2.1. Synthesis and Characterization
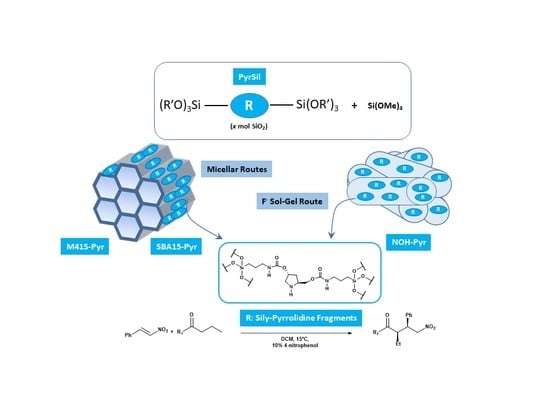
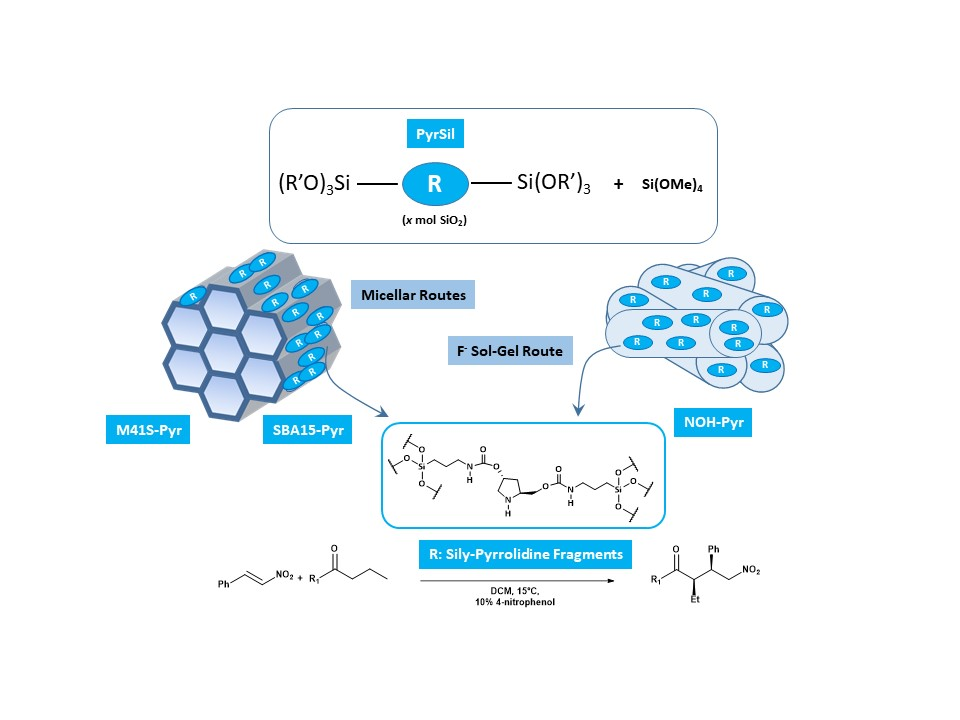
Herein we report the incorporation of pyrrolidine-type organic fragments into the structure of different mesoporous organosiliceous materials with different topologies and physico-chemical characteristics. The different properties of the synthesized hybrid materials determine the final reactivity of the organocatalytic moieties inserted and stabilized in their structure. The preparation of the hybrid materials begins with the synthesis of a bis-silylated monomer used as a precursor (PyrSil). This organosilicon precursor presents pyrrolidine-type fragments stabilized as organic bridges between the highly reactive terminal siloxane groups. Specifically, the monomer PyrSil was synthesized, starting from hydroxypyrrolidine (
1), through successive reaction steps of protection with Cl-Bnz of pyrrolidine, reduction of ester group into hydroxyl (
3) with LiAlH
4, and a final condensation reaction with isocyanate silane molecules to form the silyl organosiloxane derivative (
4). Therefore, the bis-silylated monomer, containing non-active pyrrolidine-urethane groups as organic bridges, were de-protected via reduction in the presence of a Pd/C catalyst under H
2 to provide the final bridged silsesquioxane precursor (
5), PyrSil, containing active functional pyrrolidine moieties in its composition (
Scheme 1).
Therefore, the bis-silylated monomer was used in different controlled hydrolysis and condensation synthesis processes, with and without structure-directing agents, to provide several families of organic-inorganic mesoporous materials. Specifically, different synthesis routes were explored to obtain ordered hybrid materials with MCM-41- and SBA-15-topology-types (M41S-Pyr and SBA-15-Pyr), depending on the use of long chain neutral amine surfactants or block copolymers during micellar processes together with the bis-silylated precursor. Non-ordered mesoporous hybrid solids were synthesized through a sol-gel process in a fluoride medium in the absence of structure-directing agents and under soft conditions at neutral pH conditions and at room temperature. In this case, the fluoride ions act as a mineralizing agent generating penta-coordinated organosilicon complexes as highly reactive intermediates that allowed the rapid gelation of the synthesis slurry. After the aging period at 36 °C, which allowed the assembly of the PyrSil monomers, the formation of non-ordered mesoporous structures (NOH-Pyr), containing in their framework active pyrrolidine units, was achieved (
Scheme 2).
X-ray diffraction (XRD) patterns of ordered mesoporous materials prepared in the presence of structure-directing agents showed that the long-range order of the synthesized hybrid solids decreased significantly with the increase of the amount of pyrrolidine units incorporated in the structure. Likewise, the hybrid material incorporating a 30 mol% of PyrSil in the synthesis gel did not present the diffraction band (100) characteristic of the hexagonal symmetry of the M41S-type materials prepared in the presence of long-chain surfactants. Similarly, the XRD pattern of solids prepared with block copolymers with high concentrations of pyrrolidine fragments did not display the low-angle diffraction bands characteristic of materials exhibiting a SBA-15-type structural arrangement. Nevertheless, in the specific case of the mesoporous hybrid solids obtained starting from 5 mol% of the bis-silylated PyrSil precursor, it was observed that they reasonably conserved the low angle diffraction bands indicative of the M41S- and SBA-15-type ordering. For an amount of PyrSil precursor above 5 mol% used in the synthesis procedure in the presence of structure directing agents, the progressive collapse of the mesoporous topology occurred due to the difficult assembling in an orderly manner of a high concentration of pyrrolidine fragments together with units of silicon tetrahedral along the walls of mesoporous channels (
Figure 1). In the case of hybrid materials prepared through sol-gel processes in a fluoride medium in the absence of structure-directing agents, diffractograms (not shown) did not exhibit any diffraction bands as expected for mesoporous solids without any structural ordering.
The images obtained using transmission electronic microscopy (TEM) showed how the mesoporous ordering of the hybrid materials was reasonably conserved in the samples that were prepared with up to 5 mol% of PyrSil in the synthesis gel, although an elevated disorder and poor regular distribution of internal channels was observed. Specifically, in the ordered solids, cavities of approximately 30–100 Å were detected for materials with an SBA-15 or M41S topology, within the expected mesoporous range (
Figure 2). In the non-ordered mesoporous solids, obtained through synthesis in a fluoride medium, exhibited an irregular morphology, where cavities of different diameters distributed in an irregular way were observed as expected.
Through the chemical analysis (CHNS) of the final mesoporous samples, the increase of embedded organic content according to the amount of bis-silylated monomer (PyrSil) used in the synthesis process was confirmed. The as-synthesized ordered materials with topology SBA-15 and M41S were treated with different extraction methodologies in order to eliminate surfactant molecules and block copolymers that acted as structure-directing agents, with the aim of preserving the composition and structure of the pyrrolidine units inserted in the structural framework, and without carrying out a calcination process. However, although several successive extraction treatments were carried out, it was not possible to completely remove the molecules of the structure-directing agents. The maximum number of organic templates was eliminated when the content of pyrrolidine units was more elevated, with the elimination of structure directing agents in the samples obtained with 5 mol% of PyrSil being more difficult. This matter could be associated with the poorer structuration level achieved for the samples with a higher content of organic pyrrolidine structural units in the framework that facilitated the easier elimination of internal template molecules. In the case of hybrid materials prepared in a fluoride medium in the absence of templates, all the organic content came from the units of pyrrolidine incorporated in the walls of the mesoporous solids. In fact, the calculated C/N molar ratios fully coincided with the theoretical values (C/N = 4.3 for a pyrrolidine-bridged unit) expected for the organic fragments from the PyrSil precursor, confirming that the organic units were preserved as they were initially after being incorporated into the mesoporous architectures (
Table 1).
Figure 3 shows the thermogravimetric analysis (TGA) curves of the different organic-inorganic mesoporous samples obtained, as well as the corresponding derivatives (DTA) that allowed us to establish the thermal stability of the solids. The results showed two main weight losses. The first, located around 250–300 °C, was assigned to the remaining organic molecules of the structure-directing agents (surfactants or block copolymers (I)), which were not completely eliminated during the extraction process. Fragments of the silyl-propyl-pyrrolidine units inserted in the structure, coming from the PyrSil monomer precursor, could also be included in this first weight loss due to their decomposition during the thermal treatment. The second weight loss (II) was attributed to the included pyrrolidine fragments inserted into the structural frameworks. Specifically, this second weight loss was established in the temperature range of 300–500 °C, providing evidence for the thermal stability of the solids obtained. In the case of the as-synthesized ordered mesoporous materials, before the extraction process, these two weight losses were better defined (see
Supplementary Information). In all cases, an initial loss assigned to hydration water retained at low temperatures was observed, as well as a final loss (located around 550 °C) that was attributed to the hydroxylation water generated at high temperatures due to condensation phenomena existing between silanol groups present on the surface or in structure defects of the solids. In the case of the hybrid materials obtained without organic templates, only one weight loss was observed at 300–500 °C corresponding to silyl-pyrrolidine units present in the framework. It is noteworthy that the organic content was always higher when it was estimated using elemental analysis (CHNS) than when using thermogravimetric analysis (TGA) since in the latter, the oxygenated species contained in the hybrid materials are taken into account (
Table 1).
Not only the presence, but also the integrity, of the pyrrolidine units inserted into the structure of the solids was confirmed through the results obtained using nuclear magnetic resonance (NMR) spectroscopy. Specifically, the
13C NMR spectra showed, in all types of mesoporous hybrid materials, the chemical shifts, which were assigned to the carbon atoms from the bis-silylated precursor (PyrSil), including the atoms directly connected to the silicon atoms (
Figure 4). These results corroborated the fact that the organic units present in the solids remained intact with the same composition as in the PyrSil precursor used during the synthesis process. However, in the extracted solids, some bands could be attributed to the rest of the structure-directing agent molecules (surfactants and block copolymers) that remained in the internal channels of the materials.
In addition, it was further confirmed through the
29Si NMR spectra that the active organic silyl-pyrrolidine fragments were actually covalently inserted into the walls of the porous materials. In all cases, the chemical shifts located in the range between −60 and −80 ppm were assigned to the T-type species of silicon atoms, such as T
1 (C-Si(OH)
2(OSi)), T
2 (C-Si(OH)(OSi)
2), and T
3 (C-Si(OSi)
3), were observed, together with the conventional Q-type silicon atom signals corresponding to the tetrahedral silicon units resulting from the condensation of tetramethyl-orthosilicate (TMOS) or tetraethyl-orthosilicate (TEOS) used in the synthesis processes. These results corroborated the fact that the propyl-silyl-pyrrolidine groups were covalently inserted into the structural framework of the solids obtained through an effective hydrolysis and condensation of the terminal siloxane groups of the bis-silylated precursor with the purely siliceous units (
Figure 5). However, the presence of chemical shifts in
13C NMR spectra assigned to terminal ethoxide groups present in the starting silyl-derivative precursors were indicative that the condensation phenomenon had only partially occurred (
Figure 4). Furthermore, integrated values of chemical shifts corresponding to T-type silicon atoms corroborated that approximately 5, 10, and 30 mol% of silyl-pyrrolidine units were effectively incorporated in the network through the different synthesis processes (see
Table S1 and
29Si Cross Polarization (CP)/MAS NMR spectra of hybrid materials in
Supplementary Information). When comparing with the
29Si NMR spectrum of the monomer used as a precursor, it was observed that the chemical shifts of the T-type silicon atoms moved from a range of -40 to -60 ppm to -60 to -80 ppm that supported the successful integration of pyrrolidine fragments into the network of the solids (inset of
Figure 5a).
Infrared spectroscopy was also useful to confirm the presence and integrity of the pyrrolidine units inserted covalently in the structure of the different types of prepared mesoporous hybrid materials (
Figure 6). In all cases, the bands associated with the stretching vibrations were observed in the Fourier-transform infrared (FTIR) spectra (ν(–NH–): shoulder at 2620 cm
−1) and bending vibrations (δ(–NH–): 1559 and 1643 cm
−1) of the secondary amines present in the organic bridges included in the bis-silylated precursor (PyrSil) used in the synthesis processes, also including the cyclic aliphatic amines directly present in the pyrrolidine units. In addition, bands due to stretching vibrations attributed to the carbamate groups, ν(–NH–CO–O–), were also detected at 1703 cm
–1. The –CH
2– units included in the propyl chains located in the PyrSil precursor were observed through the existence of symmetric and asymmetric vibration bands, characteristic of alkylic groups linked to amino groups (ν(–N–CH
2–): 2897 and 2958 cm
−1; δ(–N–CH
2–): 1383 and 1447 cm
−1). In this same range, vibrations due to the rest of the surfactant or block copolymer molecules that remained in the mesoporous channels, after the extraction processes, were also observed at 1500 and 1450 cm
−1, δ(–N–CH
2–), corresponding to the presence of hexadecylamine and P123 block copolymers in M41S- and SBA-15-type hybrid materials, respectively. Specifically, we observed the signals due to P123 at 2880 and 2835 cm
−1, characteristic of alkyl groups bonded to ether groups, ν(O–CH
2–), and bending vibrations, δ(–CH
3–), at 1395 and 1365 cm
−1. However, these vibrations were practically nonexistent, confirming the high effectiveness of extraction treatments (see the FTIR spectra in
Supplementary Information, Figure S5). Additionally, a wide band centered at 3647 cm
−1 was detected in all spectra associated with the surface silanol groups (Si–OH) that are usually found in organic silicates with a high density of structure defects. The vibration band observed at 950 cm
−1 was also due to external silanols.
In the range of the infrared spectra corresponding to the vibrations characteristic of the structure framework, the band of the Si–C vibration located at 790 cm
−1 was observed, confirming the existence of bonds with a covalent nature established between the tetrahedral SiO
4 units and the bis-silylated fragments containing pyrrolidine groups. Within this range of vibration, the conventional bands of the Si–O–Si groups (458 and 1080 cm
−1) were also observed, which were the main structural components of the prepared organosiliceous hybrid materials, regardless of the achieved structural ordering level (
Figure 6). Therefore, spectroscopic results (NMR and IR) clearly showed both the integrity of the pyrrolidine units and their effective incorporation through covalent bonds in the structure of the different porous hybrid materials.
The textural properties of the different hybrid materials (specific surface area and free porous volume) were studied using the nitrogen adsorption isotherms (
Figure 7). The results showed how the materials obtained in the fluoride medium, without structure-directing agents, exhibited characteristic isotherms of non-ordered porous solids with a marked change of slope at high relative pressures (P/P
0 ≈ 0.3–0.4), due to the existence of high-diameter pores in the internal mesoporous channels existing in the organic-inorganic framework. In this family of materials, the Brunauer-Emmett-Teller (BET) surface and total volume decreased as the concentration of pyrrolidine units inserted in its structure increased due to the greater difficulty in assembly between the several structure units when the number of organic fragments was more elevated. This fact caused the collapsing of the structure for the materials prepared with 30 mol% of PyrSil as an initial precursor. In the case of non-ordered hybrid materials, the BET, surface, and the porous volume ranged from 636 m
2/g to 5 m
2/g when the amount of silyl-pyrrolidine units oscillated from 5–30 mol% of total silicon, respectively (
Table 2).
In contrast, organic-inorganic solids prepared in the presence of structure-directing agents (surfactants and block copolymers) exhibited the characteristic type IV isotherms of ordered mesoporous materials overall in the solids with a SBA-15 topology more than M41S-type materials. These isotherms showed a marked change of slope at relative pressures P/P
0 between approximately 0.5–0.6, indicating the existence of internal channels with diameters within the range of the mesopore. However, this change of slope in the isotherms was attenuated in the M41S family showed a marked loss of structural order in comparison with SBA-15-type materials at the same level of pyrrolidine incorporation, which was corroborated using X-ray diffraction (
Figure 1). As it happens in the case of non-ordered materials, it was observed that the specific surface area and the pore volume decreased markedly as the concentration of inserted pyrrolidine groups was higher, up to the point of collapse in the solids obtained by using 30 mol% of PyrSil as the initial bis-silylated precursor during the synthesis process. In particular, the BET surface area for M41S- and SBA-15-type hybrid materials oscillated between 160–317 m
2/g and 125–285 m
2/g, respectively. In both cases, the materials with the highest presence of silyl-pyrrolidine fragments exhibited the lowest BET surface due to the difficulties in assembling organic and inorganic units through hydrolysis and condensation processes. It is remarkable that solids obtained in the presence of 5 mol% of PyrSil as a monomer precursor showed BET surfaces areas similar to those obtained with 10 mol% of PyrSil. Additionally, we can observe that the materials with the highest percentage of PyrSil fragments showed BET surfaces higher than non-ordered materials, probably due to the better-achieved organization due to the presence of structure-directing agents. The results evidenced how the presence of pyrrolidine fragments in the framework strongly influenced the final textural properties of the porous hybrid solids.
From the distribution of the Barrett-Joyner-Halenda (BJH) pore diameter, it was observed that the internal pores of the different materials exhibited diameters between 75–110 Å for SBA-15-type and 30–40 Å for M41S-type and non-ordered hybrid materials such as it was expected for this type of mesoporous solids, with the broadest pore size distribution for M41S-type materials. In the three families, it was observed that the pore sizes obtained were slightly higher than in the equivalent pure silica solids, indicating the inclusion of voluminous silyl-pyrrolidine fragments into the framework to obtain internal pores with higher diameters.
2.2. Catalysis
We explore the catalytic performance of the three families of organosiliceous materials with different structuration level, order, and textural properties in the asymmetric Michael addition of butyraldehyde and β-nytrostyrene. Recently, this catalytic process was successfully performed using amino acids adsorbed on inorganic oxides as laponite-type supports that formed active hybrid organic-inorganic chiral heterogeneous catalysts [
28]. The preparation of carbonyl and nitrocarbonyl compounds through the asymmetric Michael addition reaction of carbonyl compounds and nitroolefins is a versatile and useful tool in organic synthesis, normally being carried out using homogeneous and heterogeneous catalytic systems containing prolinol active units [
29,
30,
31,
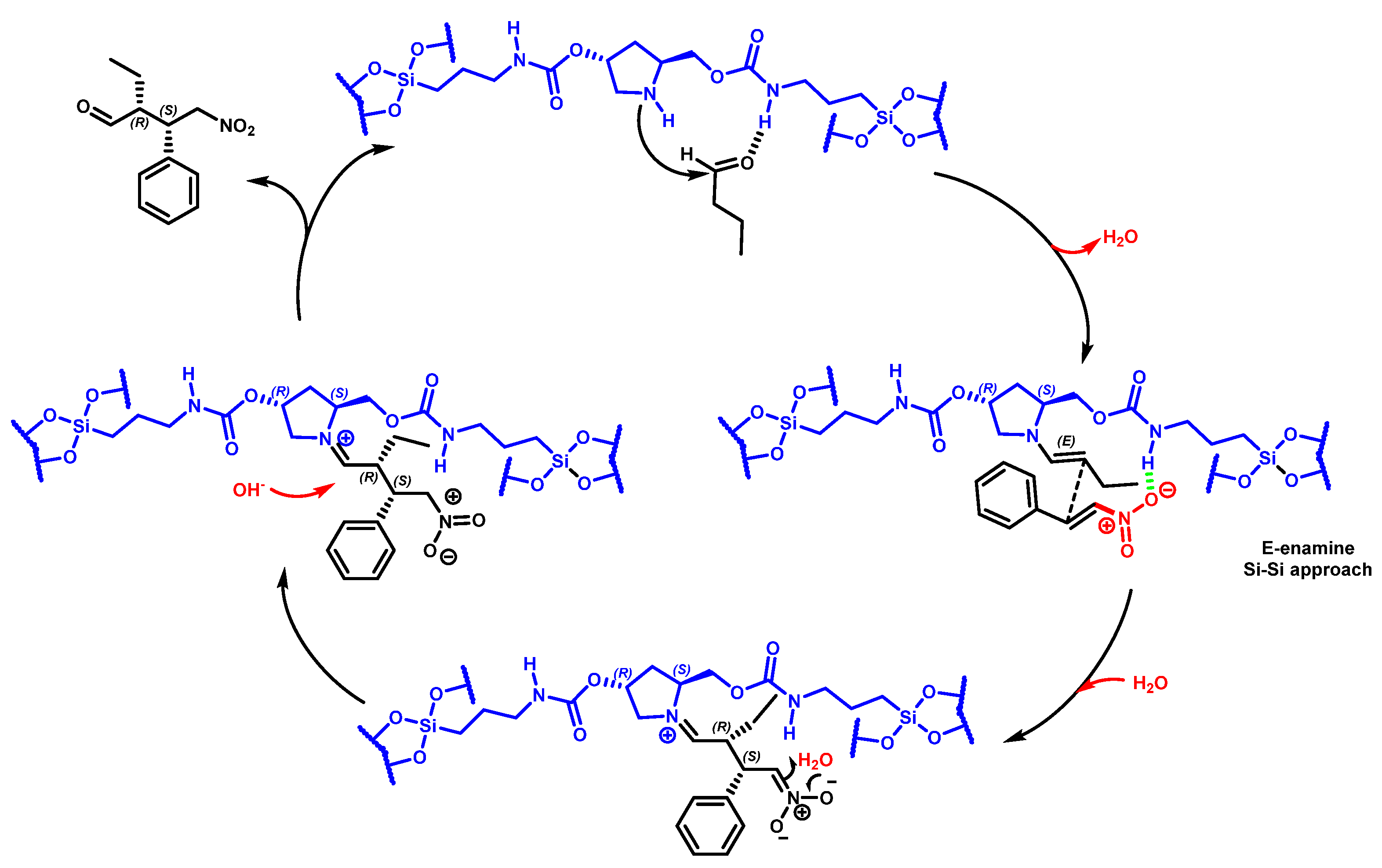
32]. The reaction mechanism widely accepted involves, first, the formation of the enamine via the condensation of a chiral amine and a carbonyl substrate. The following enamine addition onto the nitro alkene compound generates an iminium intermediate. The hydrolysis of the iminium delivers the Michael product with the regeneration of the initial asymmetric amine catalyst. The geometry of enamine is determined by the catalyst architecture, and E-enamine was thermodynamically preferred. Moreover, the chiral substituent on the catalytic solid influences the facial selectivity of the Michael addition via steric shielding and/or electronic interaction. For the Michael addition mechanism, the less-hindered SiSi transition state through an anti-enamine was usually favored for aldehydes, while the ReRe transition state through syn-enamine was usually preferred for ketones. Considering all-these mechanistic insights and the enantioselectivity reaction product, we previously reported a suggested mechanism of chiral amine-catalyzed Michael addition through E-enamine formation in the presence of pyrrolidine-carbamate units embedded in the framework (
Scheme 3) [
33].
The catalytic behavior of the three families of materials with different contents of PyrSil and topology for the chiral Michael addition between butyraldehyde and β-nitrostyrene (
Scheme 4) was studied in optimized reaction conditions, 20 mol% of chiral catalyst at 15 °C in dichloromethane (DCM) [
33]. In all cases, the (2R,3S)-2-methyl-4-nitro-3-phenylbutanal was the main isomer (syn).
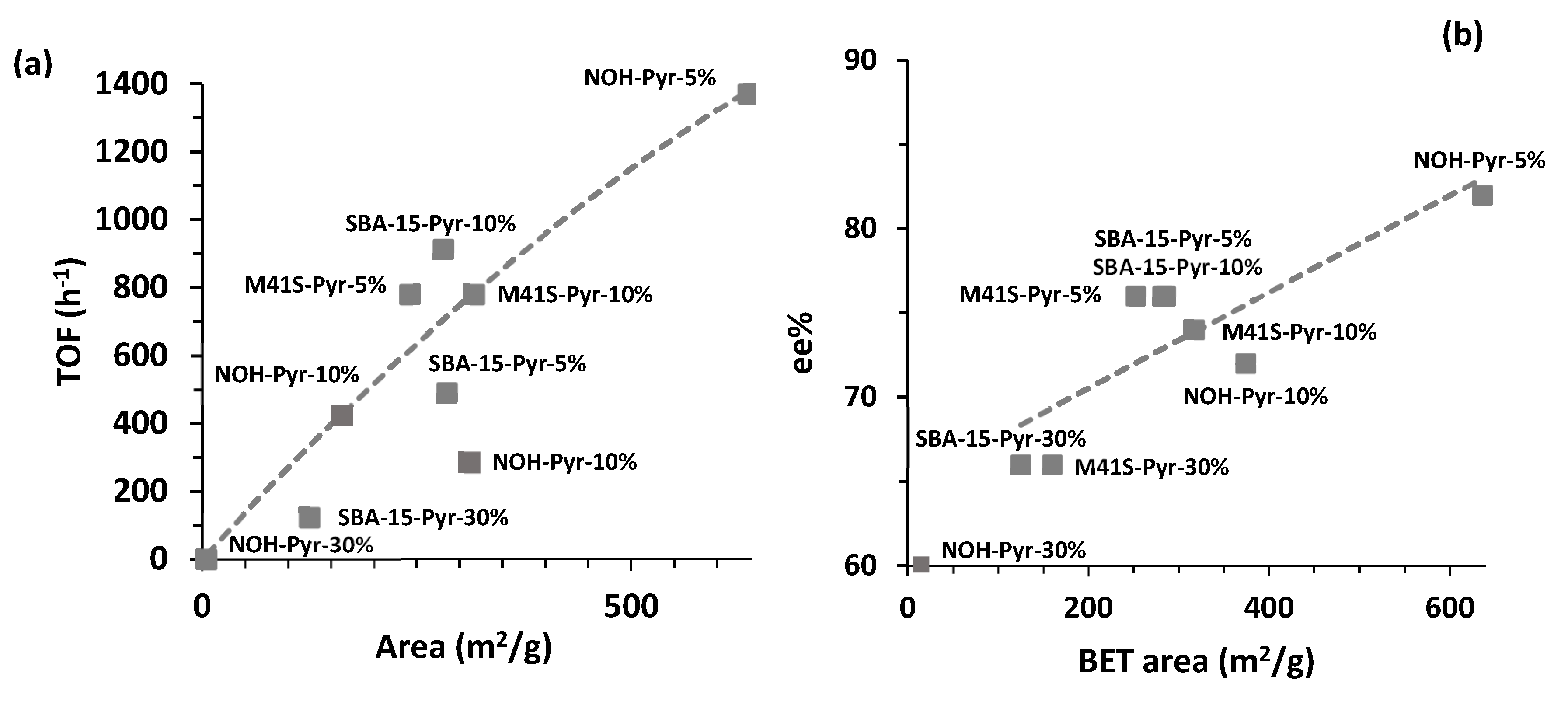
From the nitrogen adsorption isotherms, it was concluded that materials obtained in a fluoride medium without structure-directing agents exhibited isotherms of non-ordered porous materials with high-diameter pores centered at 40 Å. Moreover, it was observed that the BET surface and total volume decreased with an increase of included pyrrolidine units in the structure producing the structural collapse at 30 mol% of PyrSil. The catalytic results reported in
Table 3 and
Figure 8 show that the catalytic activity of the non-ordered porous materials clearly decayed with an increase of PyrSil content due the decrease of BET area, producing diffusion constraints and tended to zero with the collapse of the structure. Likewise, the best results in terms of activity and enantioselectivity were achieved with the NOH-Pyr-5% catalyst, which presented the highest BET area (
Table 3, entry 1), providing a (2R,3S)-2-methyl-4-nitro-3-phenylbutanal yield of 96% and an enantiomeric excess (ee) of 82%.
For hybrid materials obtained in the presence of structure-directing agents, surfactants, and block copolymers with SBA-15 and M41S topologies from the nitrogen adsorption isotherms, it was observed that the specific surface area and the pore volume decreased markedly when the amount of pyrrolidine groups inserted in the framework increased. Then, in both cases, the materials with the highest content of silyl-pyrrolidine fragments exhibited the lowest BET surface area. Moreover, it was notable that the solids obtained with 5 or 10 mol% of PyrSil monomers exhibited similar BET area, and lower than it could be expected, probably due to the incomplete removal of surfactant and block copolymer molecules that partially blocked the pores and accessibility to active chiral centers. SBA-15- and M41S-type materials with 30 mol% of PyrSil fragments showed BET surface areas higher than non-ordered material, probably due to the better-achieved organization because of the presence of structure-directing agents. The catalytic performance of the SBA-15- and M41S-type materials was in agreement with the adsorption results. Indeed, the initial rate and the yield decreased when the BET surface of the materials decreased, i.e., when the accessibility to the active centers was reduced. With these families of materials, the best catalytic performances were obtained in the presence of M41S-Pyr-5% and SBA-15-Pyr-10% with 94% and 99% yield, respectively, and a 76% ee (
Table 3, entries 1 and 5). It is important to notice that ee was lower than in the presence of NOH-Pyr-5% used as a catalyst (
Table 3, entry 1).
These results show that the void shapes and sizes present in the structure of hybrid materials controlled the diffusion of reactants and products, as well as confined the transition states and reactive intermediates. Therefore, the differences in initial reaction rates and enantioselectivity exhibited by the different asymmetric materials was reflective of the differences in the geometry and accessibility to the chiral active center. Likewise, the general trend of the catalytic results obtained in the presence of the different types of mesoporous materials is illustrated in
Figure 9 where turnover frequency (TOF) calculated at 0.5 h was plotted versus BET area. It is noticeable that the catalytic activity of the synthesized hybrid materials was dependent on the BET area and the accessibility to the active centers, as expected. A maximum was observed in the presence of NOH-Pyr-5%, which exhibited a 636 m
2/g BET area. A similar tendency was observed for the enantioselectivity process. In
Figure 9, it is also clearly shown that ee% increased with the accessibility to active sites. In contrast, catalytic data of the homogeneous pyrrolidine precursor (PyrSil) for the Michael addition between butyraldehyde and β-nitrostyrene achieved a 78% ee after 9 h of reaction time with a 96% of Michael adducts yield. This result confirmed that the chiral organosilyl derivative, containing pyrrolidine moieties, was successfully incorporated to the hybrid material without loss of its asymmetric catalytic properties.
Furthermore, the stability and recyclability of the different heterogeneous chiral catalysts was evaluated for four consecutive recycles (
Figure 10 and
Table S2). The reactions were quenched using filtration and the PyrSil hybrid materials were washed with solvent and dried before reuses. The catalytic results in terms of yield and ee for the asymmetric Michael addition of butyraldehyde to β-nitrostyrene showed a slight and constant decrease of the yield with increased uses (2–3% by run). This slow loss of yield was attributed to the strong adsorption of reactant and products that could not be completely removed by washing the catalyst after each run as it can be observed from the
13C NMR spectra (
Figure S7) or by elemental analysis (
Table S3). However, the enantio- and diasteroselectivity of each materials were fully preserved. In contrast,
13C NMR spectra showed the hybrid material’s structure was maintained over the uses, evidencing the robustness of the prepared catalysts.
By considering the catalytic performance of the different families of PyrSil materials and structural properties, NOH-Pyr-5% hybrid material was definitively the best hybrid catalyst to perform asymmetric Michael addition with a good ee.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}