In-situ Quantification of Nanoparticles Oxidation: A Fixed Energy X-ray Absorption Approach

 , ,
, ,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results & Discussion
2.1. Nanoparticles Characterisation
2.2. Design, Realisation and Testing of the Electrochemical Cell
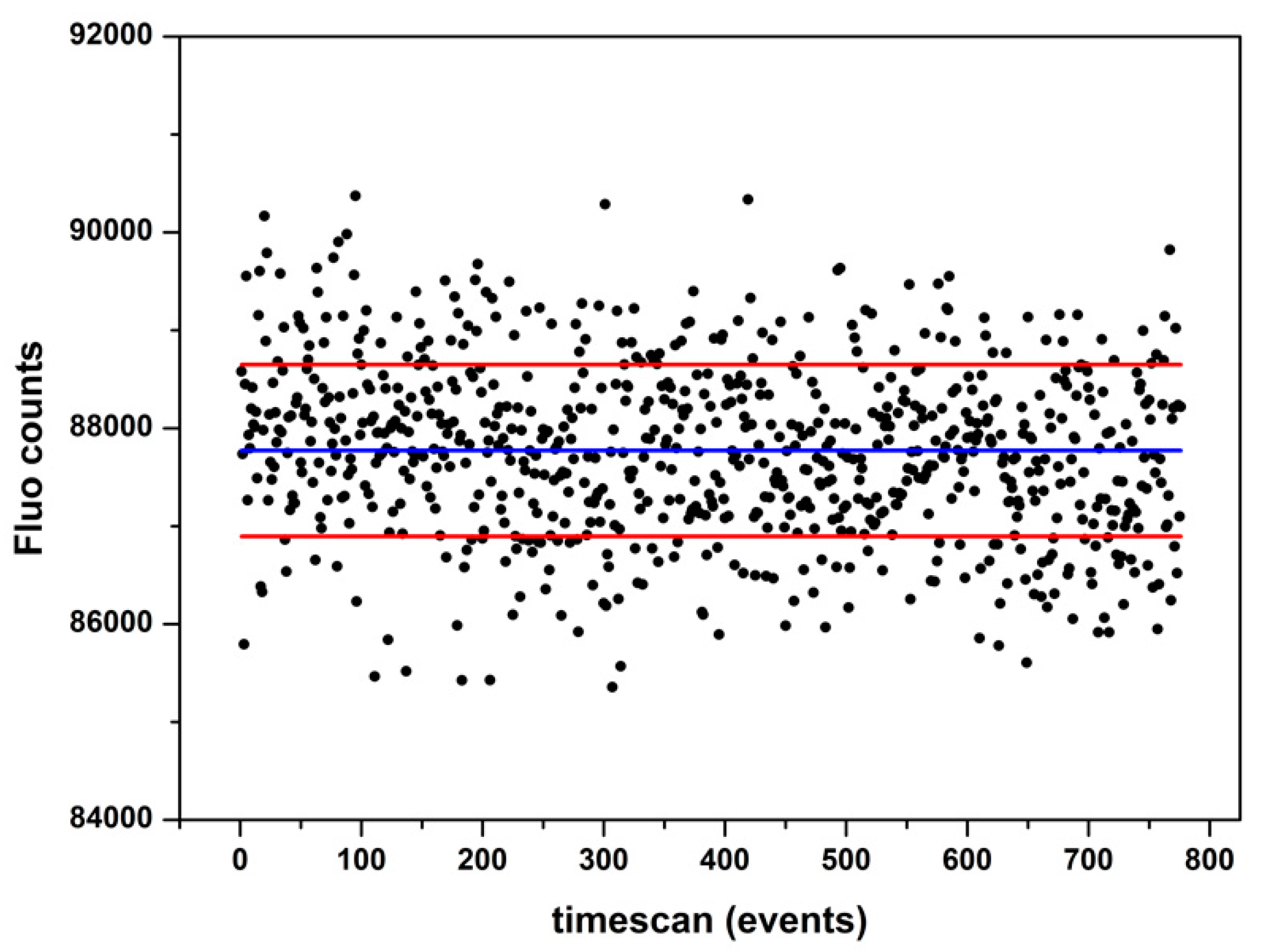
2.3. XAS Analysis
- (1) The normalised spectral intensity at 24,370 eV decreases linearly with the increase of the Pd(II) content (while at 24,350 eV the trend is opposite, with a decrease of the signal with the increase of Pd(II) content), and
- (2) Palladium electrochemical reduction/oxidation occurs through a two-electrons process [53]. Hence, in this context, Palladium speciation varies only between metallic Pd and Pd(II) species (oxides or hydroxypalladiates).
3. Materials and Methods
3.1. Chemicals and Catalyst Preparation
3.2. Beamline Set-Up
3.3. Electrochemical Set-Up
3.4. SEM, TEM and XRD Acquisitions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meirer, F.; Weckhuysen, B.M. Spatial and temporal exploration of heterogeneous catalysts with synchrotron radiation. Nat. Rev. Mater. 2018, 3, 324–340. [Google Scholar] [CrossRef]
- Fabbri, E.; Abbott, D.F.; Nachtegaal, M.; Schmidt, T.J. Operando X-ray absorption spectroscopy: A powerful tool toward water splitting catalyst development. Curr. Opin. Electrochem. 2017, 5, 20–26. [Google Scholar] [CrossRef]
- Frenkel, A.I.; Rodriguez, J.A.; Chen, J.G. Synchrotron techniques for in situ catalytic studies: Capabilities, challenges, and opportunities. ACS Catal. 2012, 2, 2269–2280. [Google Scholar] [CrossRef]
- Fracchia, M.; Ghigna, P.; Vertova, A.; Rondinini, S. Time-Resolved X-ray Absorption Spectroscopy in (Photo) Electrochemistry Time-Resolved X-ray Absorption Spectroscopy in (Photo) Electrochemistry. Surfaces 2018, 1, 138–150. [Google Scholar] [CrossRef]
- Koningsberger, D.C.; Mojet, B.L.; van Dorssen, G.E.; Ramaker, D.E. XAFS spectroscopy; fundamental principles and data analysis. Top. Catal. 2000, 10, 143–155. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, J.; Won, S.O.; Chae, K.; Sharma, S.K.; Kumar, S. Introduction to X-Ray Absorption Spectroscopy and Its Applications in Material Science. In Handbook of Materials Characterization; Sharma, S., Ed.; Springer: Cham, Switzerland, 2018; pp. 497–548. [Google Scholar]
- Niwa, H.; Horiba, K.; Harada, Y.; Oshima, M.; Ikeda, T.; Terakura, K.; ichi Ozaki, J.; Miyata, S. X-ray absorption analysis of nitrogen contribution to oxygen reduction reaction in carbon alloy cathode catalysts for polymer electrolyte fuel cells. J. Power Sources 2009, 187, 93–97. [Google Scholar] [CrossRef]
- Friebel, D.; Miller, D.J.; O’Grady, C.P.; Anniyev, T.; Bargar, J.; Bergmann, U.; Ogasawara, H.; Wikfeldt, K.T.; Pettersson, L.G.M.; Nilsson, A. In situ X-ray probing reveals fingerprints of surface platinum oxide. Phys. Chem. Chem. Phys. 2011, 13, 262–266. [Google Scholar] [CrossRef]
- McBreen, J. The application of synchrotron techniques to the study of lithium-ion batteries. J. Solid State Electrochem. 2009, 13, 1051–1061. [Google Scholar] [CrossRef]
- Erickson, E.M.; Thorum, M.S.; Vasić, R.; Marinković, N.S.; Frenkel, A.I.; Gewirth, A.A.; Nuzzo, R.G. In situ electrochemical X-ray absorption spectroscopy of oxygen reduction electrocatalysis with high oxygen flux. J. Am. Chem. Soc. 2012, 134, 197–200. [Google Scholar] [CrossRef]
- Friebel, D.; Louie, M.W.; Bajdich, M.; Sanwald, K.E.; Cai, Y.; Wise, A.M.; Cheng, M.-J.; Sokaras, D.; Weng, T.-C.; Alonso-Mori, R.; et al. Identification of highly active Fe sites in (Ni,Fe)OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 2015, 137, 1305–1313. [Google Scholar] [CrossRef]
- Zitolo, A.; Ranjbar-Sahraie, N.; Mineva, T.; Li, J.; Jia, Q.; Stamatin, S.; Harrington, G.F.; Lyth, S.M.; Krtil, P.; Mukerjee, S.; et al. Identification of catalytic sites in cobalt-nitrogen-carbon materials for the oxygen reduction reaction. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ginder-Vogel, M.; Landrot, G.; Fischel, J.S.; Sparks, D.L. Quantification of rapid environmental redox processes with quick-scanning x-ray absorption spectroscopy (Q-XAS). Proc. Natl. Acad. Sci. USA 2009, 106, 16124–16128. [Google Scholar] [CrossRef] [Green Version]
- Minguzzi, A.; Lugaresi, O.; Locatelli, C.; Rondinini, S.; D’Acapito, F.; Achilli, E.; Ghigna, P. Fixed energy X-ray absorption voltammetry. Anal. Chem. 2013, 85, 7009–7013. [Google Scholar] [CrossRef] [PubMed]
- Rondinini, S.; Lugaresi, O.; Achilli, E.; Locatelli, C.; Minguzzi, A.; Vertova, A.; Ghigna, P.; Comninellis, C. Fixed Energy X-ray Absorption Voltammetry and Extended X-ray Absorption fine Structure of Ag nanoparticle electrodes. J. Electroanal. Chem. 2016, 766, 71–77. [Google Scholar] [CrossRef]
- Rondinini, S.; Minguzzi, A.; Achilli, E.; Locatelli, C.; Agostini, G.; Pascarelli, S.; Spinolo, G.; Vertova, A.; Ghigna, P. The dynamics of pseudocapacitive phenomena studied by Energy Dispersive X-Ray Absorption Spectroscopy on hydrous iridium oxide electrodes in alkaline media. Electrochim. Acta 2016, 212, 247–253. [Google Scholar] [CrossRef]
- D’acapito Francesco, D.R. De LISA Beamline (BM08-ESRF) Annual Report 2016. Available online: https://www.researchgate.net/publication/313161233_LISA_annual_report_2016 (accessed on 2 June 2019).
- Minguzzi, A.; Ghigna, P.; Rondinini, S.; Chimica, D.; Golgi, V. α- and γ-FeOOH: Stability, Reversibility, and Nature of the Active Phase under Hydrogen Evolution. ACS Appl. Energy Mater. 2018, 1, 1716–1725. [Google Scholar]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915. [Google Scholar] [CrossRef]
- Liang, Z.X.; Zhao, T.S.; Xu, J.B.; Zhu, L.D. Mechanism study of the ethanol oxidation reaction on palladium in alkaline media. Electrochim. Acta 2009, 54, 2203–2208. [Google Scholar] [CrossRef]
- Miller, H.A.; Lavacchi, A.; Vizza, F.; Marelli, M.; Di Benedetto, F.; D’Acapito, F.; Paska, Y.; Page, M.; Dekel, D.R. A Pd/C-CeO2 Anode Catalyst for High-Performance Platinum-Free Anion Exchange Membrane Fuel Cells. Angew. Chem. Int. Ed. Engl. 2016, 55, 6004–6007. [Google Scholar] [CrossRef]
- Tsui, L.K.; Zafferoni, C.; Lavacchi, A.; Innocenti, M.; Vizza, F.; Zangari, G. Electrocatalytic activity and operational stability of electrodeposited Pd-Co films towards ethanol oxidation in alkaline electrolytes. J. Power Sources 2015, 293, 815–822. [Google Scholar] [CrossRef]
- Wang, L.; Bambagioni, V.; Bevilacqua, M.; Bianchini, C.; Filippi, J.; Lavacchi, A.; Marchionni, A.; Vizza, F.; Fang, X.; Shen, P.K. Sodium borohydride as an additive to enhance the performance of direct ethanol fuel cells. J. Power Sources 2010, 195, 8036–8043. [Google Scholar] [CrossRef]
- Bianchini, C.; Shen, P.K. Palladium-Based Electrocatalysts for Alcohol Oxidation in Half Cells and in Direct Alcohol Fuel Cells. Chem. Rev. 2009, 109, 4183–4206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bellini, M.; Bevilacqua, M.; Fornasiero, P.; Lavacchi, A.; Miller, H.A.; Wang, L.; Vizza, F. Direct Alcohol Fuel Cells: Toward the Power Densities of Hydrogen-Fed Proton Exchange Membrane Fuel Cells. ChemSusChem 2014, 8, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.P.R.; Matos, B.R.; Radtke, C.; Santiago, E.I.; Fonseca, F.C.; Amico, S.C.; Malfatti, C.F. Synthesis and performance of palladium-based electrocatalysts in alkaline direct ethanol fuel cell. Int. J. Hydrog. Energy 2016, 41, 6457–6468. [Google Scholar] [CrossRef]
- Chen, Y.X.; Lavacchi, A.; Miller, H.A.; Bevilacqua, M.; Filippi, J.; Innocenti, M.; Marchionni, A.; Oberhauser, W.; Wang, L.; Vizza, F. Nanotechnology makes biomass electrolysis more energy efficient than water electrolysis. Nat. Commun. 2014, 5, 4036. [Google Scholar] [CrossRef] [PubMed]
- Bambagioni, V.; Bianchini, C.; Chen, Y.; Filippi, J.; Fornasiero, P.; Innocenti, M.; Lavacchi, A.; Marchionni, A.; Oberhauser, W.; Vizza, F. Energy Efficiency Enhancement of Ethanol Electrooxidation on Pd–CeO2/C in Passive and Active Polymer Electrolyte-Membrane Fuel Cells. ChemSusChem 2012, 5, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Grdeń, M.; Łukaszewski, M.; Jerkiewicz, G.; Czerwiński, A. Electrochemical behaviour of palladium electrode: Oxidation, electrodissolution and ionic adsorption. Electrochim. Acta 2008, 53, 7583–7598. [Google Scholar] [CrossRef]
- Wang, L.; Lavacchi, A.; Bellini, M.; D’Acapito, F.; Di Benedetto, F.; Innocenti, M.; Miller, H.A.; Montegrossi, G.; Zafferoni, C.; Vizza, F. Deactivation of Palladium Electrocatalysts for Alcohols Oxidation in Basic Electrolytes. Electrochim. Acta 2015, 177, 100–106. [Google Scholar] [CrossRef]
- Montegrossi, G.; Giaccherini, A.; Berretti, E.; Di Benedetto, F.; Innocenti, M.; D’Acapito, F.; Lavacchi, A. Computational speciation models: A tool for the interpretation of spectroelectrochemistry for catalytic layers under operative conditions. J. Electrochem. Soc. 2017, 164, 3690–3695. [Google Scholar] [CrossRef]
- Grdeń, M. Electrochemical quartz crystal microbalance studies of a palladium electrode oxidation in a basic electrolyte solution. Electrochim. Acta 2009, 54, 909–920. [Google Scholar] [CrossRef]
- Grdeń, M.; Kotowski, J.; Czerwiński, A. The study of electrochemical palladium behavior using the quartz crystal microbalance. J. Solid State Electrochem. 2000, 4, 273–278. [Google Scholar] [CrossRef]
- Ayyappan, S.; Srinivasa Gopalan, R.; Subbanna, G.N.; Rao, C.N.R. Nanoparticles of Ag, Au, Pd, and Cu produced by alcohol reduction of the salts. J. Mater. Res. 1997, 12, 398–401. [Google Scholar] [CrossRef]
- Nguyen, V.L.; Nguyen, D.C.; Hirata, H.; Ohtaki, M.; Hayakawa, T.; Nogami, M. Chemical synthesis and characterization of palladium nanoparticles. Adv. Nat. Sci. Nanosci. Nanotechnol. 2010, 1, 035012. [Google Scholar] [CrossRef]
- Asset, T.; Serov, A.; Padilla, M.; Roy, A.J.; Matanovic, I.; Chatenet, M.; Maillard, F.; Atanassov, P. Design of Pd-Pb Catalysts for Glycerol and Ethylene Glycol Electrooxidation in Alkaline Medium. Electrocatalysis 2018, 9, 480–485. [Google Scholar] [CrossRef]
- Lenarda, A.; Bellini, M.; Marchionni, A.; Miller, H.A.; Montini, T.; Melchionna, M.; Vizza, F.; Prato, M.; Fornasiero, P. Nanostructured carbon supported Pd-ceria as anode catalysts for anion exchange membrane fuel cells fed with polyalcohols. Inorg. Chim. Acta 2018, 470, 213–220. [Google Scholar] [CrossRef]
- Marchionni, A.; Bevilacqua, M.; Bianchini, C.; Chen, Y.-X.; Filippi, J.; Fornasiero, P.; Lavacchi, A.; Miller, H.; Wang, L.; Vizza, F. Electrooxidation of Ethylene Glycol and Glycerol on Pd-(Ni-Zn)/C Anodes in Direct Alcohol Fuel Cells. ChemSusChem 2013, 6, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.; Baranton, S.; Coutanceau, C. Electro-oxidation of glycerol at Pd based nano-catalysts for an application in alkaline fuel cells for chemicals and energy cogeneration. Appl. Catal. B Environ. 2010, 93, 354–362. [Google Scholar] [CrossRef]
- Garcia, A.C.; Birdja, Y.Y.; Tremiliosi-Filho, G.; Koper, M.T.M. Glycerol electro-oxidation on bismuth-modified platinum single crystals. J. Catal. 2017, 346, 117–124. [Google Scholar] [CrossRef]
- Yasuhiro, I.; Kiyotaka, A.; Mizuki, T. (Eds.) “Reflection XAFS”. In XAFS Techniques for Catalysts, Nanomaterials, and Surfaces; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic Properties of Palladium. Platin. Met. Rev. 2013, 57, 127–136. [Google Scholar] [CrossRef]
- Achilli, E.; Minguzzi, A.; Visibile, A.; Locatelli, C. short communications 3D-printed photo-spectroelectrochemical devices for in situ and in operando X-ray absorption spectroscopy investigation. J. Synchrotron. Radiat. 2016, 23, 622–628. [Google Scholar] [CrossRef]
- Henke, B.L.; Gullikson, E.M.; Davis, J.C. X-Ray Interactions: Photoabsorption, Scattering, Transmission, and Reflection at E = 50-30,000 eV, Z = 1-92. At. Data Nucl. Data Tables 1993, 54, 181–342. [Google Scholar] [CrossRef] [Green Version]
- Grden, M.; Czerwinski, A. EQCM studies on Pd—Ni alloy oxidation in basic solution. J. Solid State Electrochem. 2008, 12, 375–385. [Google Scholar] [CrossRef]
- Cookson, B.J. The Preparation of Palladium Nanoparticles. Plat. Met. Rev. 2012, 56, 83–98. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, J.; Kim, D.H. Investigation on the enhanced catalytic activity of a Ni-promoted Pd/C catalyst for formic acid dehydrogenation: Effects of preparation methods and Ni/Pd ratios. RSC Adv. 2018, 8, 2441–2448. [Google Scholar] [CrossRef]
- Waser, J.; Levy, H.A.; Peterson, S.W. The structure of PdO. Acta Crystallogr. 1953, 6, 661–663. [Google Scholar] [CrossRef]
- Moore, W.J.; Pauling, L. The Crystal Structures of the Tetragonal Monoxides of Lead, Tin, Palladium, and Platinum. J. Am. Chem. Soc. 1941, 63, 1392–1394. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: Amsterdam, The Netherlands, 1997; ISBN 9780750633659. [Google Scholar]
- Torapava, N.; Elding, L.I.; Mändar, H.; Roosalu, K.; Persson, I. Structures of polynuclear complexes of palladium(ii) and platinum(ii) formed by slow hydrolysis in acidic aqueous solution. Dalton Trans. 2013, 42, 7755–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, F.; Veronesi, G.; Capella, S.; Bellis, D.; Charlet, L.; Cedola, A.; Belluso, E. New insights on the biomineralisation process developing in human lungs around inhaled asbestos fibres. Sci. Rep. 2017, 7, 44862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, W.P.; Robinson, S.D.; Swars, K. Pd Palladium: Palladium Compounds; Griffith, W.P., Swars, K., Eds.; Springer: Berlin/Heidelberg, Germany, 1989; ISBN 978-3-662-09190-6. [Google Scholar]
- Vayenas, C.G.; White, R.E. Modern Aspects of Electrochemistry 51; Springer International Publishing: Basel, Switzerland, 2011. [Google Scholar]
- d’Acapito, F.; Lepore, G.O.; Puri, A.; Laloni, A.; La Mannna, F.; Dettona, E.; De Luisa, A.; Martin, A. The LISA beamline at ESRF. J. Synchrotron. Radiat. 2019, 26, 551–558. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Giaccherini, A.; Cinotti, S.; Guerri, A.; Carlà, F.; Montegrossi, G.; Vizza, F.; Lavacchi, A.; Felici, R.; Di Benedetto, F.; Innocenti, M. Operando SXRD study of the structure and growth process of Cu2S ultra-thin films. Sci. Rep. 2017, 7, 1615. [Google Scholar] [CrossRef] [PubMed]
- Giaccherini, A.; Russo, F.; Carlà, F.; Guerri, A.; Picca, R.A.; Cioffi, N.; Cinotti, S.; Montegrossi, G.; Passaponti, M.; Di Benedetto, F.; et al. Operando SXRD of E-ALD deposited sulphides ultra-thin films: Crystallite strain and size. Appl. Surf. Sci. 2018, 432, 53–59. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berretti, E.; Giaccherini, A.; Montegrossi, G.; D’Acapito, F.; Di Benedetto, F.; Zafferoni, C.; Puri, A.; Lepore, G.O.; Miller, H.; Giurlani, W.; et al. In-situ Quantification of Nanoparticles Oxidation: A Fixed Energy X-ray Absorption Approach. Catalysts 2019, 9, 659. https://doi.org/10.3390/catal9080659
Berretti E, Giaccherini A, Montegrossi G, D’Acapito F, Di Benedetto F, Zafferoni C, Puri A, Lepore GO, Miller H, Giurlani W, et al. In-situ Quantification of Nanoparticles Oxidation: A Fixed Energy X-ray Absorption Approach. Catalysts. 2019; 9(8):659. https://doi.org/10.3390/catal9080659
Chicago/Turabian StyleBerretti, Enrico, Andrea Giaccherini, Giordano Montegrossi, Francesco D’Acapito, Francesco Di Benedetto, Claudio Zafferoni, Alessandro Puri, Giovanni Orazio Lepore, Hamish Miller, Walter Giurlani, and et al. 2019. "In-situ Quantification of Nanoparticles Oxidation: A Fixed Energy X-ray Absorption Approach" Catalysts 9, no. 8: 659. https://doi.org/10.3390/catal9080659
APA StyleBerretti, E., Giaccherini, A., Montegrossi, G., D’Acapito, F., Di Benedetto, F., Zafferoni, C., Puri, A., Lepore, G. O., Miller, H., Giurlani, W., Innocenti, M., Vizza, F., & Lavacchi, A. (2019). In-situ Quantification of Nanoparticles Oxidation: A Fixed Energy X-ray Absorption Approach. Catalysts, 9(8), 659. https://doi.org/10.3390/catal9080659