Pharmaceutical Cocrystal Development of TAK-020 with Enhanced Oral Absorption

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cocrystal Screening
2.2.1. Conventional Reaction Crystallization (CRC)
2.2.2. Cocktail Cocrystal Grinding (CCG)
2.2.3. Saturated CCF Solution Slurry (SCS)
2.3. Cocrystal Powder Preparation
2.4. Powder X-Ray Diffractometry (PXRD)
2.5. Thermal Analysis
2.6. Dissolution Test
2.7. Intrinsic Dissolution Rate (IDR)
2.8. In Vitro Dissolution-Permeation Study
2.9. HPLC
2.10. NMR Spectroscopy
2.11. Single-Crystal X-Ray Diffraction (SCXRD)
2.12. Formulation Preparation for PK Study
2.13. In Vivo PK Study
2.14. LC−MS/MS
2.15. Statistics
3. Results and Discussion
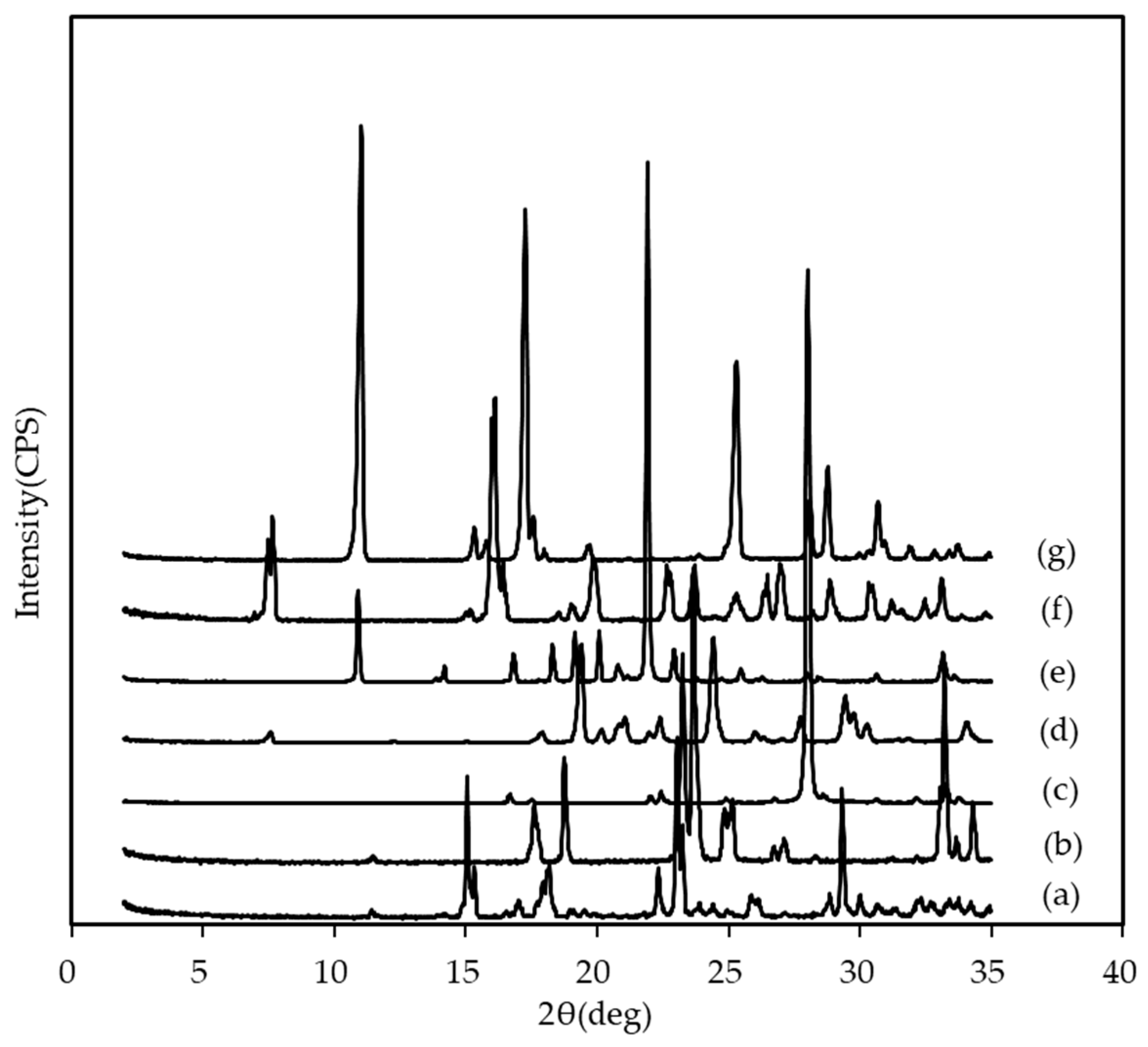
3.1. Preparation of TAK-020 Cocrystals with Three Types of Screening
3.2. In Vitro Dissolution Performance for Selecting Cocrystal Form
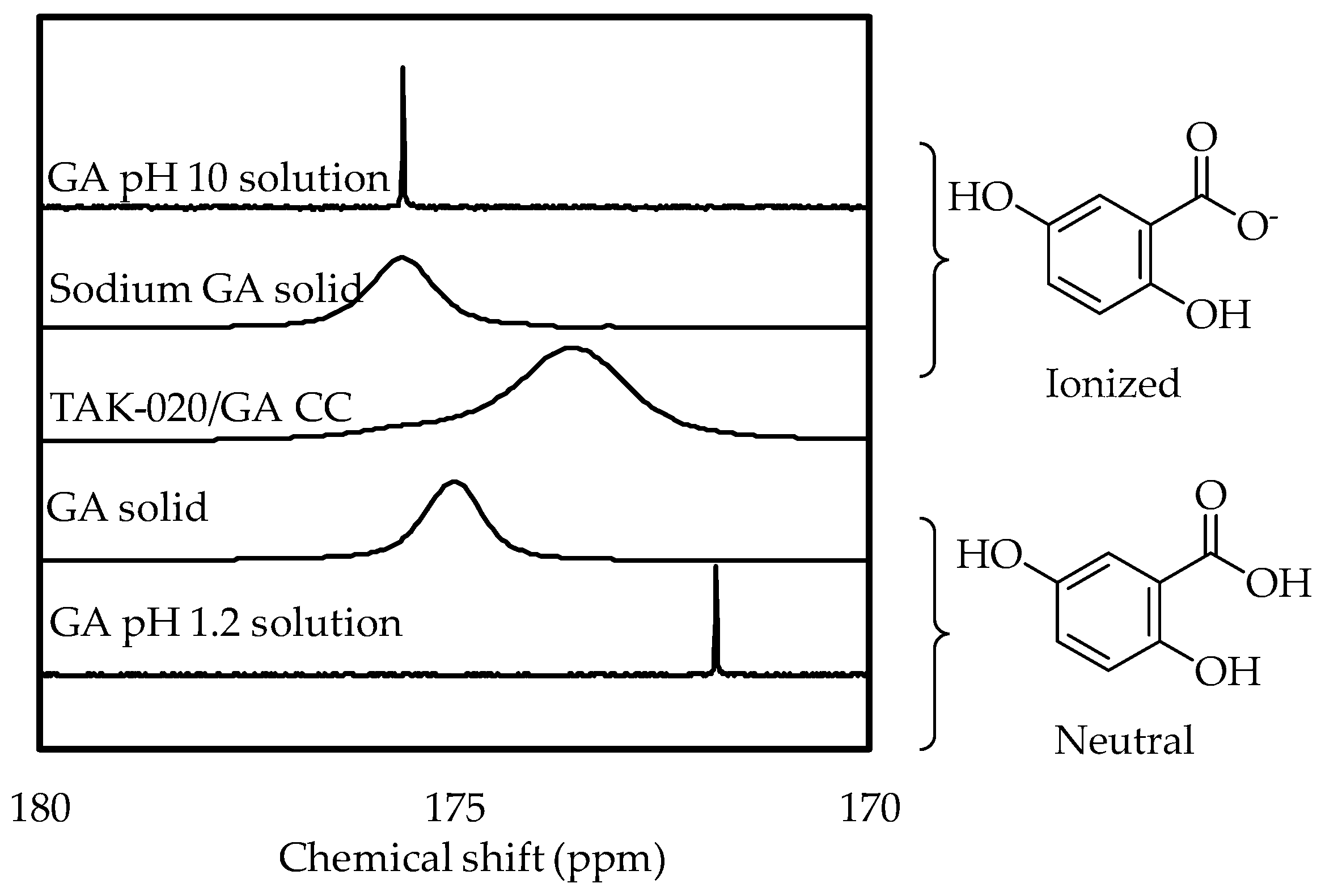
3.3. Certification of the Cocrystal Definition
3.4. PK Profiling for Determining the Optimal Drug Form
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Nakashima, S.; Yamamoto, K.; Arai, Y.; Ikeda, Y. Impact of physicochemical profiling for rational approach on drug discovery. Chem. Pharm. Bull. 2013, 61, 1228–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Vinod, P.; Sunil, P.; Sonali, P.; Tejaswini, P.; Sachinkumar, P.; Shitalkumar, P. Solubility enhancement of poorly soluble drugs by using novel techniques: A review. J. Curr. Pharm. Res. 2014, 4, 1258–1270. [Google Scholar]
- Blagden, N.; Matas, M.D.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug. Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Prohotsky, D.; Zhao, F. A survey of top 200 drugs inconsistent practice of drug strength expression for drugs containing salt forms. J. Pharm. Sci. 2012, 101, 1–6. [Google Scholar] [CrossRef]
- Fridgeirsdottir, G.A.; Harris, R.; Fischer, P.M.; Roberts, C.J. Support tools in formulation development for poorly soluble drugs. J. Pharm. Sci. 2016, 105, 2260–2269. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, Q.; Wang, J.; Mei, X. Cocrystals of baicalein with higher solubility and enhanced bioavailability. Cryst. Growth Des. 2017, 17, 1893–1901. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kojima, T.; Karashima, M.; Ikeda, Y. Physicochemical evaluation and developability assessment of coamorphouses of low soluble drugs and comparison to the co-crystals. Chem. Pharm. Bull. 2016, 64, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Karashima, M.; Kimoto, K.; Yamamoto, K.; Kojima, T.; Ikeda, Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur. J. Pharm. Sci. 2016, 107, 142–150. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. Cryst. Eng. Comm. 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Schlutheiss, N.; Newman, A. Pharmaceutical cocrystals and their physicochemical properties. Cryst. Growth. Des. 2009, 9, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, D.J.; Rodríguez-Hornedo, N. Solubility advantage of pharmaceutical cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- ter Horst, J.H.; Deij, M.A.; Cains, P.W. Discovering new co-crystals. Cryst. Growth Des. 2009, 9, 1531–1537. [Google Scholar] [CrossRef]
- Malamatari, M.; Ross, S.A.; Douroumis, D.; Velaga, S.P. Experimental cocrystal screening and solution based scale-up cocrystallization methods. Adv. Drug. Deliv. Rev. 2017, 117, 162–177. [Google Scholar] [CrossRef]
- Leung, D.H.; Lohani, S.; Ball, R.G.; Canfield, N.; Wang, Y.; Rhodes, T.; Bak, A. Two novel pharmaceutical cocrystals of a development compound –screening, scale-up, and characterization. Cryst. Growth Des. 2012, 12, 1254–1262. [Google Scholar] [CrossRef]
- Meanwell, N.A. The emerging utility of co-crystals in drug discovery and development. Annu. Rep. Med. Chem. 2008, 43, 373–401. [Google Scholar]
- Kojima, T.; Tsutsumi, S.; Yamamoto, K.; Ikeda, Y.; Moriwaki, T. High-throughput cocrystal slurry screening by use of in situ Raman microscopy. Int. J. Pharm 2010, 399, 52–59. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tsutsumi, S.; Ikeda, Y. Establishment of cocrystal cocktail grinding method for rational screening of pharmaceutical cocrystals. Int. J. Pharm 2012, 437, 162–171. [Google Scholar] [CrossRef]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Teramura, T.; Terada, K. Detection of cocrystal formation based on binary phase diagrams using thermal analysis. Pharm. Res. 2013, 30, 70–80. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Regulatory classification of pharmaceutical co-crystals guidance for industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/regulatory-classification-pharmaceutical-co-crystals (accessed on 27 January 2020).
- European Medicines Agency. Reflection paper on the use of cocrystals and other solid state forms of active substances in medicinal products. Available online: https://www.ema.europa.eu/en/use-cocrystals-active-substances-medicinal-products#current-version-section (accessed on 27 January 2020).
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical cocrystals: Regulatory and strategic aspects, design and development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.; Koida, T.; Takata, N.; Ikeda, Y.; Ono, M.; Inoue, M.; Fukami, T.; Yonemochi, E. Characterization and quality control of pharmaceutical cocrystals. Chem. Pharm. Bull. 2016, 64, 1421–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, D.P.; Zode, S.S.; Bansal, A.K. Challenges in translational development of pharmaceutical cocrystals. J. Pharm. Sci. 2017, 106, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.D.; Sabat, M.; Scorah, N.; Smith, C.; Vu, P.H. Pyridinyl and fused pyridinyl triazolone derivatives. WO 2014/164558 A1, 9 October 2014. [Google Scholar]
- Stella, J.S.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Stahl, P.H.; Wermuth, C.G. Handbook of pharmaceutical salts properties, selection, and use. Int. Union Pure Appl. Chem. 2002, 24, 21. [Google Scholar]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Iida, M.; Tada, N.; Kojima, T.; Ikeda, Y.; Moriwaki, T.; Higashi, K.; Moribe, K.; Yamamoto, K. Characterization and evaluation of miconazole salts and cocrystals for improved physicochemical properties. Int. J. Pharm. 2011, 421, 230–236. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELXL. Acta Crystallogr. Sect. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, K.; Yamamoto, M.; Kitayama, M.; Sawai, Y.; Hohokabe, M.; Iwata, K. Cocrystal, Production Method Thereof, and Medicament Containing Cocrystal. WO 2017/111179 A1, 29 June 2017. [Google Scholar]
- Alhalaweh, A.; Ali, R.H.H.; Velaga, S.P. Effects of polymer and surfactant on the dissolution and transformation profiles of cocrystals in aqueous media. Cryst. Growth Des. 2014, 14, 643–648. [Google Scholar] [CrossRef]
- Saal, C.; Becker, A. Pharmaceutical salts: A summary on doses of salt formers from the Orange Book. Eur. J. Pharm. Sci. 2013, 49, 614–623. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine, U.S. Department of Health & Human Services. Available online: https://www.nlm.nih.gov/toxnet/index.html (accessed on 27 January 2020).
- Paterson, J.R.; Blacklock, C.; Campbell, G.; Wiles, D.; Lawrence, J.R. The identification of salicylates as normal constituents of serum: A link between diet and health? J. Clin. Pathol. 1998, 51, 502–505. [Google Scholar] [CrossRef] [Green Version]
- Boreham, D.R.; Martin, B.K. The kinetics of elimination of salicylic acid and the formation of gentisic acid. Br. J. Pharmac. 1969, 37, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Needs, C.J.; Brooks, P.M. Clinical pharmacokinetics of the salicylates. Clin. Pharmacokinet. 1985, 10, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Ashidate, K.; Kawamura, M.; Mimura, D.; Tohda, H.; Miyazaki, S.; Teramoto, T.; Yamamoto, Y.; Hirata, Y. Gentisic acid, an aspirin metabolite, inhibits oxidation of low-density lipoprotein and the formation of cholesterol ester hydroperoxides in human plasma. Eur. J. Pharmacol. 2005, 513, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Tehama, B.G.; Lloyd, E.J.; Wong, M.G.; Pitt, W.R.; Montana, J.G.; Manallack, S.T.; Gancia, E. Estimation of pKa using semi-empirical molecular orbital methods. part 1: Application to phenols and carboxylic acids. Quant. Struct. Act. Relat. 2002, 21, 457–472. [Google Scholar] [CrossRef]
- Ando, S.; Kikuchi, J.; Fujimura, Y.; Ida, Y.; Higashi, K.; Moribe, K.; Yamamoto, K. Physicochemical characterization and structural evaluation of a specific 2:1 cocrystal of naproxen–nicotinamide. J. Pharm. Sci. 2012, 101, 3214–3221. [Google Scholar] [CrossRef]
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.H. 1H/15N NMR chemical shielding, dipolar 15N, 2Hcoupling and hydrogen bond geometry correlations in a novel series of hydrogen-bonded acid–base complexes of collidine with carboxylic acids. Magn. Reson. Chem. 2001, 39, S18–S29. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.E.; Desper, J. Cocrystal or salt: Does it really matter? Mol. Pharm. 2007, 4, 317–322. [Google Scholar] [CrossRef]
- O’Driscoll, C.M. Lipid-based formulations for intestinal lymphatic delivery. Eur. J. Pharm. Sci. 2002, 15, 405–415. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCF | CRC | CCG | SCS | CCF | CRC | CCG | SCS |
|---|---|---|---|---|---|---|---|
| Citric acid (CA) | - | ✓ | ✓ | Lactamide | - | ||
| Fumaric acid | - | - | - | Glycolamide | - | ||
| Glutaric acid (GA) | - | Tromethamine | - | - | |||
| Malonic acid (MoA) | - | ✓ | ✓ | Meglumine | - | - | - |
| Succinic acid | - | - | - | Glycine | (-) | ||
| Maleic acid (MeA) | - | ✓ | ✓ | l-Arginine | - | (-) | - |
| l-Malic acid (MaA) | ✓ | ✓ | l-Lysine | - | (-) | - | |
| l-Tartaric acid | - | - | - | l-Proline | (-) | ||
| Mandelic acid | ✓ | l-Tryptophan | (-) | ||||
| Benzoic acid | - | - | - | Phosphoric acid | - | ||
| Gentisic acid (GA) | - | ✓ | ✓ | l-Ascorbic acid | - | ||
| Salicylic acid (SA) | - | ✓ | ✓ | Urea | - | - | - |
| Hippuric acid | - | - | - | Saccharin | - | - | - |
| Nicotinamide | - | - | - | Ethylmaltol | - | - | |
| Benzamide | - |
| CCF Group | CCF Name | Molecular Weight (Da) | H-Acceptor | H-Donor | pKa |
|---|---|---|---|---|---|
| Tri-carboxylic acid | Citric acid | 192 | 7 | 4 | 3.1, 4.8, 6.4 |
| Di-carboxylic acid | Malonic acid | 104 | 2 | 4 | 2.8, 5.7 |
| Maleic acid | 116 | 4 | 2 | 1.9, 6.2 | |
| l-Malic acid | 134 | 5 | 3 | 3.5, 5.1 | |
| Aromatic and carboxylic acid | Mandelic acid | 152 | 3 | 2 | 3.4 |
| Gentisic acid | 154 | 4 | 3 | 2.9 | |
| Salicylic acid | 138 | 3 | 2 | 3.0, 13.8 |
| Dissolved Amount as TAK-020 (μg/mL) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Media | JP1 (pH 1.2) | JP2 (pH 6.8) | FaSSIF (pH 6.5) | FeSSIF (pH 5.0) | ||||||||
| TAK-020 | 1.6 | ± | 0.0 | 1.7 | ± | 0.1 | 3.0 | ± | 0.9 | 10.4 | ± | 0.2 |
| TAK-020/CA CC | 1.9 | ± | 0.1 | 2.2 | ± | 0.0 | 7.3 | ± | 0.3** | 30.4 | ± | 0.4** |
| TAK-020/MoA CC | 7.2 | ± | 1.0** | 15.9 | ± | 1.2** | 10.0 | ± | 0.3** | 36.0 | ± | 5.0** |
| TAK-020/MeA CC | 2.4 | ± | 0.4 | 5.7 | ± | 0.2** | 9.5 | ± | 0.9** | 25.9 | ± | 0.4** |
| TAK-020/MaA CC | 2.1 | ± | 0.0 | 5.6 | ± | 0.4** | 8.6 | ± | 0.2** | 29.2 | ± | 1.8** |
| TAK-020/MdA CC | 2.4 | ± | 0.3 | 5.0 | ± | 0.4** | 8.1 | ± | 0.3** | 27.3 | ± | 0.7** |
| TAK-020/GA CC | 7.4 | ± | 0.3** | 2.8 | ± | 0.6 | 9.4 | ± | 0.4** | 27.1 | ± | 1.2** |
| TAK-020/SA CC | 9.3 | ± | 1.0** | 3.8 | ± | 0.6* | 17.8 | ± | 0.9** | 21.1 | ± | 1.1** |
| Molecular Formula | C25H23N5O7 |
|---|---|
| Molecular weight | 505.49 |
| Temperature (K) | 100 |
| Crystal dimensions (mm) | 0.050 × 0.010 × 0.002 |
| Crystal system | Monoclinic |
| Space group | P21 (#4) |
| a (Å) | 13.6572 |
| b (Å) | 19.8263 |
| c (Å) | 8.9035 |
| α (°) | (90) |
| β (°) | 105.883 |
| γ (°) | (90) |
| V (Å3) | 2318.8 |
| Z | 4 |
| Calculated density (g/cm3) | 1.448 |
| R (I > 2.00 σ (I)) | 0.0905 |
| Rw | 0.2649 |
| Formulation | Dose (mg) | Tmax (h) | Cmax (ng/mL) | AUC0–24 h (ng·h/mL) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IR tablet | 300 | 1.6 | ± | 0.5 | 4.7 | ± | 1.0 | 30.2 | ± | 14.5 |
| TAK-020/GA CC | 100 | 1.3 | ± | 0.7 | 229.0 | ± | 106.5** | 585.6 | ± | 231.5** |
| ASD tablet | 100 | 0.4 | ± | 0.1 | 51.5 | ± | 29.8 | 130.2 | ± | 88.3 |
| Nanocry suspension | 100 | 1.4 | ± | 0.5 | 7.7 | ± | 1.8 | 59.6 | ± | 31.8 |
| LBF | 100 | 5.6 | ± | 10.3 | 8.3 | ± | 2.0 | 52.6 | ± | 20.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimoto, K.; Yamamoto, M.; Karashima, M.; Hohokabe, M.; Takeda, J.; Yamamoto, K.; Ikeda, Y. Pharmaceutical Cocrystal Development of TAK-020 with Enhanced Oral Absorption. Crystals 2020, 10, 211. https://doi.org/10.3390/cryst10030211
Kimoto K, Yamamoto M, Karashima M, Hohokabe M, Takeda J, Yamamoto K, Ikeda Y. Pharmaceutical Cocrystal Development of TAK-020 with Enhanced Oral Absorption. Crystals. 2020; 10(3):211. https://doi.org/10.3390/cryst10030211
Chicago/Turabian StyleKimoto, Kouya, Mitsuo Yamamoto, Masatoshi Karashima, Miyuki Hohokabe, Junpei Takeda, Katsuhiko Yamamoto, and Yukihiro Ikeda. 2020. "Pharmaceutical Cocrystal Development of TAK-020 with Enhanced Oral Absorption" Crystals 10, no. 3: 211. https://doi.org/10.3390/cryst10030211
APA StyleKimoto, K., Yamamoto, M., Karashima, M., Hohokabe, M., Takeda, J., Yamamoto, K., & Ikeda, Y. (2020). Pharmaceutical Cocrystal Development of TAK-020 with Enhanced Oral Absorption. Crystals, 10(3), 211. https://doi.org/10.3390/cryst10030211