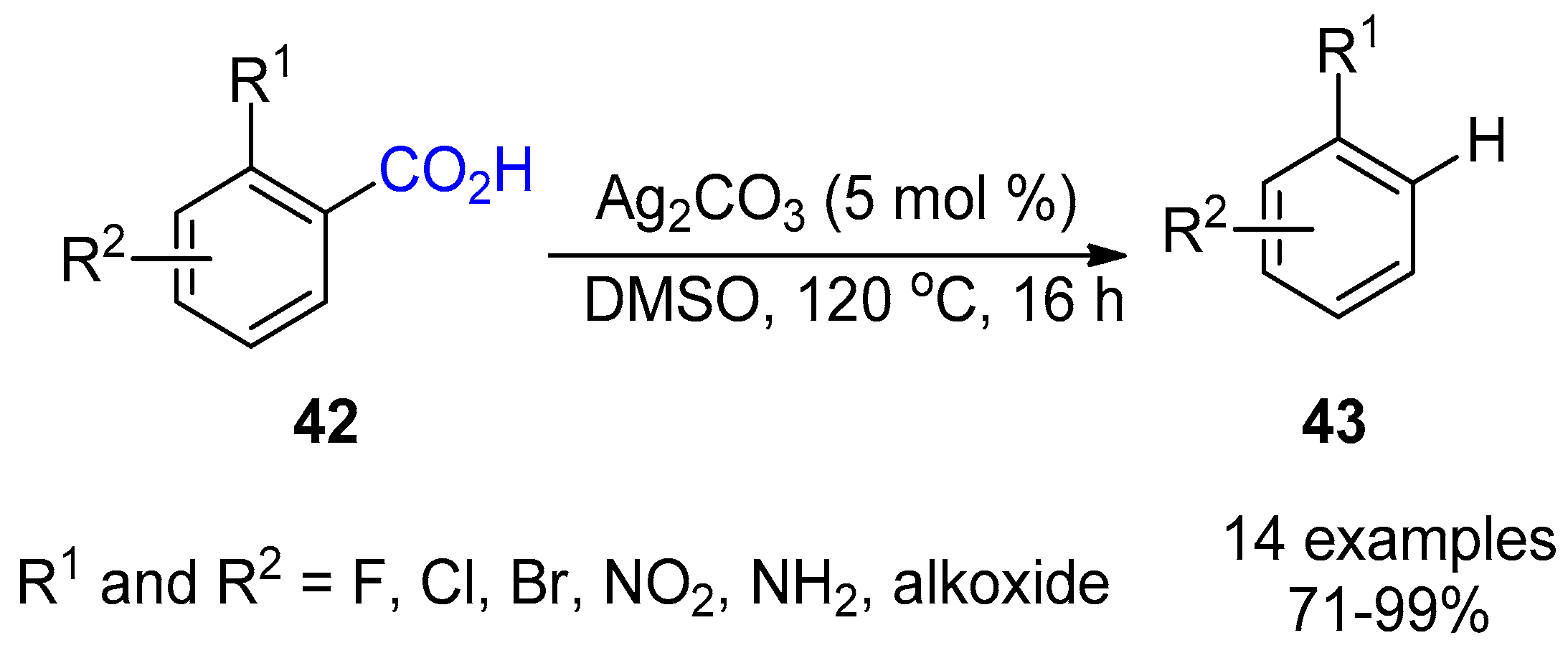
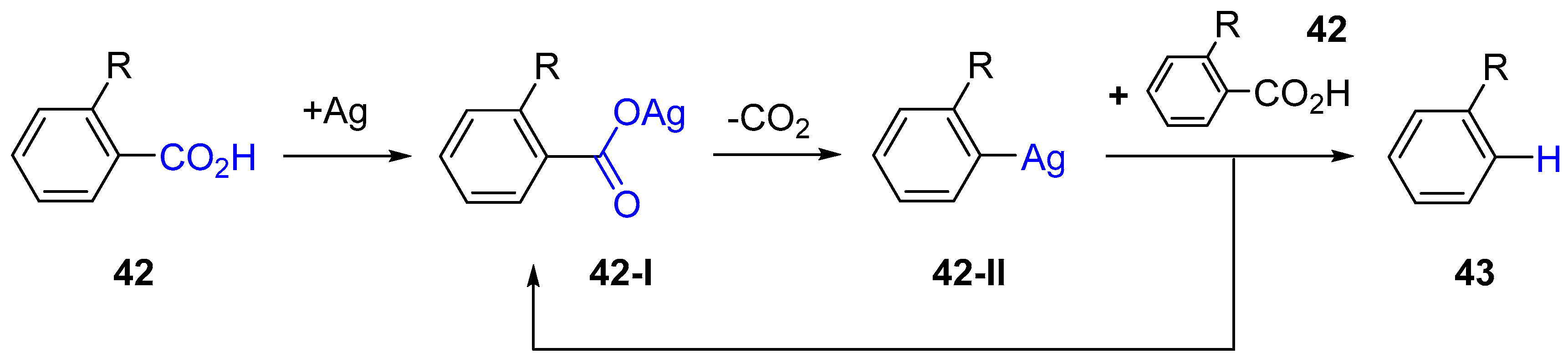
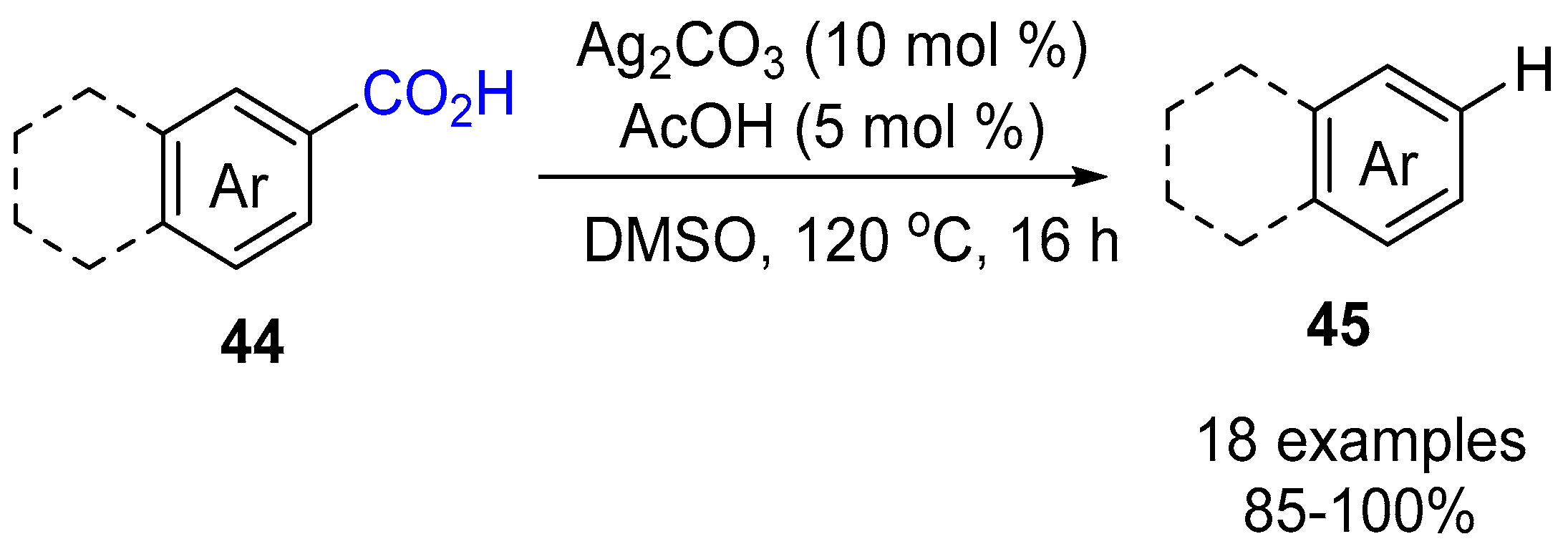
3. Functionalization of Carboxylic Acid Using Ag2CO3
3.1. Decarboxylation of Carboxylic Acids
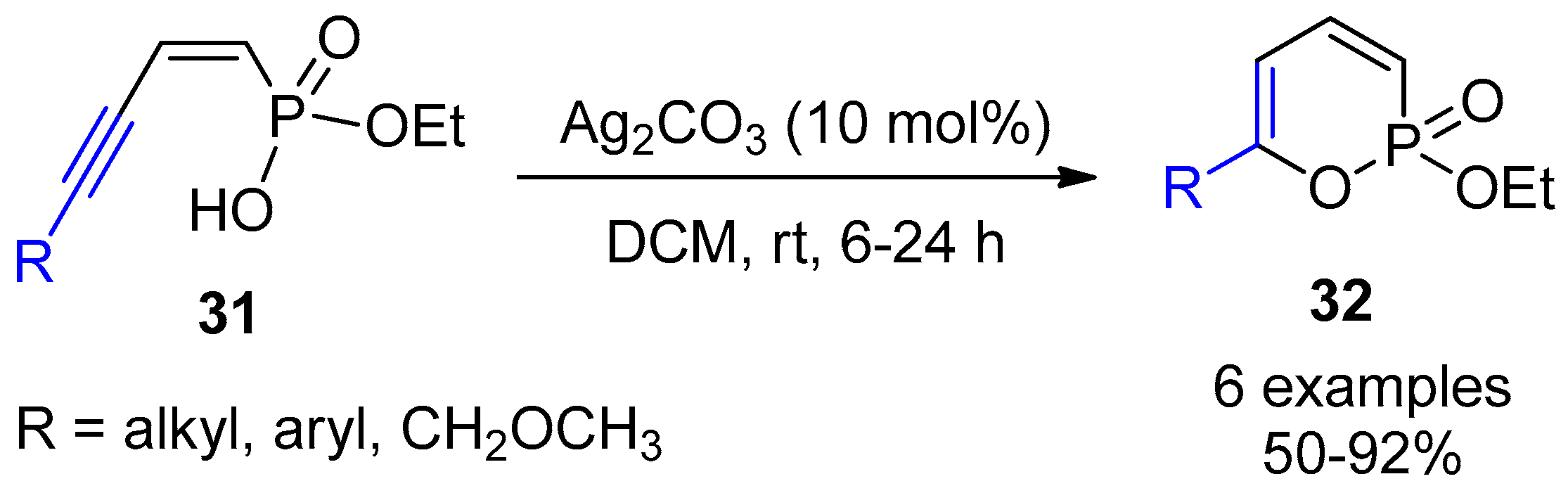
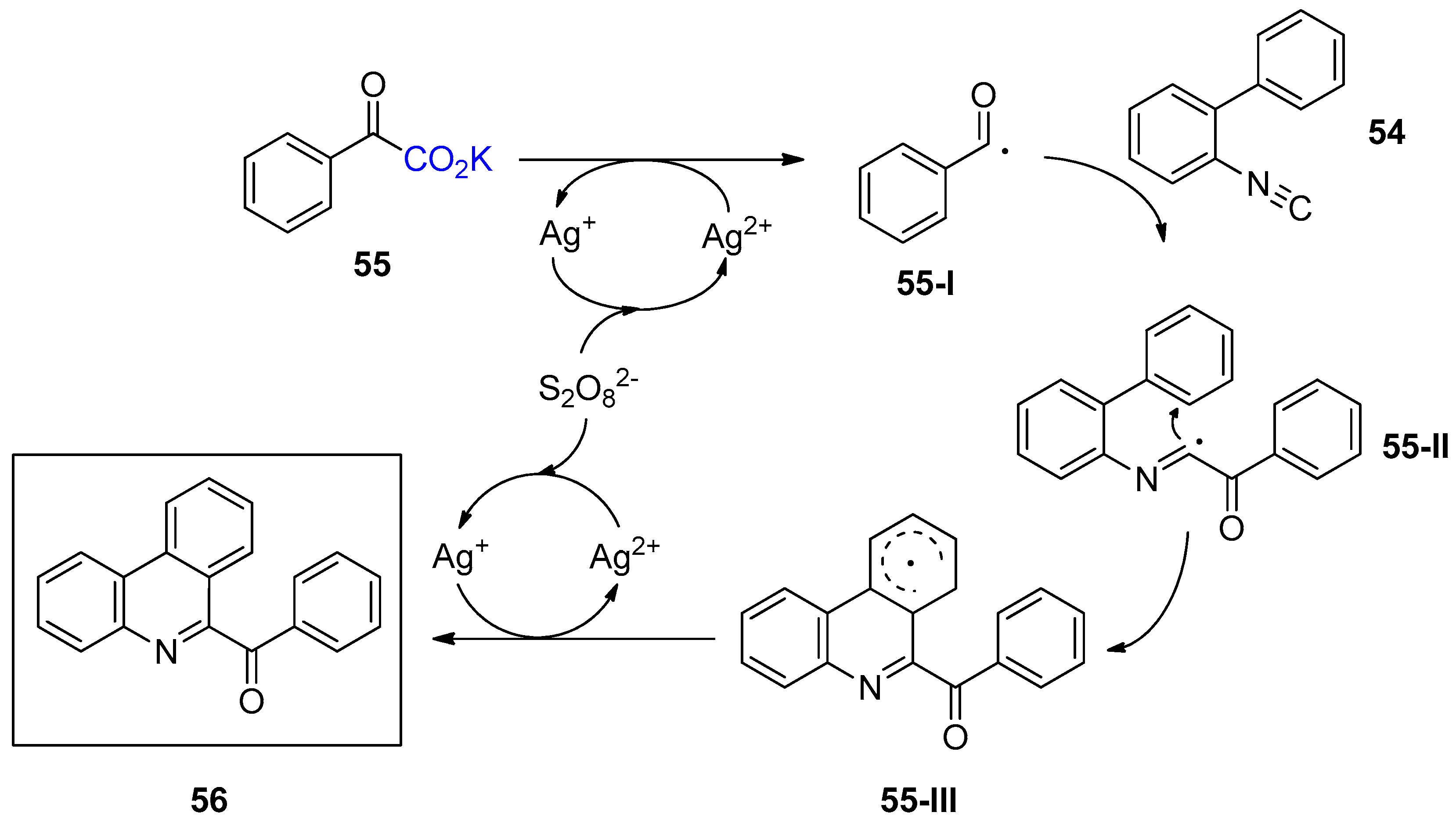
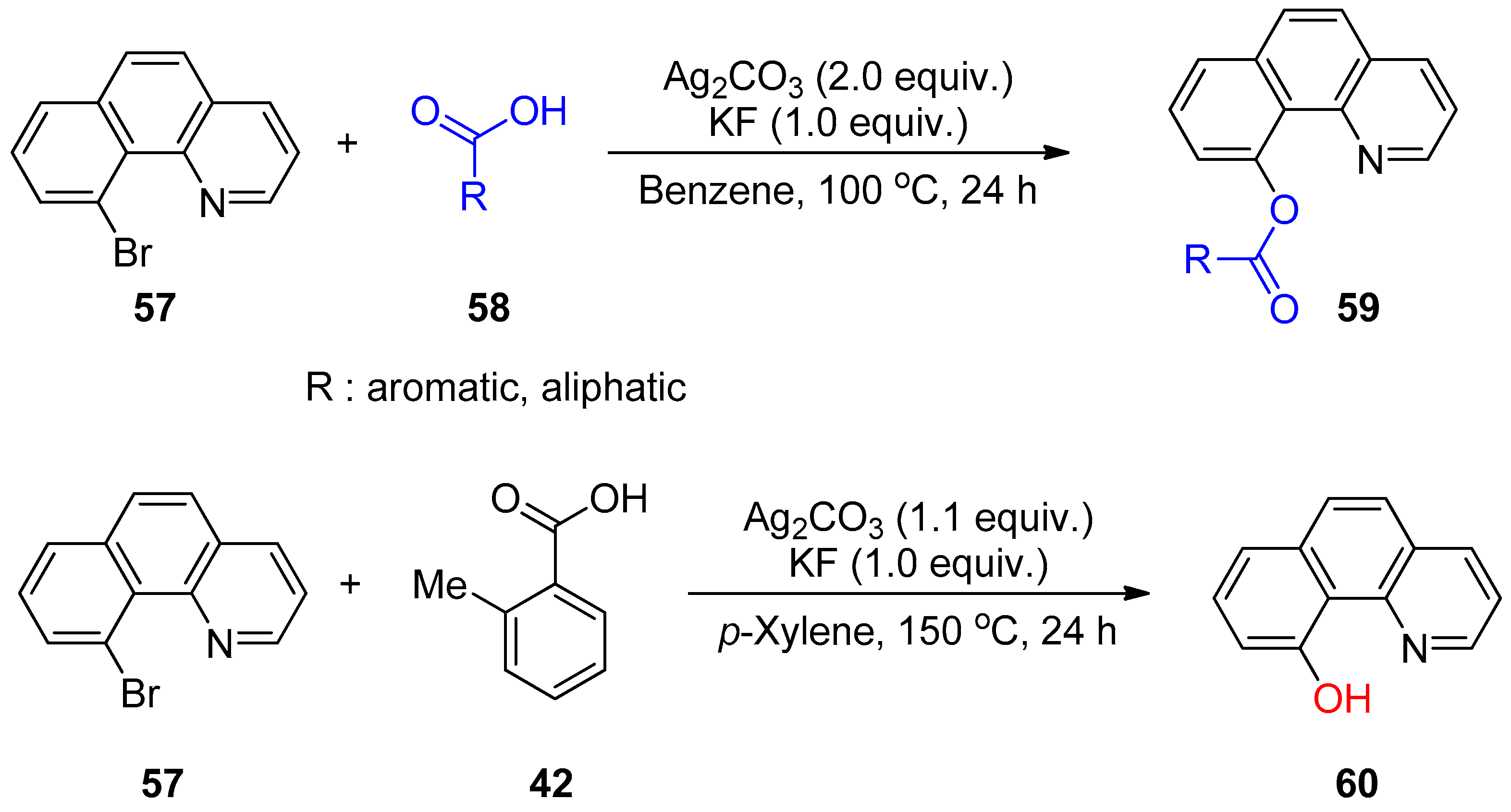
3.2. Decarboxylative Functionalization and Carboxylations
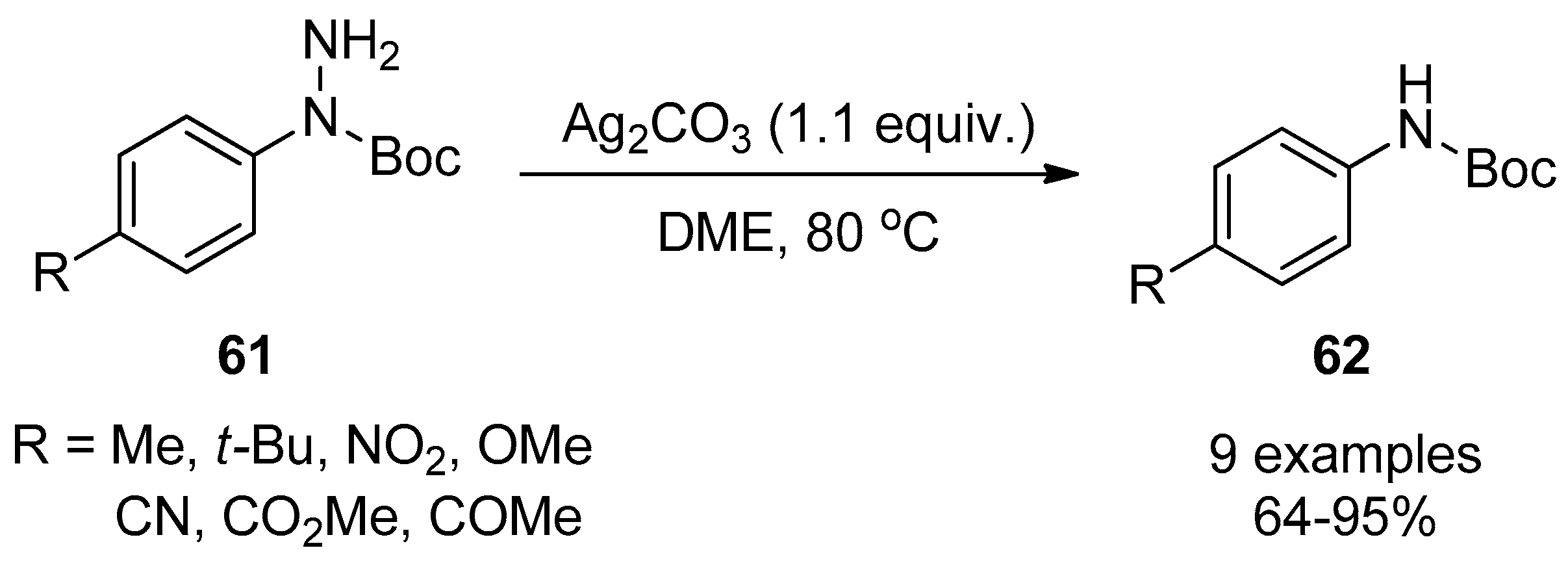
4. Miscellaneous
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Buidling Blocks and Find Chemicals, 2nd rev. and enl. ed.; WILEY-VCH: Weinheim, Germany, 2004; ISBN 3527306137. [Google Scholar]
- Patil, N.T.; Yamamoto, Y. Coinage Metal-Assisted Synthesis of Heterocycles. Chem. Rev. 2008, 108, 3395–3442. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Z.; Jiao, N. Ag-catalyzed C-H/C-C bond functionalization. Chem. Soc. Rev. 2016, 45, 4590–4627. [Google Scholar] [CrossRef] [PubMed]
- Weibel, J.M.; Blanc, A.; Pale, P. Ag-mediated reactions: Coupling and heterocyclization reactions. Chem. Rev. 2008, 108, 3149–3173. [Google Scholar] [CrossRef] [PubMed]
- Halbes-Letinois, U.; Weibel, J.M.; Pale, P. The organic chemistry of silver acetylides. Chem. Soc. Rev. 2007, 36, 759–769. [Google Scholar] [CrossRef]
- Harmata, M. Silver in Organic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; ISBN 9780470466117. [Google Scholar]
- Naodovic, M.; Yamamoto, H. Asymmetric silver-catalyzed reactions. Chem. Rev. 2008, 108, 3132–3148. [Google Scholar] [CrossRef]
- Sekine, K.; Yamada, T. Silver-catalyzed carboxylation. Chem. Soc. Rev. 2016, 45, 4524–4532. [Google Scholar] [CrossRef] [Green Version]
- Fetizon, M.; Balogh, V.; Golfier, M. Oxidations with silver carbonate/celite. V. Oxidations of phenols and related compounds. J. Org. Chem. 1971, 36, 1339–1341. [Google Scholar] [CrossRef]
- Jedinak, L.; Rush, L.; Lee, M.; Hesek, D.; Fisher, J.F.; Boggess, B.; Noll, B.C.; Mobashery, S. Use of Silver Carbonate in the Wittig Reaction. J. Org. Chem. 2013, 78, 12224–12228. [Google Scholar] [CrossRef] [Green Version]
- Boto, A.; Alvares, L. Furan and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K.C., Chattopadhyay, S.K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 97–152. ISBN 3527634894. [Google Scholar]
- Young, I.S.; Thornton, P.D.; Thompson, A. Synthesis of natural products containing the pyrrolic ring. Nat. Prod. Rep. 2010, 27, 1801–1839. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Guo, S.; Ke, J.; Hao, J.; Xu, H.; Chen, H.; Lei, A. Silver-mediated oxidative C-H/C-H functionalization: A strategy to construct polysubstituted furans. J. Am. Chem. Soc. 2012, 134, 5766–5769. [Google Scholar] [CrossRef] [PubMed]
- Moran, W.J.; Rodríguez, A. Metal-catalyzed furan synthesis. A review. Org. Prep. Proced. Int. 2012, 44, 103–130. [Google Scholar] [CrossRef] [Green Version]
- Hay, A. Oxidative Coupling of Acetylenes. J. Org. Chem. 1960, 25, 1275–1276. [Google Scholar] [CrossRef]
- He, C.; Hao, J.; Xu, H.; Mo, Y.; Liu, H.; Han, J.; Lei, A. Heteroaromatic imidazo[1,2-a]pyridines synthesis from C-H/N-H oxidative cross-coupling/cyclization. Chem. Commun. 2012, 48, 11073–11075. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; He, C.; Liu, H.; Li, M.; Lei, A. Oxidative cross-coupling/cyclization to build polysubstituted pyrroles from terminal alkynes and β-enamino esters. Chem. Commun. 2013, 49, 7549–7551. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Wu, N.; Liao, P.; Bi, X. Silver-catalyzed cross-coupling of propargylic alcohols with isocyanides: An atom-economical synthesis of 2,3-ALLENAMIDES. Chem. Eur. J. 2014, 20, 2154–2158. [Google Scholar] [CrossRef]
- Pandya, A.N.; Fletcher, J.T.; Villa, E.M.; Agrawal, D.K. Silver-mediated synthesis of indolizines via oxidative C-H functionalization and 5-endo-dig cyclization. Tetrahedron Lett. 2014, 55, 6922–6924. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, B.; Klajn, J.; Gryko, D.T. Recent advances in the synthesis of indolizines and their π-expanded analogues. Org. Biomol. Chem. 2016, 14, 7804–7828. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, S.; Shao, A.; Gao, M.; Huang, Y.; Lei, A. Silver-Mediated Selective Oxidative Cross-Coupling between C-H/P-H: A Strategy to Construct Alkynyl(diaryl)phosphine Oxide. Org. Lett. 2015, 17, 118–121. [Google Scholar] [CrossRef]
- Liu, J.; Fang, Z.; Zhang, Q.; Liu, Q.; Bi, X. Silver-catalyzed isocyanide-alkyne cycloaddition: A general and practical method to oligosubstituted pyrroles. Angew. Chem. Int. Ed. 2013, 52, 6953–6957. [Google Scholar] [CrossRef]
- Gao, M.; He, C.; Chen, H.; Bai, R.; Cheng, B.; Lei, A. Synthesis of pyrroles by click reaction: Silver-catalyzed cycloaddition of terminal alkynes with isocyanides. Angew. Chem. Int. Ed. 2013, 52, 6958–6961. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Meng, X.; Liao, P.; Liu, J.; Bi, X. Silver-catalyzed cyclization of 2-pyridyl alkynyl carbinols with isocyanides: Divergent synthesis of indolizines and pyrroles. Chem. Commun. 2014, 50, 11837–11839. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Wang, T.; Qin, C.; Jiao, N. Silver-catalyzed nitrogenation of alkynes: A direct approach to nitriles through C≡C bond cleavage. Angew. Chem. Int. Ed. 2013, 52, 6677–6680. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, J.; Zhang, L.; Liao, P.; Song, J.; Bi, X. Silver(I)-catalyzed hydroazidation of ethynyl carbinols: Synthesis of 2-azidoallyl alcohols. Angew. Chem. Int. Ed. 2014, 53, 5305–5309. [Google Scholar] [CrossRef] [PubMed]
- Barnert, K. The Chemistry of Vinyl, Allenyl, and Ethynyl Azides. In Organic Azides: Syntheses and Applications; Bräse, S., Banert, K., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2010; pp. 115–156. ISBN 0470682523. [Google Scholar]
- Liu, Z.; Liao, P.; Bi, X. General silver-catalyzed hydroazidation of terminal alkynes by combining TMS-N3 and H2O: Synthesis of vinyl azides. Org. Lett. 2014, 16, 3668–3671. [Google Scholar] [CrossRef]
- Ning, Y.; Wu, N.; Yu, H.; Liao, P.; Li, X.; Bi, X. Silver-catalyzed tandem hydroazidation/alkyne-azide cycloaddition of diynes with TMS-N3: An easy access to 1,5-fused 1,2,3-triazole frameworks. Org. Lett. 2015, 17, 2198–2201. [Google Scholar] [CrossRef]
- Kumar, Y.K.; Kumar, G.R.; Reddy, T.J.; Sridhar, B.; Reddy, M.S. Synthesis of 3-Sulfonylamino Quinolines from 1-(2-Aminophenyl) Propargyl Alcohols through a Ag(I)-Catalyzed Hydroamination, (2+3) Cycloaddition, and an Unusual Strain-Driven Ring Expansion. Org. Lett. 2015, 17, 2226–2229. [Google Scholar] [CrossRef]
- Peng, A.-Y.; Ding, Y.-X. Synthesis of 2H-1,2-Oxaphosphorin 2-Oxides via Ag2CO3-Catalyzed Cyclization of (Z)-2-Alken-4-ynylphosphonic Monoesters. Org. Lett. 2005, 7, 3299–3301. [Google Scholar] [CrossRef]
- Anastasia, L.; Xu, C.; Negishi, E.I. Catalytic and selective conversion of (Z)-2-en-4-ynoic acids to either 2H-pyran-2-ones in the presence of ZnBr2 or (Z)-5-alkylidenefuran-2(5H)-ones in the presence of Ag2CO3. Tetrahedron Lett. 2002, 43, 5673–5676. [Google Scholar] [CrossRef]
- Tan, X.C.; Liang, Y.; Bao, F.P.; Wang, H.S.; Pan, Y.M. Silver-mediated C-H bond functionalization: One-pot to construct substituted indolizines from 2-alkylazaarenes with alkynes. Tetrahedron 2014, 70, 6717–6722. [Google Scholar] [CrossRef]
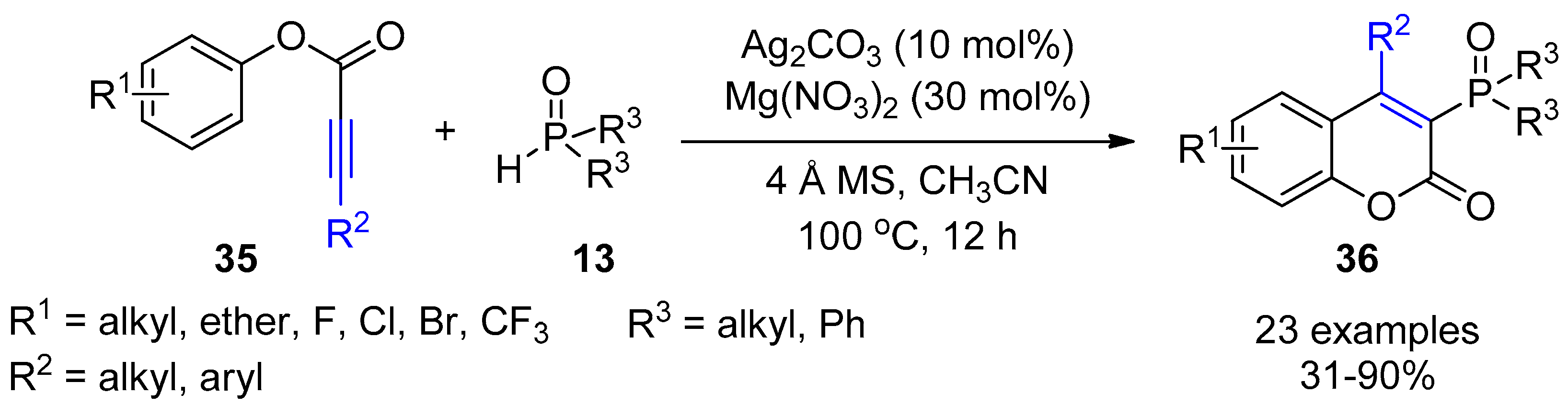
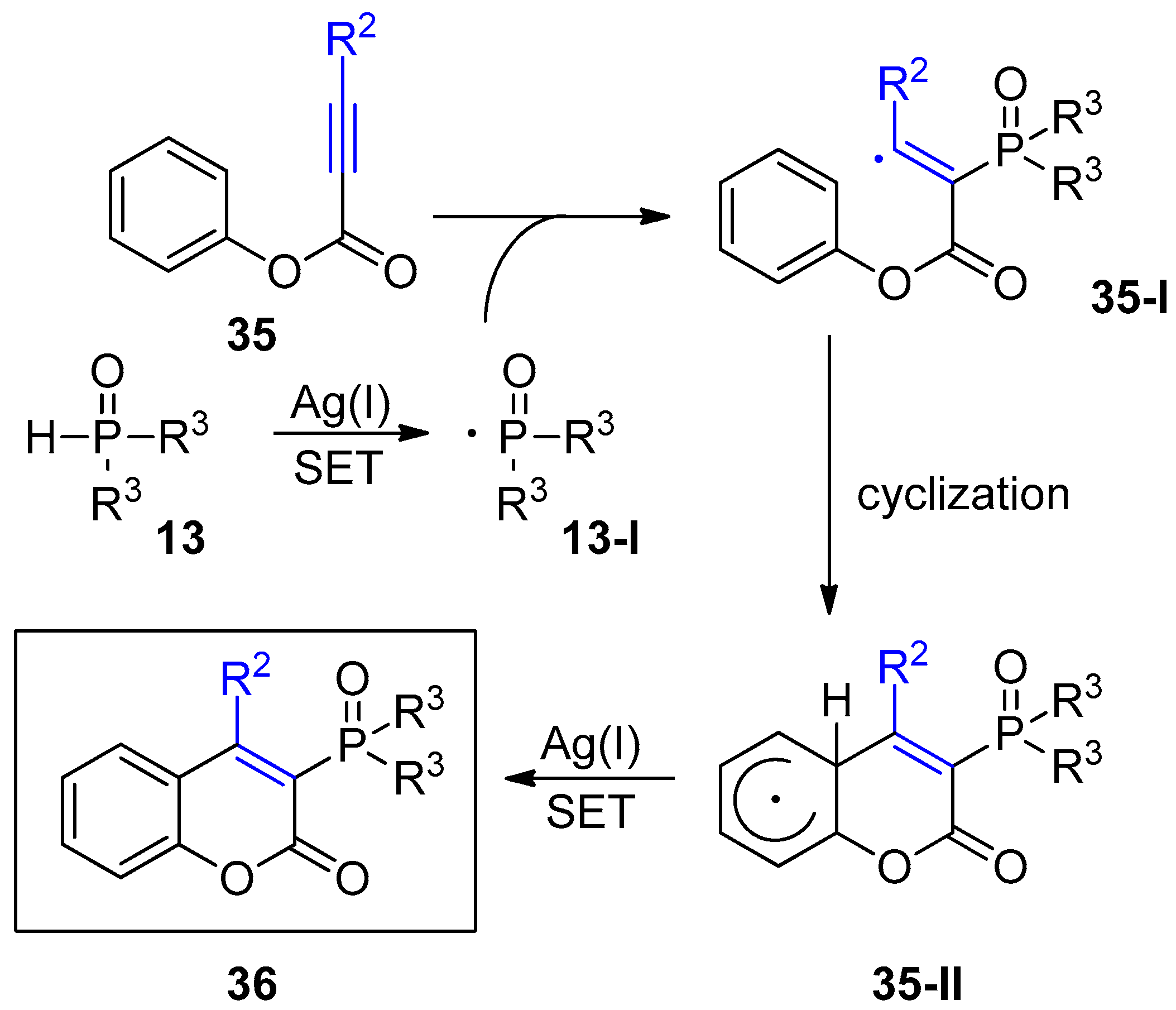
- Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Silver-catalyzed synthesis of 3-phosphorated coumarins via radical cyclization of alkynoates and dialkyl H -phosphonates. Org. Lett. 2014, 16, 3356–3359. [Google Scholar] [CrossRef] [PubMed]
- Ahmar, S.F.E. Expedient Synthesis of Complex γ-Butyrolactones from 5-(1-Arylalkylidene) Meldrum’s Acids via Sequential Conjugate Alkynylation/Ag(I)-Catalyzed Lactonization. Org. Lett. 2014, 16, 5748–5751. [Google Scholar] [CrossRef] [PubMed]
- Suresh Kumar, E.; Etukala, J.; Ablordeppey, S. Indolo[3,2-b]quinolines: Synthesis, Biological Evaluation and Structure Activity-Relationships. Mini-Rev. Med. Chem. 2008, 8, 538–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Curran, D.P. Synthesis of Carbocyclic and Heterocyclic Fused Quinolines by Cascade Radical Annulations of Unsaturated N-Aryl Thiocarbamates, Thioamides, and Thioureas. Org. Lett. 2003, 5, 1765–1768. [Google Scholar] [CrossRef]
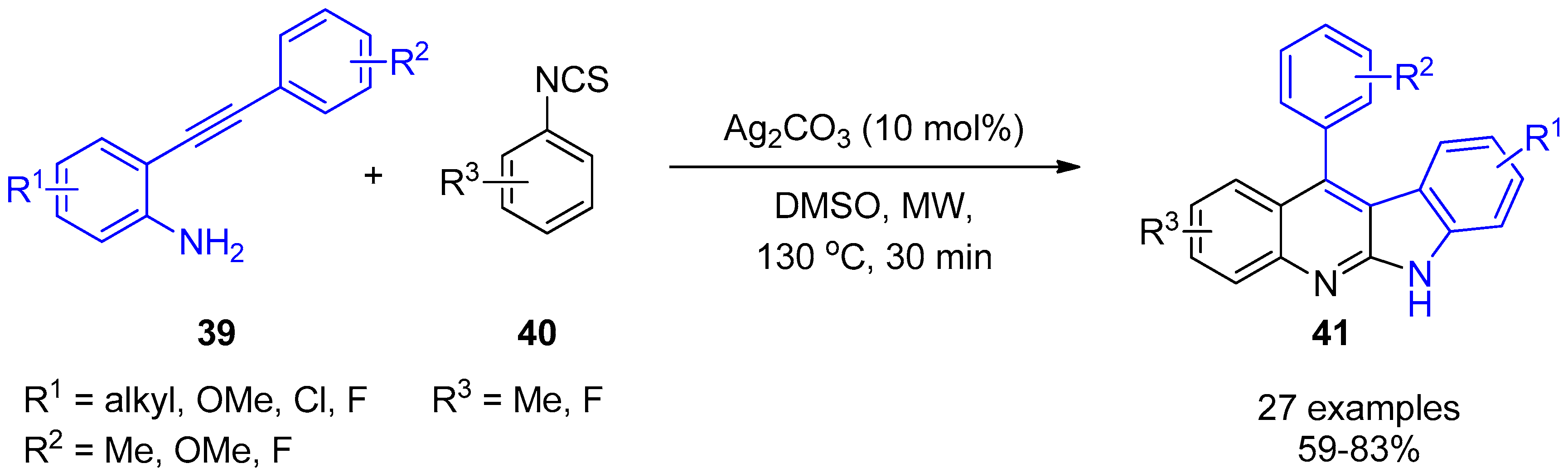
- Ali, W.; Dahiya, A.; Pandey, R.; Alam, T.; Patel, B.K. Microwave-Assisted Cascade Strategy for the Synthesis of Indolo[2,3-b]quinolines from 2-(Phenylethynyl)anilines and Aryl Isothiocynates. J. Org. Chem. 2017, 82, 2089–2096. [Google Scholar] [CrossRef]
- Cornella, J.; Sanchez, C.; Banawa, D.; Larrosa, I. Silver-catalysed protodecarboxylation of ortho-substituted benzoic acids. Chem. Commun. 2009, 7176–7178. [Google Scholar] [CrossRef]
- Lu, P.; Sanchez, C.; Cornella, J.; Larrosa, I. Silver-catalyzed protodecarboxylation of heteroaromatic carboxylic acids. Org. Lett. 2009, 11, 5710–5713. [Google Scholar] [CrossRef]
- Jafarpour, F.; Jalalimanesh, N.; Olia, M.B.A.; Kashani, A.O. Silver-catalyzed facile decarboxylation of coumarin-3-carboxylic acids. Tetrahedron 2010, 66, 9508–9511. [Google Scholar] [CrossRef]
- Hartwig, J.F. Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism. Angew. Chem. Int. Ed. 1998, 37, 2046–2067. [Google Scholar] [CrossRef]
- Luo, Y.; Pan, X.; Wu, J. Silver-catalyzed decarboxylative halogenation of carboxylic acids. Tetrahedron Lett. 2010, 51, 6646–6648. [Google Scholar] [CrossRef]
- Suresh, R.; Kumaran, R.S.; Senthilkumar, V.; Muthusubramanian, S. Silver catalyzed decarboxylative acylation of pyridine-N-oxides using α-oxocarboxylic acids. RSC Adv. 2014, 4, 31685–31688. [Google Scholar] [CrossRef]
- Cheng, K.; Zhao, B.; Qi, C. Silver-catalyzed decarboxylative acylation of arylglyoxylic acids with arylboronic acids. RSC Adv. 2014, 4, 48698–48702. [Google Scholar] [CrossRef]
- Shang, R.; Liu, L. Transition metal-catalyzed decarboxylative cross-coupling reactions. Sci. China Chem. 2011, 54, 1670–1687. [Google Scholar] [CrossRef]
- Liu, J.; Fan, C.; Yin, H.; Qin, C.; Zhang, G.; Zhang, X.; Yi, H.; Lei, A. Synthesis of 6-acyl phenanthridines by oxidative radical decarboxylation-cyclization of α-oxocarboxylates and isocyanides. Chem. Commun. 2014, 50, 2145–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, K.; Park, H.; Kim, S.; Kim, M. Temperature-controlled acyloxylations and hydroxylations of bromoarene by a silver salt. Tetrahedron Lett. 2016, 57, 781–783. [Google Scholar] [CrossRef]
- Leblanc, Y.; Fitzsimmons, B.J. [4+2] Cycloaddition reaction of bis (trichloroethyl) azodicarboxylate and glycals: Preparation of a C1-C1 2-amino disaccharide. Tetrahedron Lett. 1989, 30, 2889–2892. [Google Scholar] [CrossRef]
- Trimble, L.A.; Vederas, J.C. Amination of chiral enolates by dialkyl azodiformates. Synthesis of .alpha.-hydrazino acids and .alpha.-amino acids. J. Am. Chem. Soc. 1986, 108, 6397–6399. [Google Scholar] [CrossRef]
- Lee, K.S.; Lim, Y.K.; Cho, C.G. Ag+ mediated deaminations of N-Boc aryl hydrazines for the efficient synthesis of N-Boc aryl amines. Tetrahedron Lett. 2002, 43, 7463–7464. [Google Scholar] [CrossRef]
- Hili, R.; Yudin, A.K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2006, 2, 284–287. [Google Scholar] [CrossRef]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef] [PubMed]
- Louillat, M.L.; Patureau, F.W. Oxidative C-H amination reactions. Chem. Soc. Rev. 2014, 43, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Lee, S.Y.; Chang, S. Silver-mediated direct amination of benzoxazoles: Tuning the amino group source from formamides to parent amines. Angew. Chem. Int. Ed. 2009, 48, 9127–9130. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Mukhopadhyay, C. Silver-mediated Cα(sp3)-H functionalization of primary amines: An oxidative C-N coupling strategy for the synthesis of two different types of 1,2,4,5-tetrasubstituted imidazoles. Eur. J. Org. Chem. 2015, 2015, 1246–1256. [Google Scholar] [CrossRef]
- Youn, S.W.; Ko, T.Y.; Jang, M.J.; Jang, S.S. Silver(I)-mediated C-H amination of 2-alkenylanilines: Unique solvent-dependent migratory aptitude. Adv. Synth. Catal. 2015, 357, 227–234. [Google Scholar] [CrossRef]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Liao, P.; Zhang, L.; Tu, T.; Bi, X. Silver-Catalyzed Cross-Coupling of Isocyanides and Active Methylene Compounds by a Radical Process. Angew. Chem. Int. Ed. 2015, 54, 10618–10622. [Google Scholar] [CrossRef]
- Sun, W.B.; Xue, J.F.; Zhang, G.Y.; Zeng, R.S.; An, L.T.; Zhang, P.Z.; Zou, J.P. Silver-Catalyzed Direct Csp2-H Phosphorylation of Indoles Leading to Phosphoindoles. Adv. Synth. Catal. 2016, 358, 1753–1758. [Google Scholar] [CrossRef]
- Fang, G.; Liu, J.; Fu, J.; Liu, Q.; Bi, X. Silver-Catalyzed Cascade Reaction of β-Enaminones and Isocyanoacetates to Construct Functionalized Pyrroles. Org. Lett. 2017, 19, 1346–1349. [Google Scholar] [CrossRef]
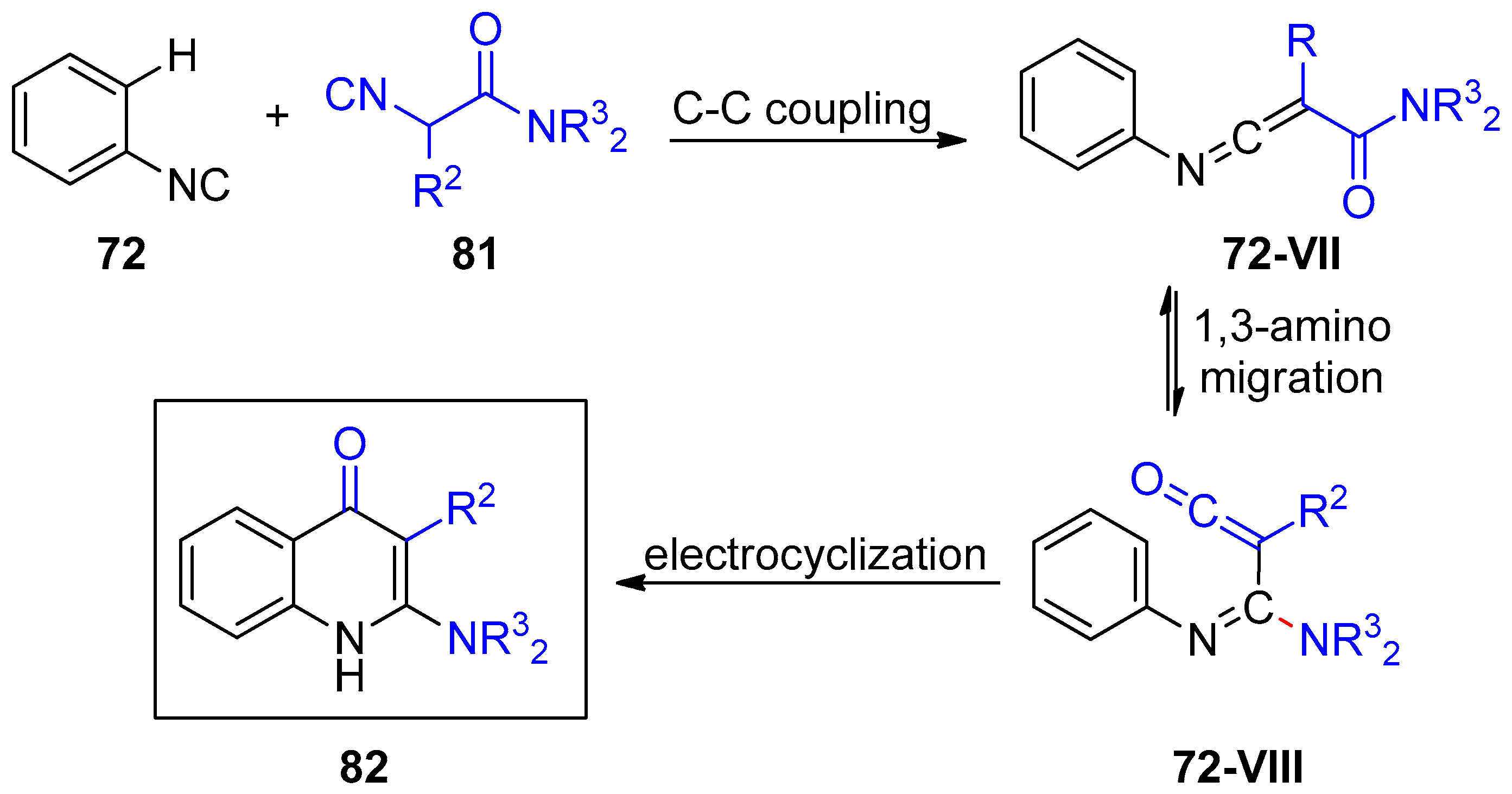
- Hu, Z.; Dong, J.; Men, Y.; Lin, Z.; Cai, J.; Xu, X. Silver-Catalyzed Chemoselective [4+2] Annulation of Two Isocyanides: A General Route to Pyridone-Fused Carbo- and Heterocycles. Angew. Chem. Int. Ed. 2017, 56, 1805–1809. [Google Scholar] [CrossRef]
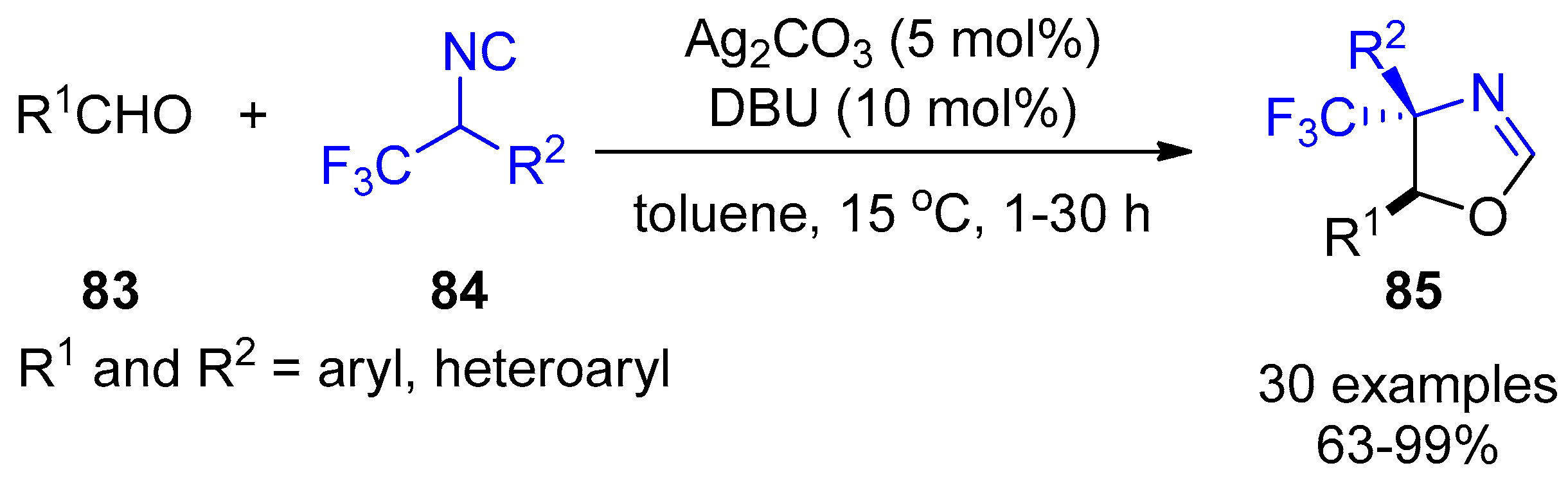
- Zhang, X.; Wang, X.; Gao, Y.; Xu, X. Silver-catalyzed formal [3+2]-cycloaddition of α-trifluoromethylated methyl isocyanides: A facile stereoselective synthesis of CF3-substituted heterocycles. Chem. Commun. 2017, 53, 2427–2430. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, H.; Wang, X.; Zhi, S.; Kan, Y.; Wang, C. Synthesis of Pyrrole via a Silver-Catalyzed 1,3-Dipolar Cycloaddition/Oxidative Dehydrogenative Aromatization Tandem Reaction. J. Org. Chem. 2017, 82, 4194–4202. [Google Scholar] [CrossRef] [PubMed]



















































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, K.; Jwa, D.G.; Lee, H.-E.; Kim, H.J.; Kim, C.; Kim, M. Recent Organic Transformations with Silver Carbonate as a Key External Base and Oxidant. Catalysts 2019, 9, 1032. https://doi.org/10.3390/catal9121032
Yoo K, Jwa DG, Lee H-E, Kim HJ, Kim C, Kim M. Recent Organic Transformations with Silver Carbonate as a Key External Base and Oxidant. Catalysts. 2019; 9(12):1032. https://doi.org/10.3390/catal9121032
Chicago/Turabian StyleYoo, Kwangho, Dong Gyun Jwa, Ha-Eun Lee, Hyun Jin Kim, Cheoljae Kim, and Min Kim. 2019. "Recent Organic Transformations with Silver Carbonate as a Key External Base and Oxidant" Catalysts 9, no. 12: 1032. https://doi.org/10.3390/catal9121032
APA StyleYoo, K., Jwa, D. G., Lee, H. -E., Kim, H. J., Kim, C., & Kim, M. (2019). Recent Organic Transformations with Silver Carbonate as a Key External Base and Oxidant. Catalysts, 9(12), 1032. https://doi.org/10.3390/catal9121032