
The Face-to-Face σ-Hole⋯σ-Hole Stacking Interactions: Structures, Energies, and Nature


Abstract
:
1. Introduction
2. Computational Details
3. Results and Discussion
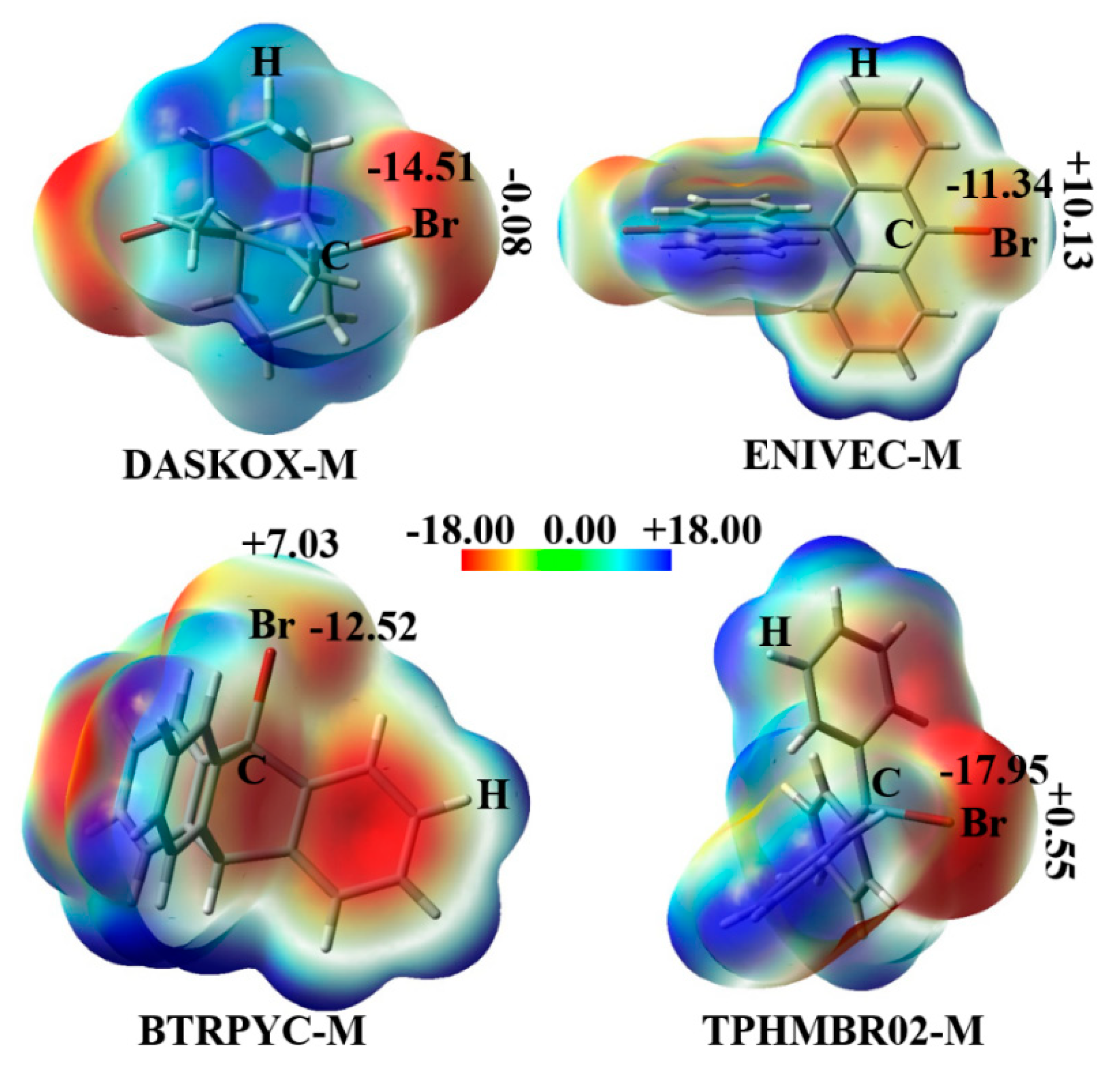
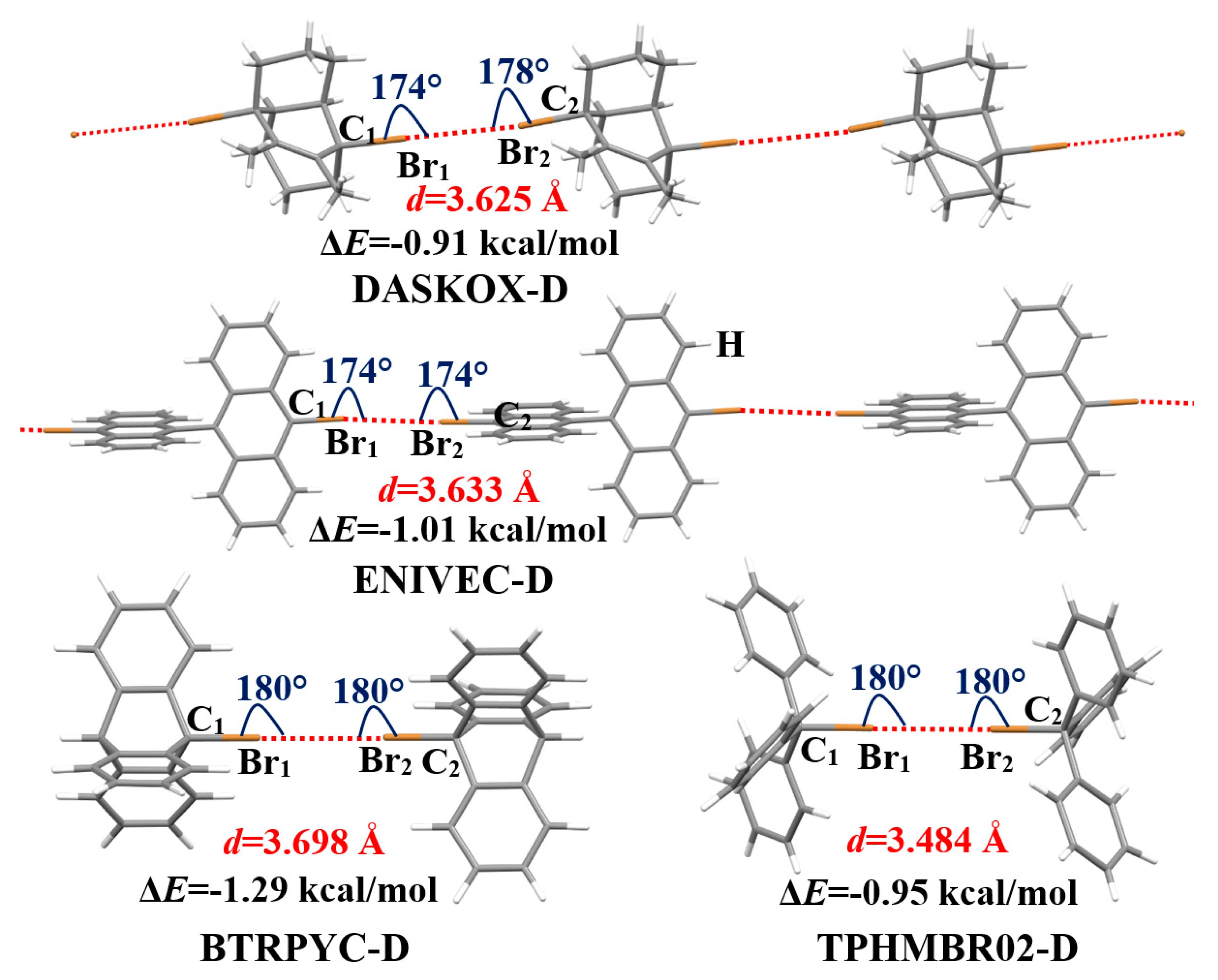
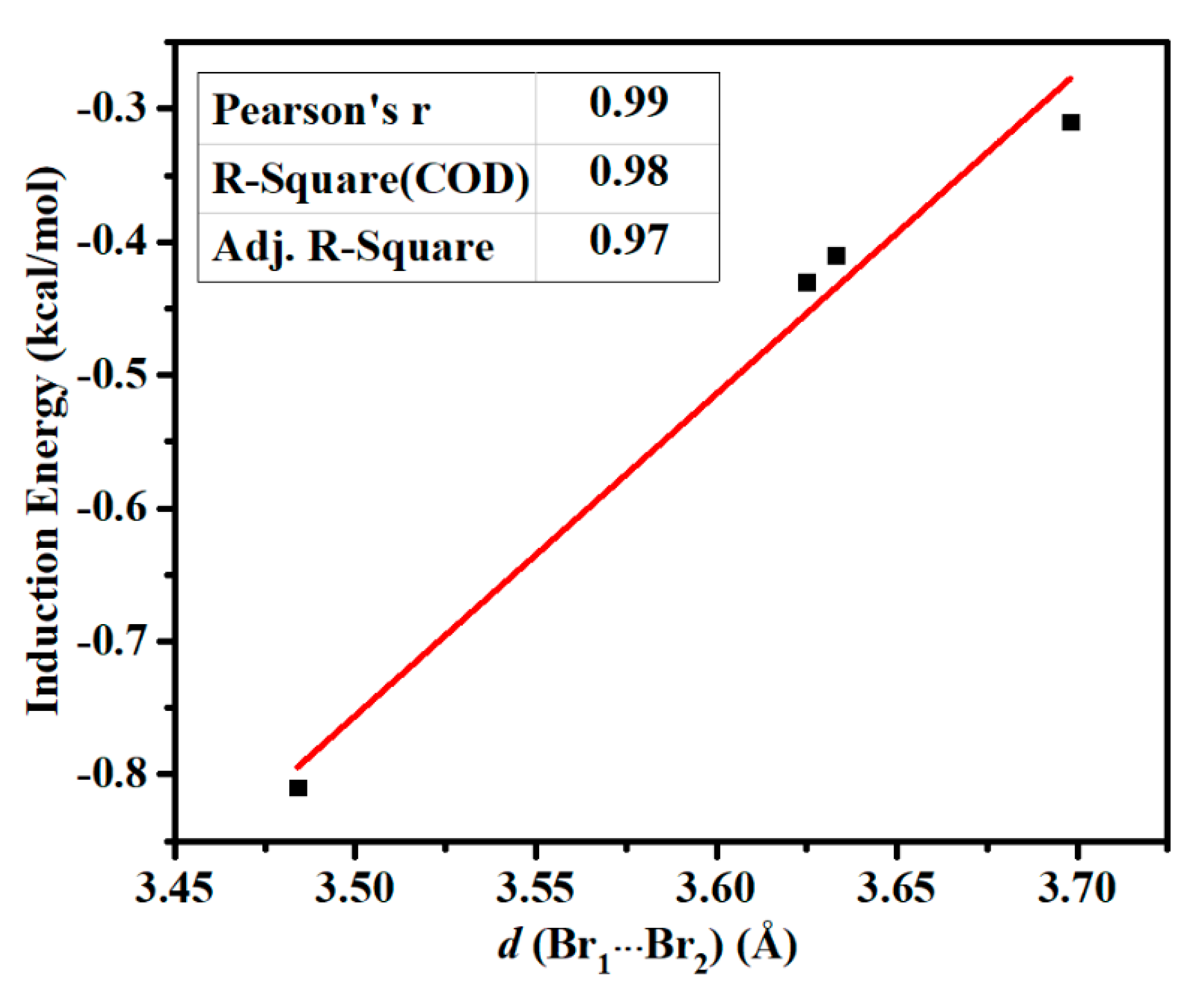
3.1. Structures and Interaction Energies
3.2. Energy Component Analysis
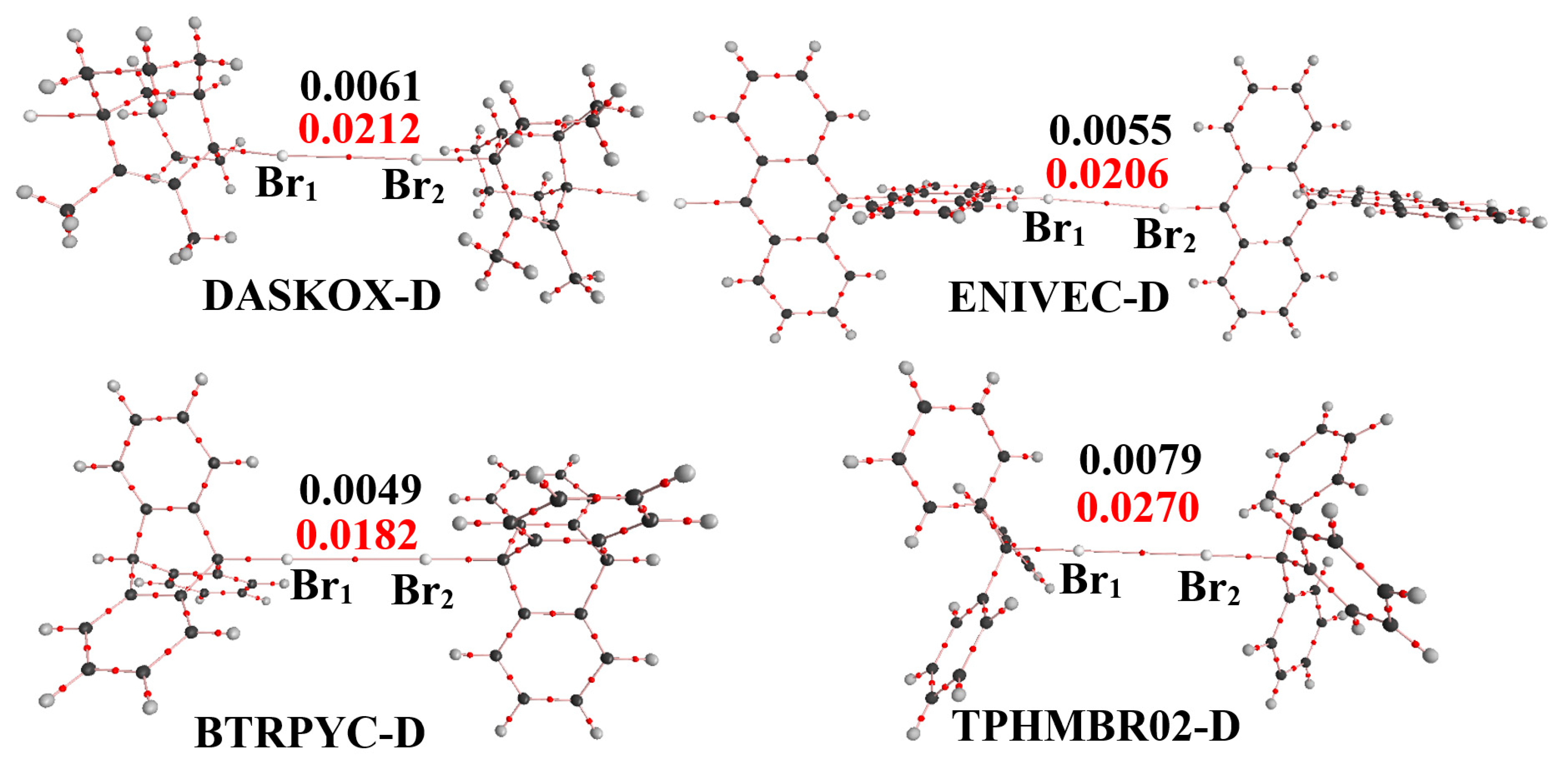
3.3. AIM Analysis
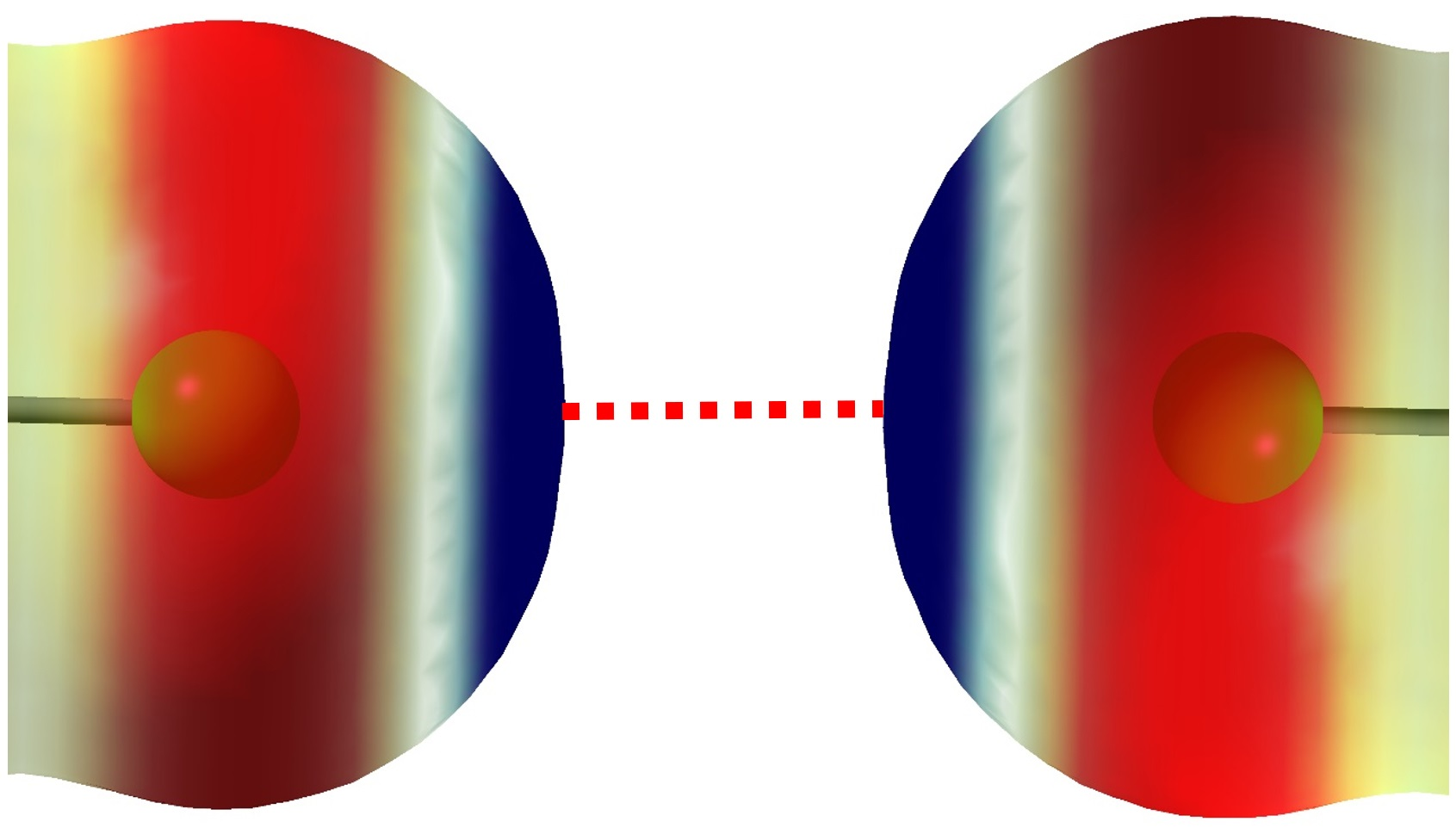
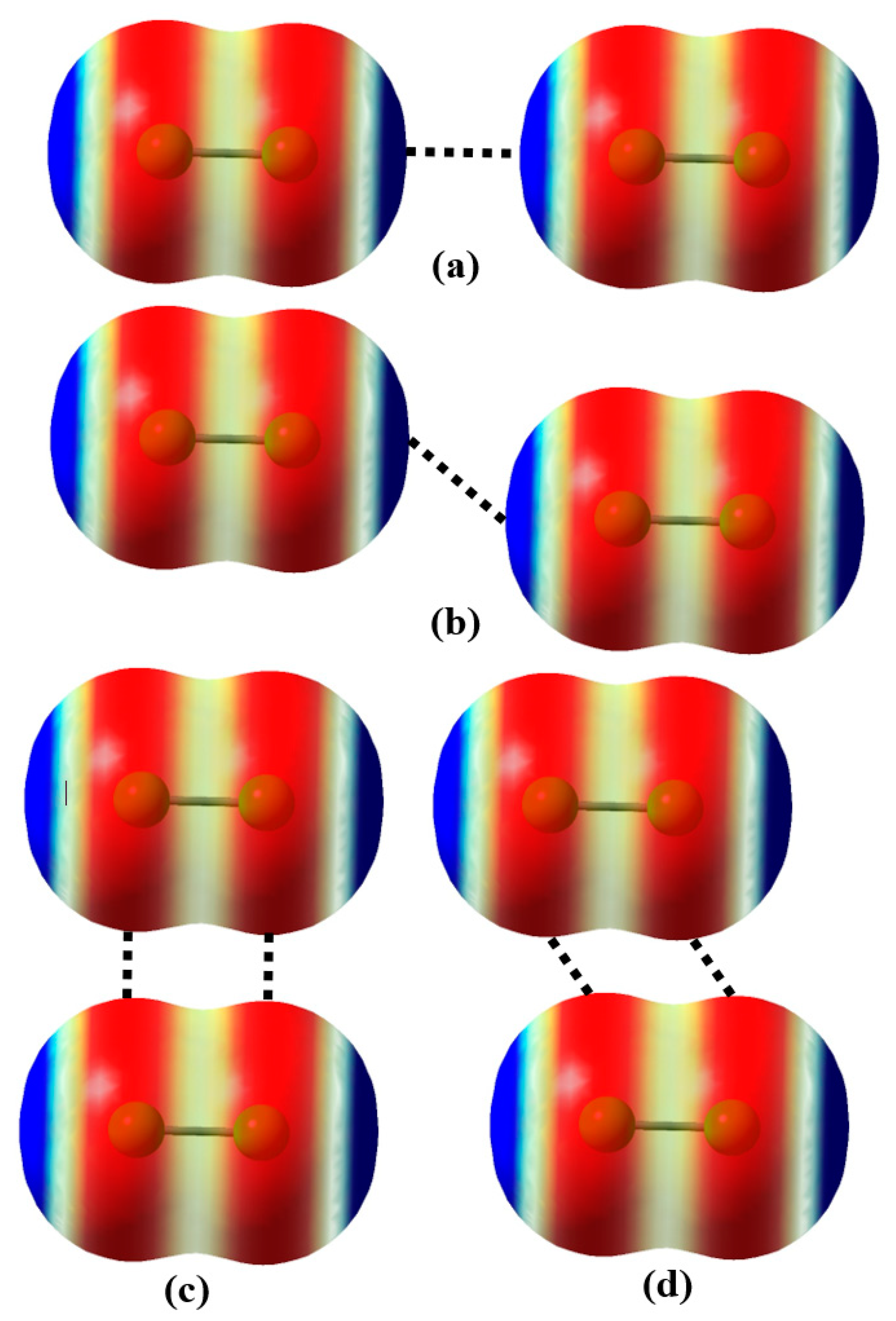
3.4. Confinement of the Face-to-Face σ-Hole⋯σ-Hole Stacking Interaction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-Holes and Electrostatically-Driven Interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef]
- Turunen, L.; Erdélyi, M. Halogen Bonds of Halonium Ions. Chem. Soc. Rev. 2020, 49, 2688–2700. [Google Scholar] [CrossRef]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen Bonding—A Novel Interaction for Rational Drug Design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef]
- Gao, L.; Zeng, Y.; Zhang, X.; Meng, L. Comparative Studies on Group III σ-Hole and π-Hole Interactions. J. Comput. Chem. 2016, 37, 1321–1327. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. On the Ability of Pnicogen Atoms to Engage in both σ and π-Hole Complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25, 152. [Google Scholar] [CrossRef] [Green Version]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Montero-Campillo, M.M.; Sanz, P.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Alkaline-Earth (Be, Mg and Ca) Bonds at the Origin of Huge Acidity Enhancements. Phys. Chem. Chem. Phys. 2018, 20, 2413–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W. The σ-Hole⋯σ-Hole Stacking Interaction: An Unrecognized Type of Noncovalent Interaction. J. Chem. Phys. 2020, 153, 214302. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond. Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond. Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Doering, W.v.E.; Schmidhauser, J.C. Synthesis of Doubly Orthogonal Hexa-1,3,5-trienes: 11,12-Dimethylbicyclo[5.3.2]dodeca-1,6,11-triene. J. Am. Chem. Soc. 1984, 106, 5025–5026. [Google Scholar] [CrossRef]
- Sarkar, M.; Samanta, A. 10,10’-Dibromo-9,9’-bianthryl. Acta Cryst. 2003, E59, o1764–o1765. [Google Scholar] [CrossRef]
- Palmer, K.J.; Templeton, D.H. Crystal and Molecular Structure of 1-Bromotriptyeene BrC20H13. Acta Cryst. 1968, B24, 1048–1052. [Google Scholar] [CrossRef]
- Dunand, A.; Gerdil, R. X-ray Structure and Crystal Packing Analysis of Triphenylbromomethane, C19H15Br. Acta Cryst. 1984, B40, 59–64. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-Hole Revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Noncovalent π⋯π Interaction between Graphene and Aromatic Molecule: Structure, Energy, and Nature. J. Chem. Phys. 2014, 140, 094302. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene⋯Naphthalene Complex: A more Challenging System than the Benzene Dimer for newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6⋯C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Li, M.M.; Wang, Y.B.; Zhang, Y.; Wang, W. The Nature of the Noncovalent Interactions between Benzene and C60 Fullerene. J. Phys. Chem. A 2016, 120, 5766–5772. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000-A Program to Analyze and Visualize Atoms in Molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Hohenstein, E.G.; Sherrill, C.D. Density Fitting and Cholesky Decomposition Approximations in Symmetry-Adapted Perturbation Theory: Implementation and Application to Probe the Nature of π-π Interactions in Linear Acenes. J. Chem. Phys. 2010, 132, 184111. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Parrish, R.M.; Sherrill, C.D.; Turney, J.M.; Schaefer, H.F. Efficient Evaluation of Triple Excitations in Symmetry-Adapted Perturbation Theory via Second-Order Møller-Plesset Perturbation Theory Natural Orbitals. J. Chem. Phys. 2011, 135, 174017. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Wavefunction Methods for Noncovalent Interactions. WIREs Comput. Mol. Sci. 2012, 2, 304–326. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E., III; Hohenstein, E.G.; Bozkaya, U.; Sokolov, Y.A.; Di Remigio, R.; Richard, R.M.; et al. PSI4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. A 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Duan, J.; Sherrill, C.D. Origin of the Surprising Enhancement of Electrostatic Energies by Electron-Donating Substituents in Substituted Sandwich Benzene Dimers. J. Am. Chem. Soc. 2011, 133, 13244–13247. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Rebek, J., Jr. Confined Molecules: Experiment Meets Theory in Small Spaces. Q. Rev. Biophys. 2020, 53, e6. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.-U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. (Ed.) Handbook of Chemistry and Physics, 87th ed.; CRC: Boca Raton, FL, USA, 2006. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Component | DASKOX-D | ENIVEC-D | BTRPYC-D | TPHMBR02-D |
|---|---|---|---|---|
| Etot | −0.95 | −1.24 | −1.59 | −1.19 |
| Eelst | −0.41 | −0.25 | −0.41 | −0.61 |
| Eexch | 1.96 | 1.29 | 1.13 | 3.01 |
| Eind | −0.43 | −0.41 | −0.31 | −0.81 |
| Edisp | −2.07 | −1.87 | −2.00 | −2.79 |
| Eelst% a | 14% | 10% | 15% | 15% |
| Eind% a | 15% | 16% | 11% | 19% |
| Edisp% a | 71% | 74% | 74% | 66% |
| Br2 | Br2⋯Br2 | (Br2⋯Br2)@C120 | |
|---|---|---|---|
| r | 2.2817 (2.2811) a | 2.2803 | 2.2882 |
| d | 3.8296 | 3.1112 | |
| ΔG | +10.90 | −77.84 | |
| EHOMO | −8.10 | −8.14 | −5.47 (−5.46) b |
| ELUMO | −3.51 | −4.01 | −4.55 (−3.85) b |
| Eg | 4.59 | 4.13 | 0.92 (1.61) b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, W. The Face-to-Face σ-Hole⋯σ-Hole Stacking Interactions: Structures, Energies, and Nature. Crystals 2021, 11, 877. https://doi.org/10.3390/cryst11080877
Zhang Y, Wang W. The Face-to-Face σ-Hole⋯σ-Hole Stacking Interactions: Structures, Energies, and Nature. Crystals. 2021; 11(8):877. https://doi.org/10.3390/cryst11080877
Chicago/Turabian StyleZhang, Yu, and Weizhou Wang. 2021. "The Face-to-Face σ-Hole⋯σ-Hole Stacking Interactions: Structures, Energies, and Nature" Crystals 11, no. 8: 877. https://doi.org/10.3390/cryst11080877
APA StyleZhang, Y., & Wang, W. (2021). The Face-to-Face σ-Hole⋯σ-Hole Stacking Interactions: Structures, Energies, and Nature. Crystals, 11(8), 877. https://doi.org/10.3390/cryst11080877